1. Introduction
The discovery in 1997 of the so-called “giant magnetocaloric effect” (GMCE) in Gd
5Si
2Ge
2 [
1] triggered a true explosion of the research effort, not only on stronger, efficient and less expensive materials, but also on magnetic refrigeration systems, which eventually could replace the conventional refrigeration machines based on the compression-expansion cycles of a fluid. Today, the most promising materials can be grouped into a few families of compounds, each one belonging to a common crystal structure and including chemically similar elements in each group. Among them, we can mention the compounds derived from MnAs [
2,
3], MnFeP
1−xAs
x [
4] and LaFe
13−xSi
x [
5]. In a conventional ferromagnetic or paramagnetic material, the maximum entropy reduction on an applied magnetic field is
kB ln(2
S + 1) per magnetic atom. This corresponds to the change from a fully disordered state with random orientation of spins at zero field to a fully ordered state with spins aligned under a strong magnetic field. The GMCE compounds have a higher entropy reduction than conventional materials for a moderate field, because they often undergo a first-order structural transition from a low magnetization phase, which we call non-magnetic and is usually paramagnetic, to a ferromagnetic one. This transition occurs spontaneously at some temperature and can be induced by a moderate external magnetic field above this temperature. However, the magnetization of the ferromagnetic phase is not near saturation at the practical temperatures for room temperature refrigeration, so that the maximum magnetic entropy change that can be induced is below the theoretical limit.
The entropy reduction is not necessarily limited to the magnetic entropy. This limit can be overcome, because the total entropy is a sum of three contributions:
where
Sm is the magnetic contribution, coming from the population of the unpaired spin states, essentially due to the inner electrons,
Se is the electronic contribution, due to the conduction electrons, and
Sph is the phonon contribution, due to the atomic vibrations in the lattice. In a first-order transition, the entropy is discontinuous, and the more stable phase is the one with the lowest free energy for a given temperature and field. An external field reduces the free energy of the magnetic phase by −
MB, which allows increasing its stability at temperatures at which the stable phase is non-magnetic in the absence of the field. The total entropy change produced by a field change at constant temperature is denoted Δ
ST. It is quite often and wrongly called “magnetic entropy change”; but actually, all of the three terms in
Equation (1) contribute, and sometimes, Δ
Sm is not even the most relevant term. On increasing the external field in a paramagnetic substance, Δ
Sm < 0, because the magnetic dipoles tend to order orienting towards the field direction, but Δ
Se and Δ
Sph can be positive or negative.
Compounds exhibiting an inverse magnetocaloric effect [
6] (IMCE) behave differently upon a field change, because the ferromagnetic phase occurs above the zero field transition temperature,
Tt, and consequently, the total entropy is higher than that of the non-magnetic phase. Below
Tt, an external magnetic field applied isothermally can convert the non-magnetic phase into a magnetic one, with a positive entropy increment that corresponds to Δ
ST > 0, with Δ
Sm ≤ 0 and Δ
Se + Δ
Sph > 0. Both, the phonon and the electronic entropy changes can give important contributions to Δ
ST. Strong magnetostructural correlations have been reported in these Heusler alloys [
7], and sharp increases in the electrical conductivity have been found in the transition to the ferromagnetic austenite [
8], indicating a high increase in the density of electronic states at the Fermi level. A study case with a main electronic entropy term is Mn
3GaC. In this compound, the low temperature phase, with no neat spontaneous magnetization, is magnetically ordered. Δ
Sm at the transition is very small, and consequently, Δ
ST is nearly independent of the field if it reaches a threshold value [
9], which, in turn, increases when the temperature decreases from
Tt. The consequence is the very interesting feature that, near
Tt, even a low magnetic field can produce the full entropy change, while for typical or, even, for GMCE compounds, a strong magnetic field is necessary to produce a significant entropy change.
The Heusler alloys, Ni
50Co
yMn
25+z−yM
25−z, with M = Sn, In, Sb, offer the possibility of having a similar behavior to Mn
3GaC, but near room temperature and with a stronger entropy change. They are derived from the stoichiometric Ni
2MnGa and have been widely studied, because of their property of shape memory controlled by the application of magnetic field. On heating, these compounds undergo a first-order transition from a low symmetry phase called “martensite” (M) to another cubic phase called “austenite” (A) at a temperature
TMA. This transition has a large latent heat and, consequently, a strong entropy increment, typically near 25–30 J/kg·K. On the other hand, the transition is usually broad and has a wide thermal hysteresis, with the reverse transition at
TAM < TMA, so that four temperatures are given as characteristic parameters: the M to A starting transition temperature on heating,
As, the finishing temperature of this transition,
Af, the A to M starting transition temperature,
Ms, and its finishing temperature,
Mf, with
Mf < TAM < Ms and
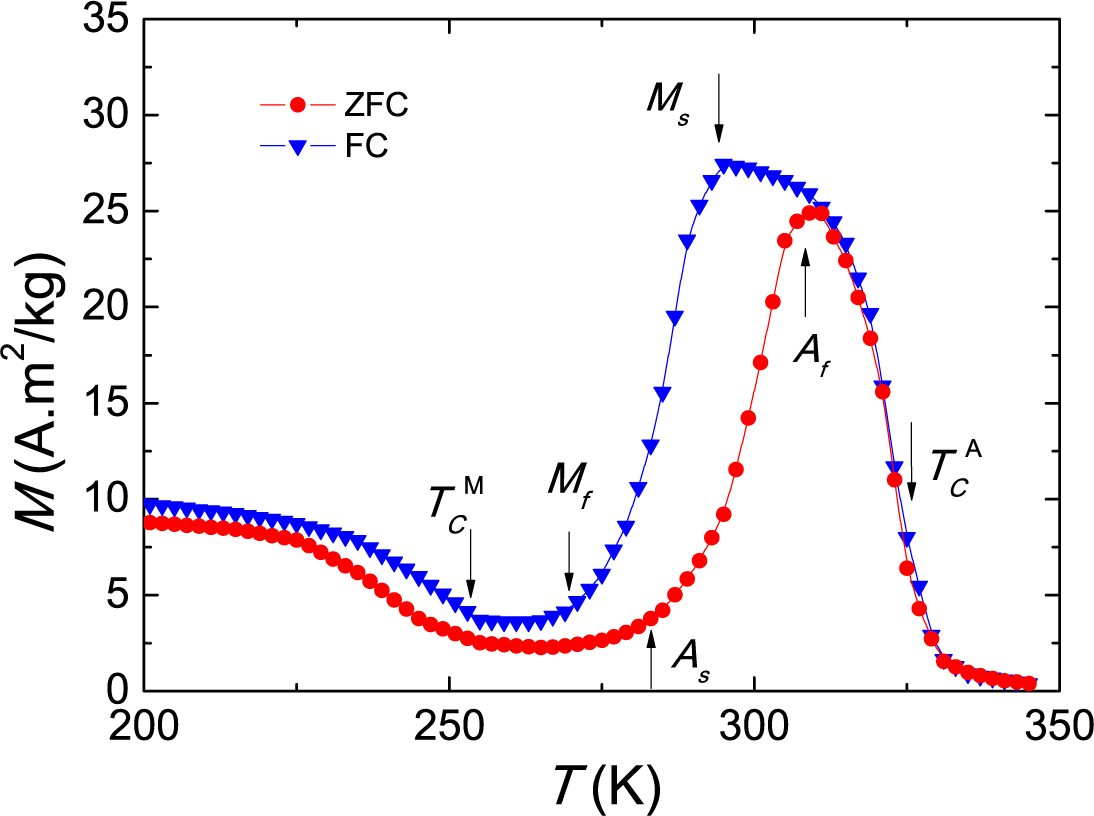
As < TMA < Af. In addition to this, the M phase has a lower magnetization than the A phase, as seen in
Figure 1. The respective Curie points are denoted as
and
. For a general picture of the dependence of these temperatures with the composition, we refer to the compositional phase diagram given in [
6,
10]. While
Ms and
Mf decrease and
increases strongly on decreasing the Sn content,
remains nearly constant. We chose the value
z = 12 in order to get
. On the other hand, the addition of a small amount of Co increases
and decreases
Ms and
Mf [
11,
12]. The parameter
y = 1 has been chosen with the aim of having
(
Figure 1). This would allow one to have a strong IMCE for the transition between paramagnetic martensite and ferromagnetic austenite when a field is applied to the martensite phase between
and
As.
Actually, a strong IMCE has been reported in several compounds, but the various results do not agree and are even contradictory, depending on the technique used to determine Δ
ST and on the experimental protocol. These protocols, taken as the sequence of fields and temperatures applied to the sample, are not usually reported in detail. For instance, in Ni
43Mn
46Sn
11, [
13] reports, for a field change from 2 T to 5 T, a maximum Δ
ST,max = 2.6 J/kg·K at 184 K from heat capacity data, but Δ
ST,max = 46.9 J/kg·K at 190 K from isothermal magnetization computed via the Maxwell relation. For Ni
50CoMn
36Sn
13, Δ
ST,max ≈ 12 J/kg·K from magnetization at constant fields on heating and on cooling, for a field change from 0 to 2 T [
14], but the Clausius–Clapeyron equation predicts Δ
ST,max ≈ 26 J/kg·K. Similar discrepancies between the results obtained from heat capacity measurements and via the Maxwell relation have been reported for Ni
46Cu
4Mn
38Sn
12 and Ni
50CoMn
34In
15 [
15]. The direct measurements of the adiabatic temperature increment, Δ
TS, gave no inverse MCE on cooling and obtained inconsistent values with those on heating [
14]. The importance of the hysteresis and the influence of the measurement protocols have been analyzed on this type of compound [
16]. For the close composition Ni
48Co
2Mn
38Sn
12, [
12] gives Δ
ST,max = 37.1 J/kg·K from isothermal magnetization, for a field change of 5 T.
The aim of the present work is to compare the existing results for ΔST and ΔTS with new directly measured values and results deduced from heat capacity at constant fields, CB, on the chosen compound, Ni50CoMn36Sn13. The experimental protocols are carefully considered in all cases, and the different results are explained considering the characteristics of the sample and the details of the protocols used in the determinations.
2. Measurement Protocols and Experimental Effects
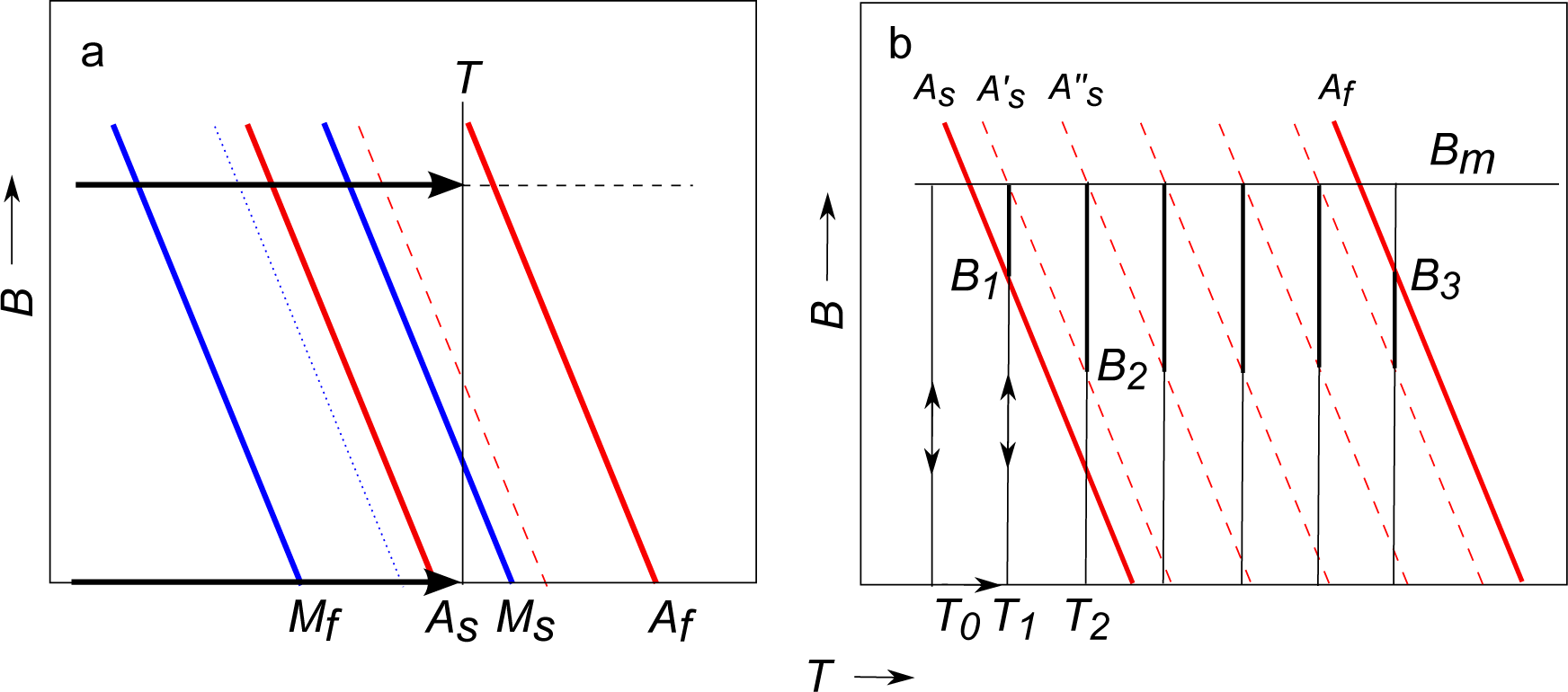
A scheme of a
B −
T phase diagram near the martensitic transition region is depicted in
Figure 2a. The continuous red lines indicate the set of points
As and
Af for the M to A conversion and the blue lines the set of points
Ms and
Mf for the transition from A to M. The specific entropy of the sample at a given temperature,
T, and field,
B, is
S(
T, B, x) =
xSA(
T, B) + (1 −
x)
SM(
T, B), being
SA, SM the specific entropies of the austenite and martensite phases and 0 ≤
x ≤ 1 the fraction of austenite. Due to the hysteresis,
x and the total entropy depend not only on
T and
B, but also on the path followed by the state point to reach a particular point in the
B −
T diagram. Therefore, the true isothermal entropy difference, Δ
ST, between two states at the same
T and different
B depends on the protocol followed by the sample to reach both states.
The sample is composed of many grains and domains, each one having slightly different compositions, microstructures or sizes, which, in turn, cause different transition temperatures,
TMA and
TAM, since they are very sensitive to the proportion Mn/Sn and their site occupancy. On the other hand, the specific latent heat for every grain is practically constant, which implies that the transition lines are nearly straight lines with the same slope for all grains, due to the Clausius–Clapeyron equation. This means that the fraction
x depends only on
T and
B for every state reached in any process involving phase conversion in only one direction, M to A or A to M. The curves of constant
x are parallel straight lines, as indicated by the red dashed lines in
Figure 2 for heating or magnetization processes. Similarly, on cooling or demagnetization processes, the constant
x curves are represented by the blue lines. The experimental phase diagram of a small piece of our compound was given in
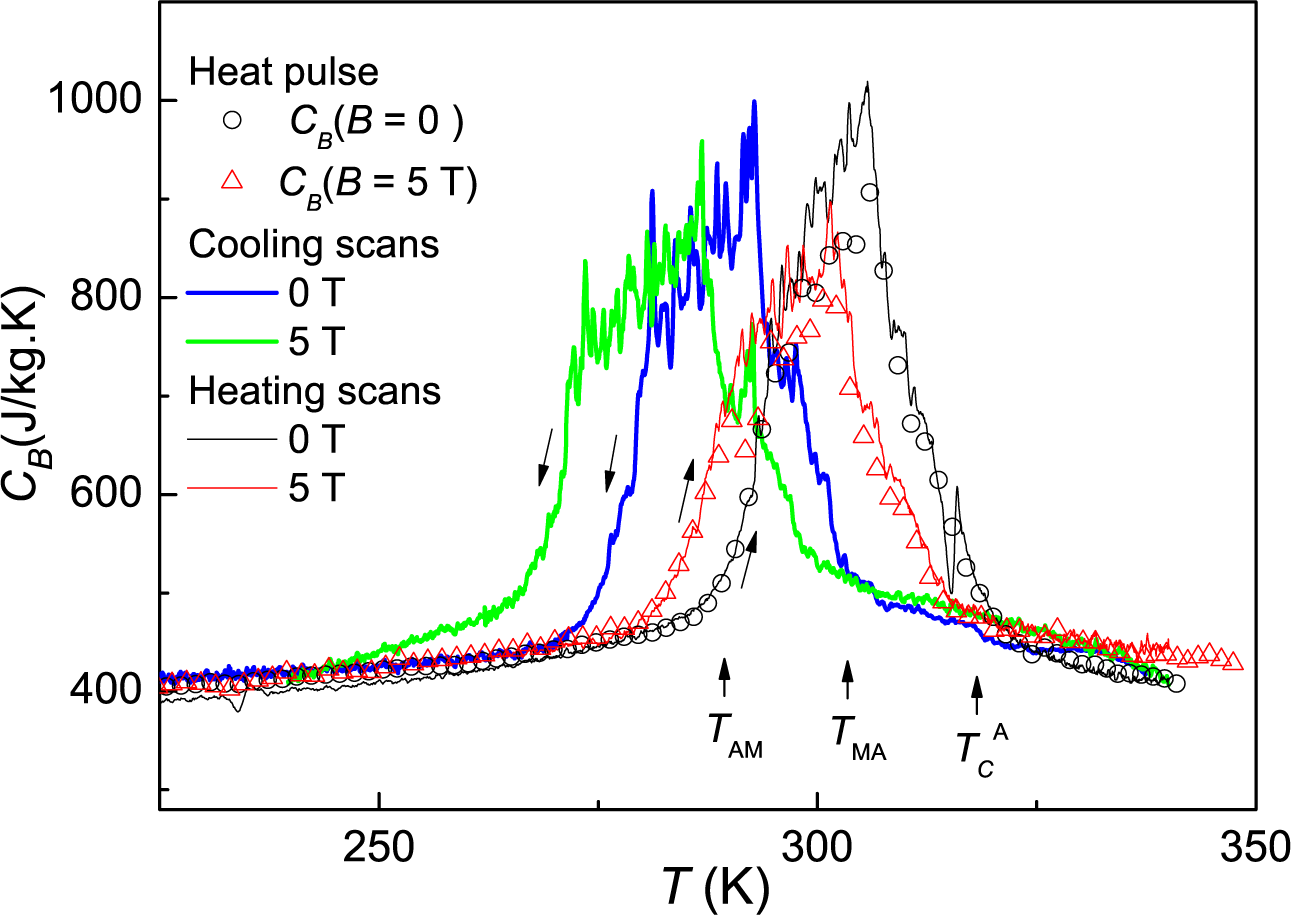
Figure 6 of [
14]. For the present sample, the width of the transition bands is broader and the transition temperatures slightly higher.
We will consider several processes, frequently used in experiments, represented by the evolution of a state point in the phase diagram. We will discuss the expected behavior for each protocol, which is applicable to compounds having a martensitic transition. This general analysis will be applied to our experimental results on Ni50CoMn36Sn13, comparing direct measurements of ΔST with values deduced from heat capacities on heating and cooling and results from isofield magnetization data.
2.1. Protocol a: Isofield Heating
The simplest protocol is heating at a constant field starting from a low temperature. This is used in heat capacity determinations, as is indicated by the arrows at zero and
B fields in
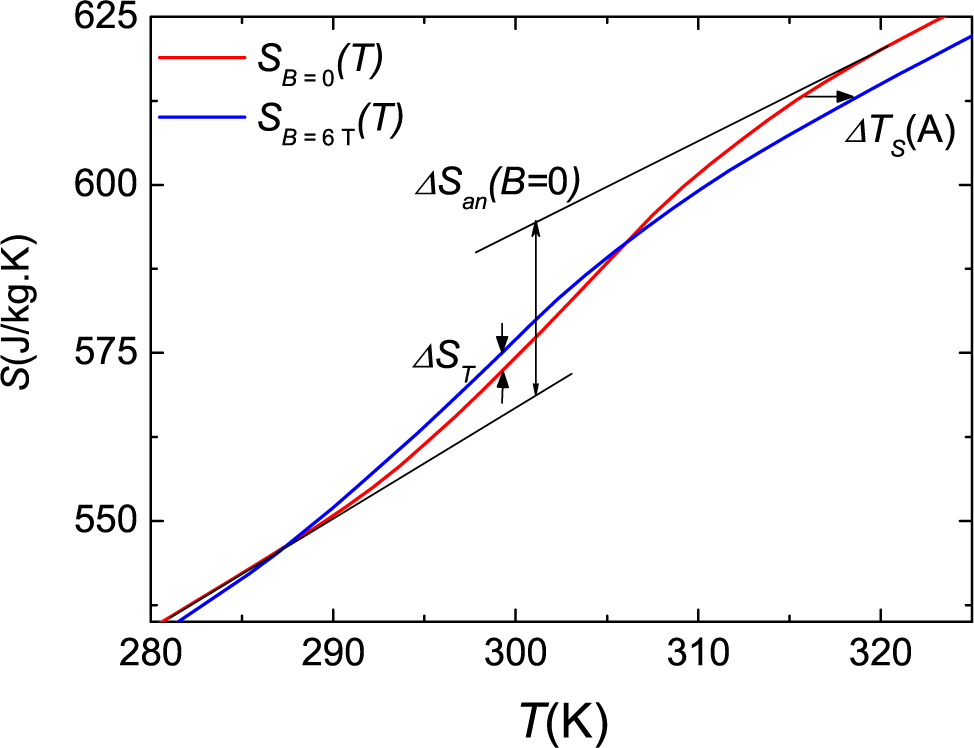
Figure 2a. The integration of
CB/T gives the entropy increment with respect to some arbitrary temperature of reference,
T0. Knowing the entropy difference for different fields at
T0 from direct measurements, as seen below in protocol
c, one can compute the entropies at any temperature and field and calculate the difference. The values for Δ
ST are obtained as the entropy difference between the sample heated at a field
B and the sample heated at zero field. This corresponds to the isothermal entropy increment upon increasing the field from zero to
B for the sample previously heated up to
T at
B = 0.
Leaving apart experimental errors, this procedure introduces other small errors when going through first-order transitions due to the irreversibility of the hysteretic processes. There is an irreversible entropy production at the transition, and the supplied heat is lower than the product of the entropy increment times the temperature. The irreversible entropy is proportional to the thermal hysteresis
δSi/Δ
ST = (
TMA −
TAM)/(
TMA +
TAM), as given in [
17], being in our compound around 0.025, considering the values reported below in
Table 1. Therefore, this is a small correction in the present case, where huge differences are observed among the results from different ways of deducing Δ
ST.
This protocol can also be used for magnetization measurements. The procedure to determine the entropy change is to compute numerically (
∂M/∂T)
B at different fields. Then, Δ
ST is computed by numerical integration of the Maxwell relation:
giving the entropy increment for an isothermal process, from magnetization data at constant
B values. In a real isothermal process along the transition region, for each field increment, δ
B, a small part of the sample undergoes the transition from M to A. The remaining part changes its entropy according to the normal MCE effect for a mixture formed by the phase fractions (1 −
x) of M and
x of A. The Maxwell relation gives exactly the entropy increment for the non-converted fractions. The finite difference approximation for the partial derivative is mathematically equivalent to the classical Clausius–Clapeyron equation for the phase converted fraction. However, this classical equation must be modified for a first-order transition according to the following equation given in [
6]:
where (
Tt, Bt) is the transition point for each particular sample grain and Δ
M, Δ
S are the corresponding jumps of magnetization and entropy, respectively, at this point. The irreversibility affects this equation through the derivative of the dissipated energy,
Ediss. This term is claimed to be negligible for Heusler alloys [
6]. It cannot be easily measured from the isothermal hysteresis loop of the magnetization, because the required magnetic field for the realization of fully reversible M to A conversion would be about 45 T at the most favorable temperature of 270 K for the title compound, as discussed later, taking into account the hysteresis, the width of the transition and the field dependence of
Tt. The dissipated energy can be estimated through the hysteresis loop of the entropy as one half of ∮
S(
T)
dT, being this integral approximately the product of the entropy jump times the hysteresis. The derivative amounts to about 10% of Δ
S. Therefore, the entropy increments can be corrected using the modified Clausius–Clapeyron
Equation (3), rather than the classical one, but it is also a minor correction compared to errors of orders of magnitude and even of sign for the case studied here and will not be considered, for the sake of simplicity.
A process of continuous cooling at a constant field is completely similar to the previous process, but the transition occurs between the blue lines of
Figure 2a.
2.2. Protocol b: Isothermal Magnetization
This is the procedure most frequently used to obtain magnetization data.
Figure 2b shows the path followed by the state point. The detailed procedure starts with the sample at low temperature and zero field. The field is increased isothermally up to some maximum value
Bm and then decreased again to zero. Meanwhile,
M is measured while changing
B on magnetization and on demagnetization. Then, the sample is heated at zero field to the next temperature and the procedure repeated. For the state evolution sketched in
Figure 2b following this protocol, the results of Δ
ST correspond to the normal MCE of the martensite up to a temperature
T0, where the last magnetization process without phase transformation takes place. At the next temperature,
T1, a fraction of the M phase converts into A when the state point goes into the transition band, limited by the red solid lines, which occurs for fields above
B1 in the upper part of the isotherm indicated by a thicker segment in
Figure 2b. The resulting Δ
ST is the sum of the normal negative MCE of the mixed martensite and austenite phases plus the positive transition entropy of the converted fraction of sample along the band between the
As and
lines.
On demagnetization at
T1, the converted A fraction does not transform back into the M phase unless
Bm is stronger than 15 T, as will be seen later, since the inverse transition of this fraction takes place along a band like the previous one displaced 15 K below, due to the thermal hysteresis. Therefore, on demagnetization, the true entropy variation corresponds to the normal MCE of the A + M mixed phase. Then, the sample is heated at zero field to a higher temperature,
T2, where it is magnetized and demagnetized again. At this new temperature, the phase conversion does not start when the state point enters the full transition band, but at
B2, when it reaches the transition line of the last sample grains converted to the A phase in the previous isotherm. Consequently, only a small fraction of the sample is converted at each isotherm. In the case of usual GMCE compounds, there is a narrow transition band width resulting in a total phase conversion for almost every temperature through the transition region, both on magnetization and on demagnetization, and giving Δ
ST independent of the temperature interval
T2 −
T1. However, for broad transitions, as happens in the present compound, the higher the temperature interval, the greater the converted fraction. It is also higher if the transition band for the whole sample is narrower. For equal intervals, the field at which the phase conversion starts,
B2, is approximately the same for all isotherms, and
Bm −
B2 is independent of
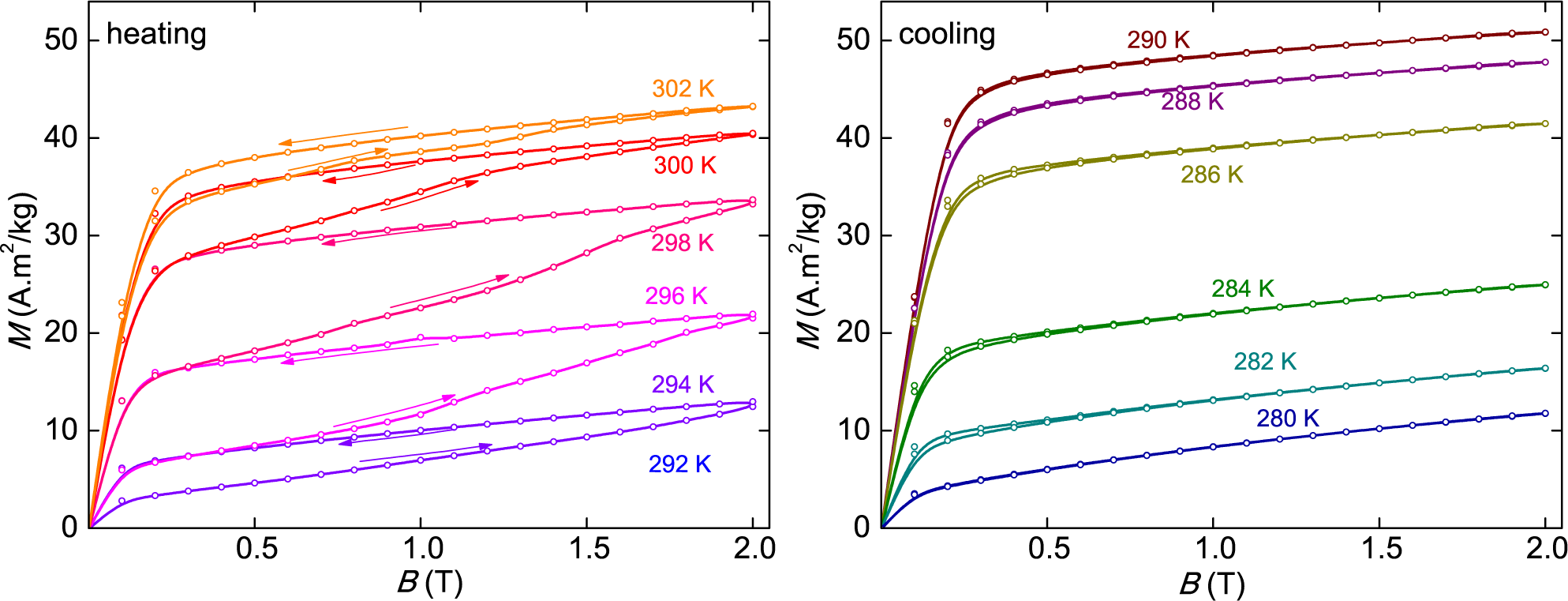
Bm. This behavior of the present sample was clearly observed in our magnetic measurements, shown in
Figure 3 (heating), and it is even more evident in the magnetization of the closely-related compound, Ni
50Mn
39.5In
10.5, taken at smaller temperature intervals, but reported without a correct explanation in
Figure 3 of [
18]. The measurements reported in that study indicate that the magnetization curve at each temperature, between 200 K and 210 K, overlaps the demagnetization curve of the previous temperature up to
B2 ≈ 9 kOeand then goes to higher values between
B2 and
Bm. Considering that the sample, structurally being in the state of M, A or in a mixture of both states, has normal MCE and that only a small portion suffers phase conversion in each isotherm, the resulting Δ
ST can be negative or positive, depending on the temperature interval and the width of the transition band. In any case, Δ
ST is much lower than the total transition entropy jump for the sample determined from heat capacity or from the Clausius–Clapeyron equation.
Finally, the phase conversion ends when the state point exits the transition band, when crossing the Af line at B3. At the subsequent temperatures, the entropy variation corresponds to the normal MCE of the austenite.
It is worth remarking that, according to this analysis, in the similar protocol made on cooling, there should not be any phase conversion in the magnetization-demagnetization process. The transition takes place only on the cooling step at zero field, between the blue lines of
Figure 2a. Our magnetization measurements shown in
Figure 3 (cooling) give overlapping curves on the increasing and decreasing field, without any additional change in the fraction of M due to phase conversion, corroborating these arguments. This explains why in the direct measurement of Δ
TS, given in [
14] and made always upon application of a field in a magnetization process, an IMCE is observed on heating, but a normal MCE is seen on cooling.
2.3. Protocol c: Heat Absorption on Isothermal Demagnetization
This is used for direct determinations of the absorbed heat on demagnetization, Q, to obtain the isothermal entropy change as −ΔST ≈ Q/T. This physical process is similar to protocol b, but the temperature increases are made at a constant field, Bm. Then, the field is decreased isothermally while measuring the heat absorbed to maintain T constant. The field is increased again up to Bm and the sample heated to a new temperature. Like in protocol b, there is no phase conversion from A to M on demagnetization for the field values used, but only from M to A on magnetization and on further heating to the next temperature, while the heat exchange is not recorded. As the measured quantity is the heat absorbed on demagnetization, it corresponds to the normal MCE of the mixture of the A and M phases existing at the initial point (T, Bm). Therefore, ΔST < 0, taken with the usual convention of ΔST = S(T, Bm) − S(T, 0).
2.4. Protocol d: Adiabatic Magnetization Cycles
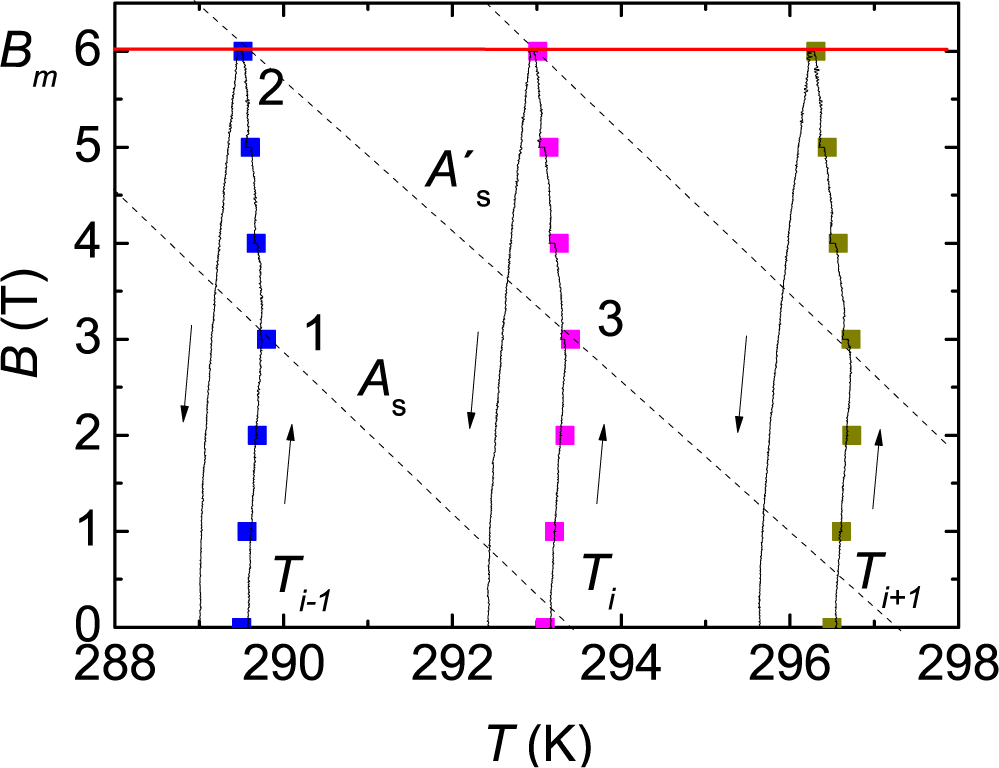
The field is increased and decreased adiabatically, and the temperature is recorded as a function of the field.
Figure 4 shows the experimental data in some processes, starting at various consecutive temperatures. The continuous lines represent the experimental dynamic processes at field rates of 0.01 T/s and −0.01 T/s, and the symbols correspond to equilibrium points as described in [
3]. In this case, when the field increases, the temperature initially increases due to the normal MCE of the pre-existent mixture of the M and A phases. Considering the adiabatic magnetization-demagnetization cycle starting at a temperature
Ti, as shown in
Figure 4, the trend changes when the phase conversion starts, which happens when the state point crosses the limiting transition line of the sample grains that did not transform in the previous magnetization runs and remained in the M state (Point 3 in
Figure 4). This line is defined by the conversion line passing through the state point reached at the maximum field of the previous magnetization process (Point 2). After that, the temperature decreases until reaching the maximum field or until the M to A phase conversion is complete. On decreasing the field, there is no phase conversion, and the slope
B −
T is always positive. This general behavior was also observed in the field cycles around the reverse martensitic transition [
19].
4. Effect of the Distribution of Transition Temperatures
In the case of compounds with normal GMCE, the application of a magnetic field induces the paramagnetic to ferromagnetic phase change at temperatures above the spontaneous zero field transition temperature, TC, and up to the transition temperature at the maximum applied field,
. Above
, the field is not strong enough to transform the sample to the ferromagnetic state. Considering that ΔST has a high value only when there is phase conversion, but is low in both the pure paramagnetic and the pure ferromagnetic phases, ΔST (T) would have a square shape for a sharp first-order transition, with a high plateau between TC and
. The up and down jumps are more or less abrupt depending on the homogeneity of the sample. However, the top of the square is essentially independent of the homogeneity, provided that it corresponds to a complete phase transformation. Moreover, ΔST at the plateau must be equal on magnetization and on demagnetization, on heating and on cooling, because S is a state function, and its variation in a closed cycle is zero, since the sample returns to the same state. For inverse GMCE materials, there is a similar behavior, with the difference that the ferromagnetic state is the stable one at higher temperatures than the non-magnetic state.
For the present compound, there are significant differences. Considering the width of the transition, ≈ 30 K, and the field dependence of the transition temperature, ≈ −1 K/T, as seen in
Figure 6 and
Table 1, a field stronger than 30 T would be necessary to achieve the complete M to A phase conversion starting with the sample in the M state. For most practical accessible fields, the transition will always be partial on isothermal magnetization and similarly on demagnetization. The width of the transition band, given by the differences
Af −
As and
Ms−
Mf, is affected by inhomogeneities in composition and temperature, internal stresses and atomic disorder. In our sample, DSC experiments on a finely powdered portion of the sample did not show any relevant change of the transition width, discarding any significant influence of the stress.
The width of the transition band explains many apparent inconsistencies in determinations of the MCE parameters, in particular Δ
ST. Looking at
Figure 2b and considering the discussion of protocol
b, it is evident that a determination of Δ
ST on an increasing field will depend on the temperature step between consecutive measurements and the width of the transition band. The values obtained from isothermal magnetization measurements (protocol
b), using the Maxwell relation, give a spurious contribution, as described in [
22], but distributed along the whole transition band, where a partial phase conversion exists. On the other hand, the heat capacity data give precise values of the entropy change upon field changes, and the isofield magnetization measurements, on heating, give also correct results. Nevertheless, a comparison of the present heat capacity results (
Figure 8) with previous deductions from isofield magnetization [
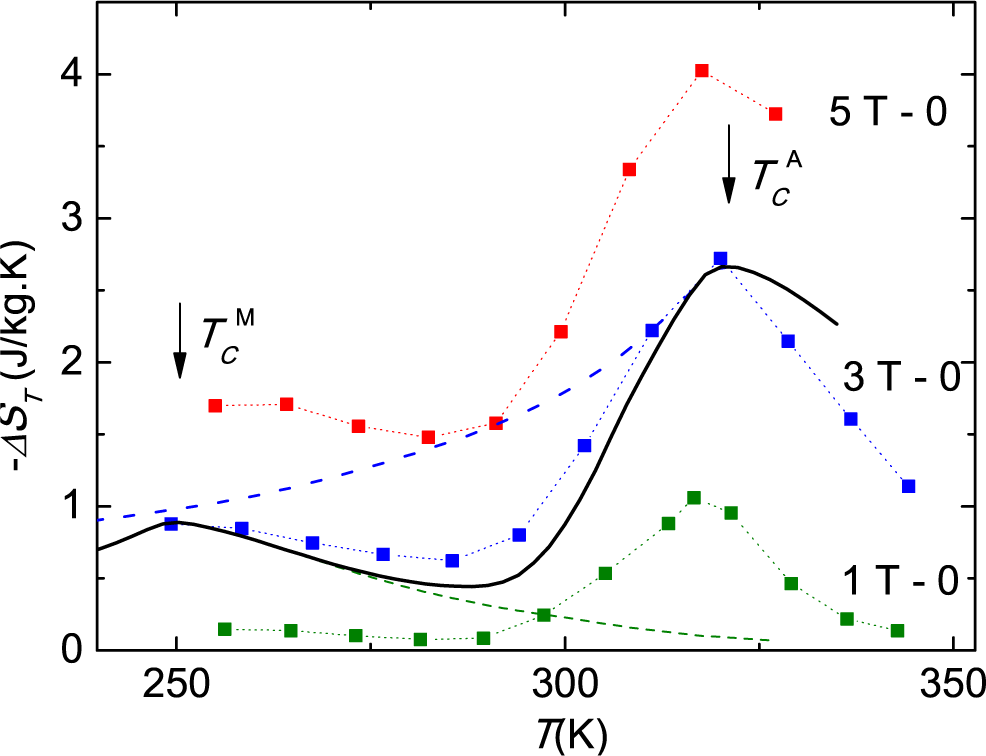
14] differ by one order of magnitude. This difference can be explained quantitatively considering the actual width of the transition for each of the samples used in the experiments. The work in [
14] reports a maximum Δ
ST (298
K) = +10.4 J/kg·K for a field increment of 2 T, calculated from isofield magnetization on heating. From the magnetization curves reported in
Figure 2 of this work, one can estimate a 40% conversion from M to A at 298 K for this field change. Considering this part of the total anomalous entropy for the M to A transition, given in
Table 1, and the negative contribution of the normal magnetic entropy change, shown in
Figure 5, one obtains Δ
ST (298
K) ≈ −0.7+0.40×30 = 11.3 J/kg·K, in good agreement with the value deduced from the magnetization measurements [
14]. The sample used in the present study is bigger and has a broader transition. Heat capacity determinations give only a maximum phase conversion of 20% for a field variation of 5 T, leading to much smaller entropy changes, as shown in
Figure 8a. Therefore, the contradiction between the quite different Δ
ST values obtained for different samples is only apparent and can be quantitatively explained considering the different transition band widths of the samples.
5. Conclusions
The apparently contradictory results for ΔST obtained from different techniques and samples have been explained when considering the path followed by the state point on the phase diagram, the measuring protocol, the hysteresis and the width of the transition for each sample.
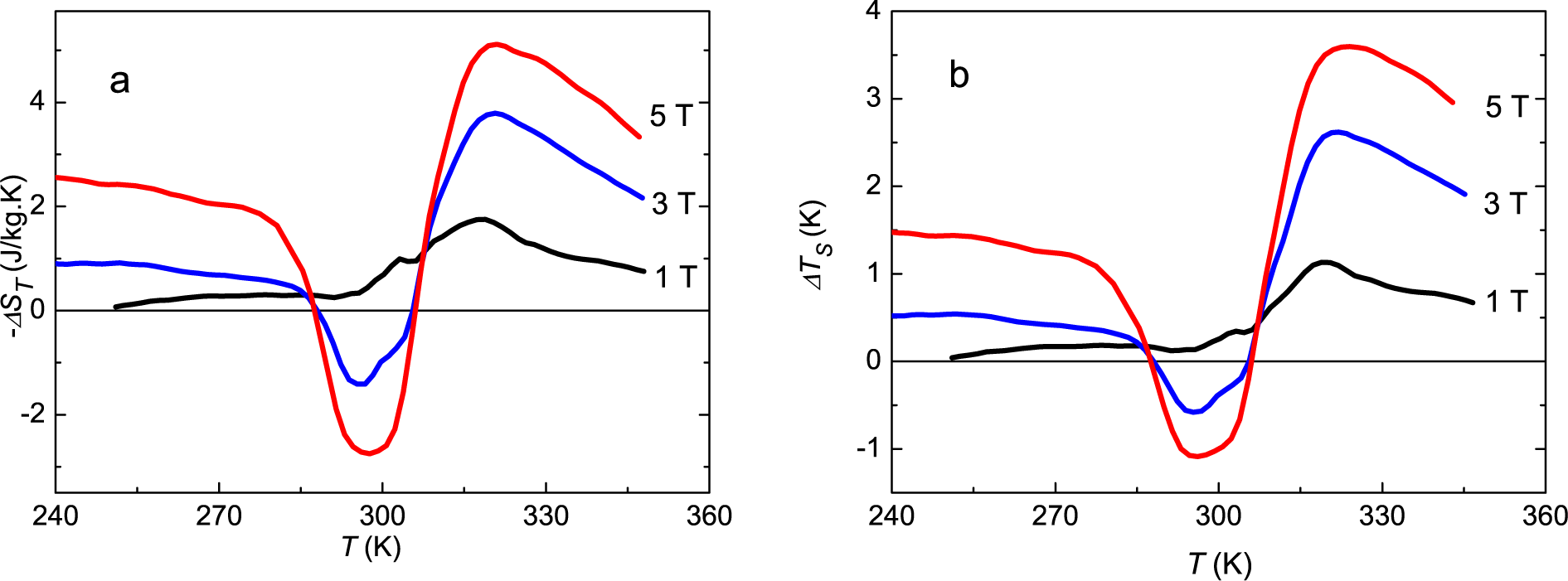
Due to the large hysteresis, the direct determination of the isothermal entropy change on demagnetization gives the opportunity to determine independently the purely magnetic entropy increment of the mixture of martensite and austenite at each temperature. In our sample, these direct determinations gave positive values for −ΔST at every temperature, like in compounds with normal MCE.
The heat capacity at a constant field allows determining the entropy change at the transition involving magnetic, phonon and electronic contributions. Any other experimental data, like magnetization, needs an explanation of the protocol followed in the experiments to be able to obtain a reliable interpretation of the results. In any case, the isothermal or adiabatic magnetization and demagnetization in the transition region induce a partial phase conversion, and the thermal effect depends on the fraction converted. Depending on the state path followed in a direct measurement of Δ
ST, a different value can be obtained for Δ
ST (296
K, Δ
B = 5 T) between −2.2 J/kg·K and +3.8 J/kg·K, corresponding to the absence of any part of the sample changing phase and to the maximum phase conversion for this field, respectively. The experimental value depends on the temperature step used in the series of measurements. The data in
Table 1 and in
Figure 5, giving results for the martensitic transition and for the magnetic entropy changes, allow one to predict the results for any other protocols with a fair approximation.
Ni
50CoMn
36Sn
13 and some other related Heusler alloys offer the opportunity to have a large entropy change with low applied fields under appropriate conditions [
23]. However, the actual compounds have serious drawbacks with regard to applications in magnetic refrigeration: (1) The sample conversion is never complete in moderate magnetic fields, due to the broad transition. Therefore, the entropy change is always lower, and, sometimes, even opposite in sign, than the expected change corresponding to the latent heat of the martensitic transition. (2) At temperatures at which the M to A transformation can be induced by a magnetic field, the large hysteresis prevents having the reverse transformation on demagnetization. Conversely, at temperatures at which the A to M transformation occurs on demagnetization, the applied field is not strong enough to achieve the M to A transformation. (3) Even if both previous difficulties were overcome with a very homogeneous sample having a sharp martensitic transition and a small hysteresis, the useful temperature span would be small. Due to the Clausius–Clapeyron equation and the relatively small magnetization change between the M to A phases compared to other GMCE materials, the transition line in a
B −
T diagram is very steep. Consequently, a given field increment produces a small shift of the martensitic transition temperature, allowing the occurrence of the transition and the GMCE only in a narrow temperature interval.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}