Estimation of Activity Interaction Parameters in Fe-S-j Systems
by
Tianhua Ju
1, 
Xueyong Ding
1,*,
Yingyi Zhang
2,*,
Weiliang Chen
1,
Xiangkui Cheng
3,
Bo Wang
1,
Jingxin Dai
1 and
Xinlin Yan
4,* 1
School of Metallurgy, Northeastern University, Shenyang 110004, China
2
School of Metallurgical Engineering, Anhui University of Technology, Maanshan 243002, China
3
Institute of International Vanadium Titanium, Panzhihua University, Panzhihua 617000, China
4
Institute of Solid State Physics, Vienna University of Technology, Wiedner Hauptstr. 8-10, 1040 Vienna, Austria
*
Authors to whom correspondence should be addressed.
Entropy 2018, 20(10), 808; https://0-doi-org.brum.beds.ac.uk/10.3390/e20100808
Submission received: 3 September 2018
/
Revised: 17 September 2018
/
Accepted: 10 October 2018
/
Published: 22 October 2018
(This article belongs to the Section Thermodynamics)
Abstract
:It is important to know the activity interaction parameters between components in melts in the process of metallurgy. However, it’s considerably difficult to measure them experimentally, relying still to a large extent on theoretical calculations. In this paper, the first-order activity interaction parameter () of j on sulphur in Fe-based melts at 1873 K is investigated by a calculation model established by combining the Miedema model and Toop-Hillert geometric model as well as considering excess entropy and mixing enthalpy. We consider two strategies, with or without using excess entropy in the calculations. Our results show that: (1) the predicted values are in good agreement with those recommended by Japan Society for Promotion of Science (JSPS); and (2) the agreement is even better when excess entropy is considered in the calculations. In addition, the deviations of our theoretical results from experimental values depend on the element j’s locations in the periodic table.
1. Introduction
Sulphur is one of the most detrimental impurity elements in metallurgy that typically causes the deterioration of hot ductility [1] and the degradation of the corrosion resistance [2] of steels. The content of sulphur in steels is normally required to be extremely low. “Inclusion engineering” [3] could be one of the ways to reduce the harmful effects of sulphur [4] with a relatively low cost. However, implementation of this technique needs to well understand the basic thermodynamics behavior of sulphur in iron-based melts.
The activity interaction parameter, which is first introduced by Wagner [5] in dilute solution to account for the effects of an added alloying element on the activity coefficient of a solute, provides more useful information in the process of metallurgy computation. Previously, only first-order activity interaction parameters had been considered in Wagner’s formalism, resulting in inadequacy to describe the behavior of solutions that are “not very diluted”. This phenomenon was observed by Lupis and Elliott [6], who then proposed an introduction of higher order interaction coefficients to the mathematical apparatus. Darken [7] also observed that the Wagner’s formalism was not suited to the non-dilute solution situationnand suggested a quadratic formalism by considering the first- and second-order activity interaction parameters. Pelton and Bale [8,9] further developed Darken’s quadratic formalism by introducing the unified interaction parameter (UIP) to the formalism. In UIP, the first-order interaction parameter is identical to Wagner’s first-order interaction parameter. Therefore, the activity interaction parameter is an extremely important and fundamental thermodynamics parameter in the fields of metallurgy and materials. In addition, the activity interaction parameters also bear significant correlations among properties such as the heat of formation of the corresponding oxides and atomic number of the deoxidants [10], as well as how the solubility of one element in a liquid metal is affected by the second solute [11]. Therefore, knowing the activity interaction parameter in the Fe-S-j systems is very important to understand the thermodynamic behavior of sulphur in steel.
The activity interaction parameter can be basically determined by experimental methods. However, it is practically impossible to determine all these parameters due to the large number of potential elements for combining systems and possible technical issues. As a result, theoretical methods become most attractive approaches. In theoretical works, two methods are deserved to be mentioned since in which, only few physical parameters of constituent elements are needed. One is proposed by Ding [12,13] and the other is developed by Ueno and Waseda [14]. Ding [12,13] at the early 1990s proposed a method that, through combing the Miedema model and geometric model as well as including other thermodynamics relations, established a model to predict the activity interaction parameter and infinite dilute activity coefficient in any metal-based melt. Almost at the same time, Ueno and Waseda [14] applied the pseudopotential formalism coupled with the free energy of a hard sphere model and built a model for the activity interaction parameter in metal-based melts. The former we called as Ding method and the latter as Ueno method. In the Ueno method, the final solution formula needs to improve because it does not satisfy the Lupis reciprocal relationship [6], i.e., This problem does not exist in the Ding method.
After the Ding method, many prediction models have been established based on it. For example, Fan [15] coupled Chou’s geometric solution model with the Miedema model, Wang [16] applied Toop’s geometric model as extending method, Zhang [17] combined the Miedema model and Chou’s geometric solution model and also included excess entropy, etc. The prediction capability of the Ding method totally relies on the Miedema model. In the past, Ding coupled the Miedema model with Toop-Kholer geometric model to calculate the activity interaction parameter of solutes in Fe-based [12,18], Cu-based [18], and Co-based [18] melts, respectively, and the predicted data are in good agreement with the experimental data. In these calculations, however, data on sulphur with other solutes are not included due to the fact the physical parameters (given by Miedema et al.) of sulphur which were given by Neuhausen [19] were not available until 2003. Thence, applying the Ding method to calculate the activity interaction parameter of sulphur with other solutes has become possible.
In iron-based melts, due to the importance of sulphur for the properties of steel, many activity interaction parameters of sulphur with other solutes have been determined experimentally. The results are compiled in “Thermodynamic Data for Steelmaking” [20] edited by the Japan Society for the Promotion of Science (JSPS). However, data on the activity interaction parameters of sulphur with some important elements such as Rh, Ru, Er, Os, Re, etc. are still missing. In addition, the experimental data are usually inconsistent from different sources. For example, the given by Inoue et al [21] is −9000, however, the value given by JSPS [20] is −515. Therefore, applying the theoretical method to predict the activity interaction parameter in Fe-S-j has practical significance.
In this work, the activity interaction parameters (in which the composition coordination is expressed in mass%) in Fe-based melts were calculated by establishing a model based on the Ding method. Although many models based on the Ding’s method for activity interaction parameter calculations have been established, most of them have problems in use. The models coupled with Chou’s model [15,17], for instance, have the problem that the similarity coefficient is difficult to obtain. The models combined with the Toop/Toop-Kholer geometric model, such as Ding’s model [12] and Wang’s model [16], have mathematical difficulties in the deduction process when the solvent is chosen as an asymmetric component and one has to resort to other geometric models. For this reason, in our present work, we adopted the Toop-Hillert geometric model [22] in our model establishment.
2. Calculation Method
2.1. Basic Relations
In a ternary system, i-j-k, k is a solvent, the activity interaction parameter can be expressed as:
and:
where R and T are the gas constant and absolute temperature, respectively; is the activity interaction parameter of j on i that the composition coordinate is in a molar fraction; and are the excess Gibbs free energy and excess entropy, respectively; ΔH is the mixing enthalpy of solution.
Generally, the thermodynamics properties of a multi-component system are obtained from all the sub-binary systems with an assigned probability weights, which is called geometric model method, as follows:
Therefore, when the excess Gibbs free energy of the binaries is available, the excess Gibbs free energy of the i-j-k system, gE, can be obtained, and then the activity interaction parameter can be calculated.
In liquid binary alloys, a satisfactory equation relating SE and ΔH has been deduced by Tanaka et al. [23], based on the free volume theory and excess volumes of the alloys, as follows (supposing and i-j binary alloy):
where and are the melting points of pure elements A and B under the standard pressure respectively. Therefore, if:
then:
2.2. Miedema Model
In a binary system i-j, the mixing enthalpy ΔHy can be obtained from the Miedema model [24,25,26], which was proposed by Miedema and his colleagues for estimating the heat of formation of solid or liquid metal alloys. For simplicity, the equation is deduced as follows:
where:
where, is the atom fraction; V, , and are the basic physical parameters of elements, representing mole volume, electron density, and electronegativity, respectively; p, q, and βij are the empirical parameters defined by Miedema, and their data are correlate to the constituents. The values of molar volume, electron density, and electronegativity of elements, except for O, S, Se, Te, as well as values of all the constants, are obtainable in reference [26].
2.3. Hillert-Toop Geometric Model
The Toop-Hillert geometric model [22] is used in this work to represent the excess Gibbs free energy of a ternary system, i-j-k, from the three sub-binaries i-j, i-k, j-k. The Toop-Hillert geometric model is an asymmetric model. Hence, the exact expression depends on the selected asymmetric component. If the component i is an asymmetric component, the excess Gibbs free energy gE can be expressed as:
2.4. Calculation Model
Inserting Equations (5) and (7) into Equation (6), then expanding the formalism as Equation (8) to calculate the excess Gibbs free energy in a ternary system, finally, according to the Equation (1), the formalism for activity interaction parameter calculation is obtained:
- If the asymmetric component is solute, the activity interaction parameter can be calculated by:
- If the solvent is an asymmetric component, the activity interaction parameter is:wherehere:where the αij is identical to Equation (5).
If the mass fraction (wt.%) is used, the activity interaction parameter is often denoted as , and it can be obtained by applying the below transformation from
where and are the molecular weight of solute j and solvent k, respectively. In this paper, is used.
3. Results and Discussion
The Miedema parameters [19] of O, S, Se, Te, and Po required in the calculation of are listed in Table 1. The rule for selecting the asymmetric component is according to the criterion described in [27]. The temperature for the calculations of is 1873 K, and calculations are performed with (SE ≠ 0, case 1) or without considering (SE = 0, case 2) the excess entropy. When SE = 0, the αij is equal to 1, else it is identical to Equation (5). The calculated results and experimental values recommended by JSPS [20] are listed in Table 2.
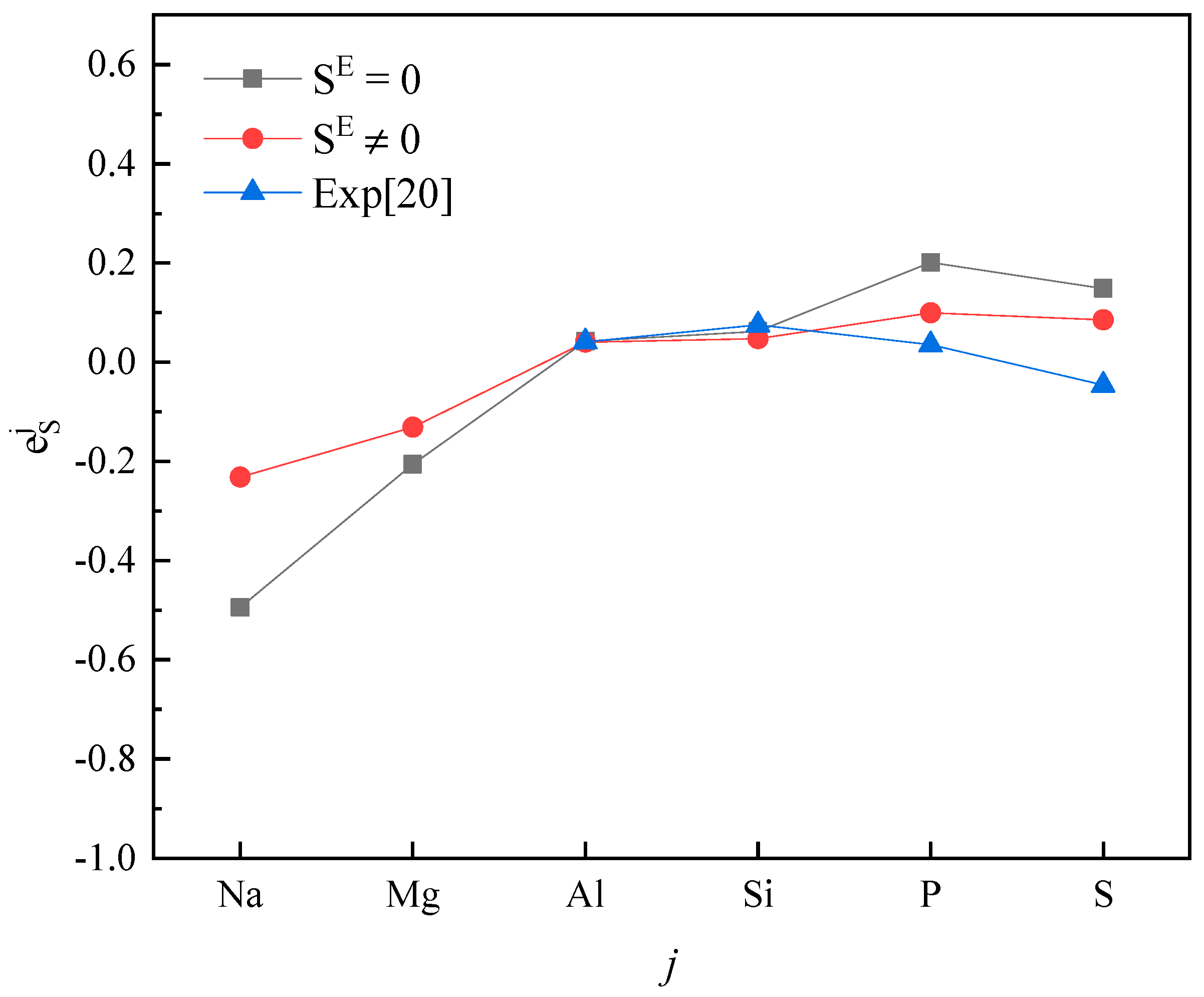
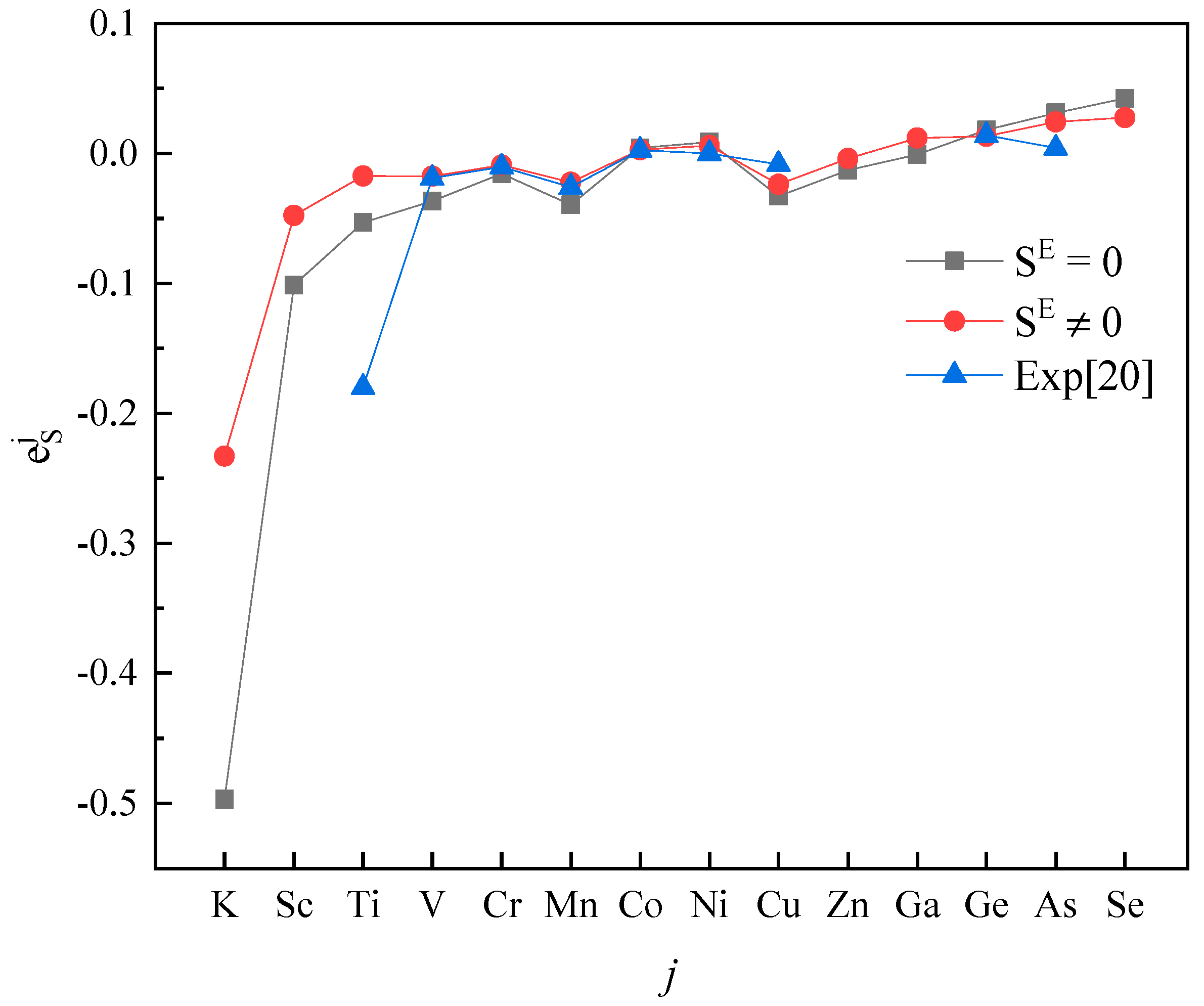
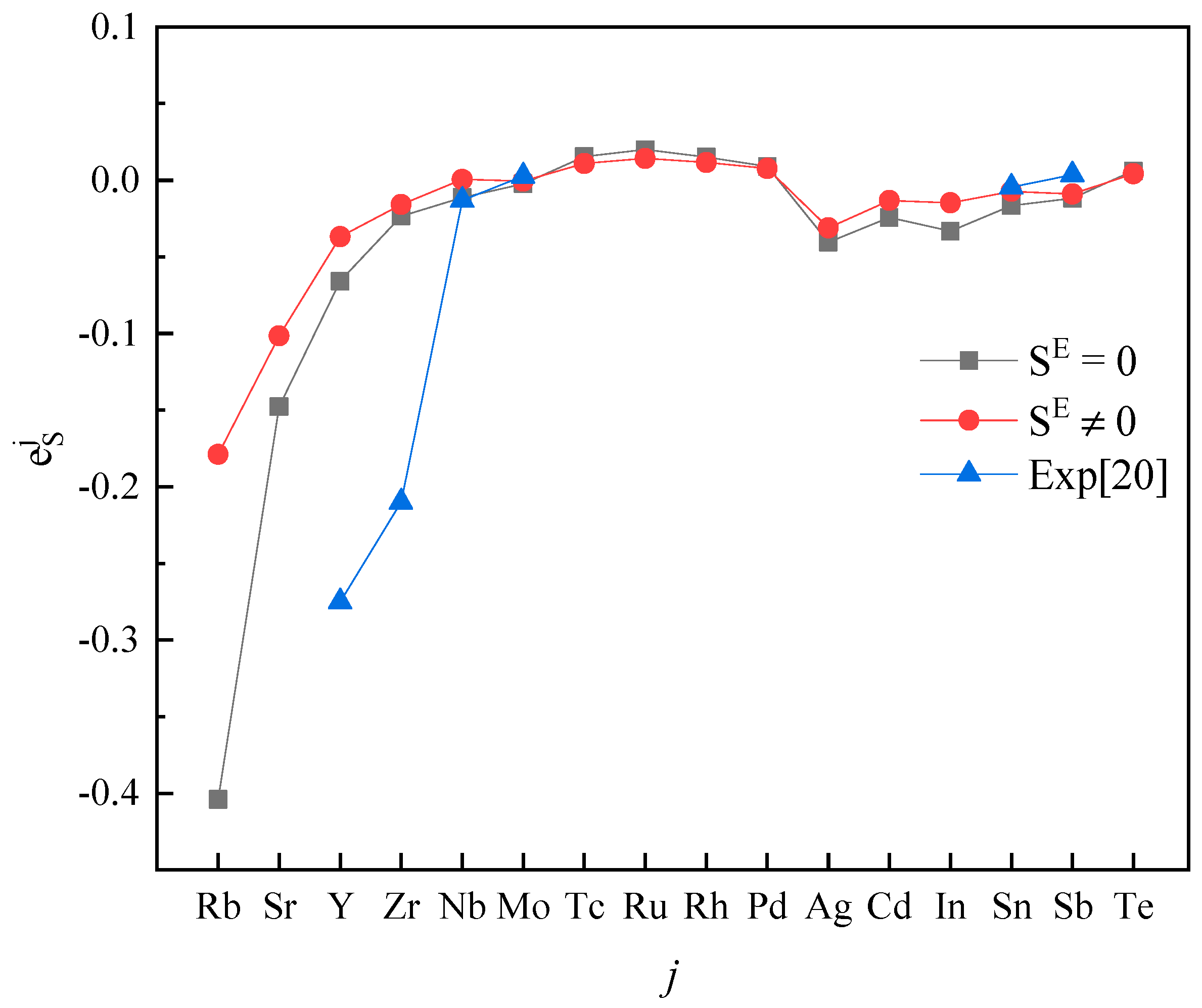
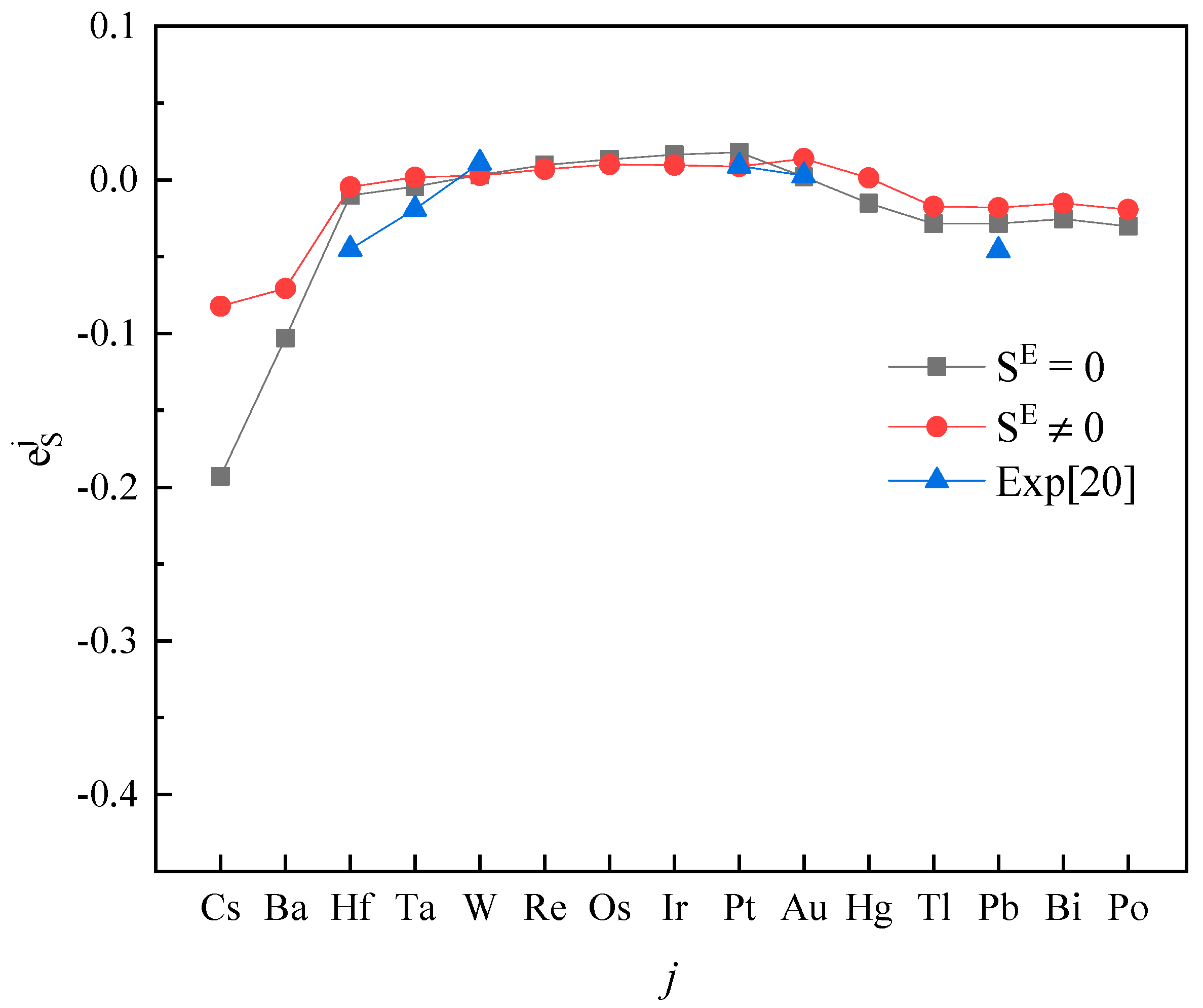
From Table 2, it’s noted that the calculated results are very encouraging. Over 85 percent of the predictions has the correct sign of compared to the experimental data in both cases. In the case 1, there are five data inconsistent in sign, in the case 2, there are only four. The absolute values are in general reasonable, except that an especially strong interaction exists between the S and j, where the absolute values are smaller than the experimental data recommended by JSPS [20], as shown for , , in Table 2. However, due to the experimental difficulties at high-temperatures, experimental values from different labs for some components are scattered. For example, measured by Taguchi et al. [28] is −22.4 ± 6.4, but by Inoue et al. [21] it is −269 ± 28, respectively; the and obtained by Wu et al. [29] are −1.56 and −2.11, and given by JSPS are −18.3 and −9.1, respectively.
Plotting calculated values (for and ) and experimental values according to incremental order of elements in each period of the periodic table, one can see the same trends among them (Figure 1, Figure 2, Figure 3, Figure 4 and Figure 5). Our results, however, differ from those by Silva [10], who showed the experimental () increases linearly with increasing atomic number. In addition, it’s obvious (Figure 1, Figure 2, Figure 3, Figure 4 and Figure 5) that a better agreement is achieved when (case 1) between theoretical values and experimental ones. Thus, theoretically, considering the excess entropy is more favorable.
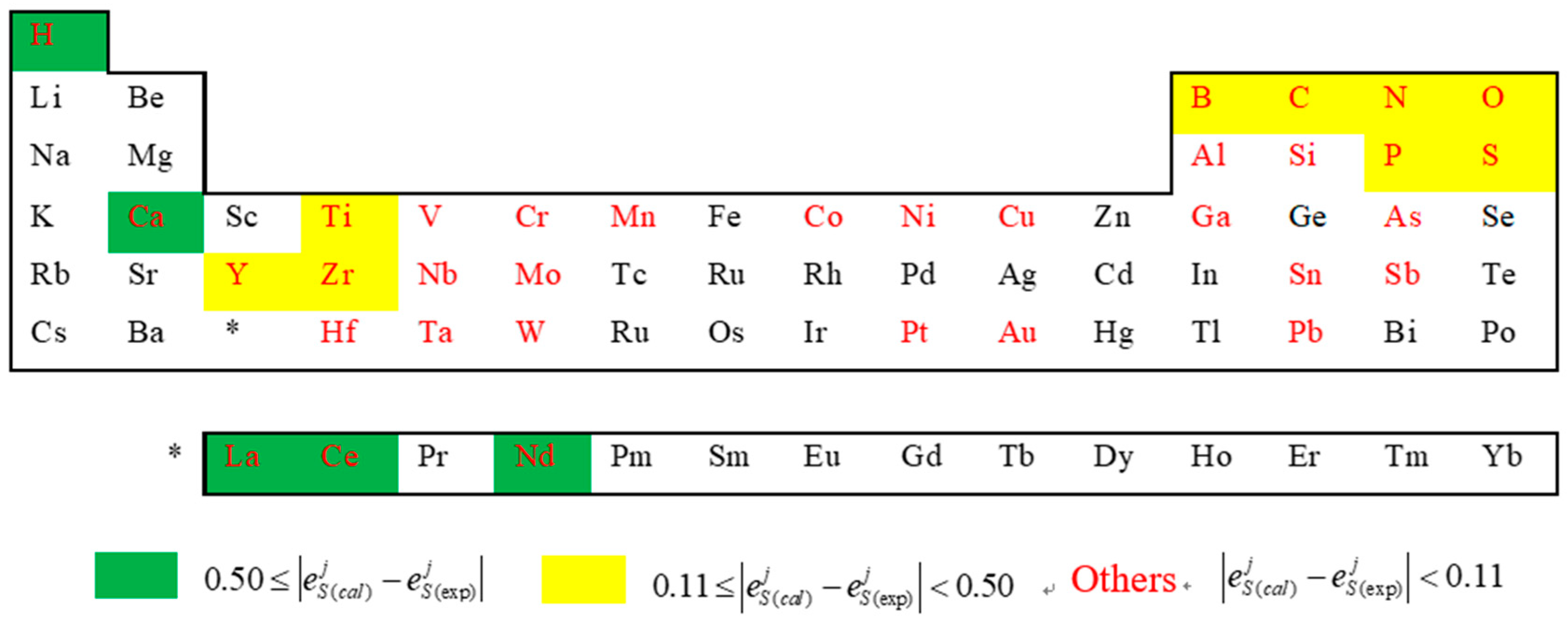
To establish our calculation method, we noticed that the precision of the calculated results heavily relies on the Miedema model and the geometric model. The Miedema model is one of the most successful models to predict the formation enthalpy of alloys. It may be owing to that more physical quantities such as electronegativity, electron density, and molar volume than other models like the Pauling electronegativity model (only electronegativity considered) have been considered [24]. However, it’s not sufficient when the constituents’ physical properties are of large differences, some minor contribution terms which are usually neglected now can’t be ignored. In the Fe-S-j system, is not only dependent on the physical property differences between sulphur and elements j, but also on the differences between elements j and iron as well as between iron and sulphur. Consequence, the elements j with large deviations between calculated and experimental values are mainly located in the periodic table far from the Fe group, especially in the left side (Figure 6), for example, the elements Ca, Ce, La, Y, Zr, etc. Moreover, the elements H, B, C, N, P, etc. are dealt with in Miedema’s model in a complicated way, and in current model, this may be also need some appropriate corrections to be made. Aiming at the deviation of the Miedema’s model, it can be improved by adding some terms such as a volume correction term [30] and an improved atomic size term [31], as well as by modifying the Miedema parameters of a specified element [32].
In addition, the energy of triplet interactions is neglected in our calculation model due to the contribution of this term to the excess Gibbs free energy gE of ternary elements is usually very small [33]. To see the influence of the geometric model, the results from the Ding’s model [12], which also includes a geometric model, are listed in Table 2 for comparison. One can see large deviations from experiments in both the present and Ding’s models. This means the deviations come mainly from the Miedema model basis instead of the geometric model. We are attempting to modify the Miedema model by adding some terms such as a volume correction term [30] and/or improving the atomic size factor [31] to optimize the calculated values. This work is now ongoing.
4. Conclusions
Because the activity interaction parameter is very important to understand the thermodynamic properties of Sulphur-contained iron-based melts, a great deal of work has been done on the experimental measurements. However, important data such as , , , etc. are still lacking. Considering the complexity of measurements and the experimental data depend strongly on the experimental techniques, in this work we employed a theoretical method and systematically calculated the activity interaction parameter in the Fe-S-j systems. Based on our study, we conclude:
(1) A model for calculating the activity interaction parameter in a ternary system was established based on the Ding’s method, wherein the Toop-Hillert model was used.
(2) The calculated results for in Fe-based melts (Fe-S-j) by current model at 1873 K, with or without considering the excess entropy, show that better results would be obtained with considering the excess entropy. And better results would be obtained for the elements j located in the middle of periodic table nearby the Fe group.
(3) The reason for the large deviations between calculated and experimental values is because of the inaccuracy of Miedema’s model when the constituents’ physical properties are of large differences.
Author Contributions
Conceptualization, X.D.; Methodology, X.D. and T.J.; Software, T.J.; Validation, W.C. and B.W.; Formal Analysis, T.J. and W.C.; Investigation, T.J. and X.D.; Resources, Y.Z.; Data Curation: T.J.; Writing-Original Draft Preparation, T.J.; Writing-Review & Editing, X.Y. and X.C.; Visualization, J.D.; Supervision, X.D.; Project Administration, X.D. and Y.Z.; Funding Acquisition, X.D.
Funding
This work was supported by the National Key R&D Program of China (2017YFB0603800, 2017YFB0603801 and 2017YFB0603802) and the National Natural Science Foundation (Grant No. 51604049).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
References
- Mintz, B. The influence of composition on the hot ductility of steels and to the problem of transverse cracking. ISIJ Int. 1999, 39, 833–855. [Google Scholar] [CrossRef]
- Ryan, M.P.; Williams, D.E.; Chater, R.J.; Hutton, B.M.; McPhail, D.S. Why stainless steel corrodes. Nature 2002, 415, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Costa e Silva, A. Thermodynamic aspects of inclusion engineering in steels. Rare Met. 2006, 25, 412–419. [Google Scholar] [CrossRef]
- Gollapalli, V.; Rao, M.B.V.; Karamched, P.S.; Borra, C.R.; Roy, G.G.; Srirangam, P. Modification of oxide inclusions in calcium-treated Al-killed high sulphur steels. Ironmak. Steelmak. 2018, 1–8. [Google Scholar] [CrossRef]
- Wagner, C. Thermodynamics of Alloys; Addison-Wesley Press: Boston, MA, USA, 1952. [Google Scholar]
- Lupis, C.H.P.; Elliott, J.F. Generalized interaction coefficients. Acta Metall. 1966, 14, 529–538. [Google Scholar] [CrossRef] [Green Version]
- Darken, L. Thermodynamics of binary metallic solutions. Trans. Met. Soc. AIME 1967, 239, 80–89. [Google Scholar]
- Pelton, A.D.; Bale, C.W. A modified interaction parameter formalism for non-dilute solutions. Metall. Trans. A 1986, 17, 1211–1215. [Google Scholar] [CrossRef]
- Bale, C.W.; Pelton, A.D. The unified interaction parameter formalism: Thermodynamic consistency and applications. Metall. Trans. A 1990, 21, 1997–2002. [Google Scholar] [CrossRef]
- Costa e Silva, A. Interaction parameters of oxygen and deoxidants in liquid iron. J. Min. Metall. Sect. B Metall. 2016, 52, 41–46. [Google Scholar] [CrossRef]
- Waseda, Y. Interaction parameters in metallic solutions estimated from liquid structure and the heat of solution at infinite dilution. High Temp. Mater. Process. 2012, 31, 203–208. [Google Scholar] [CrossRef]
- Ding, X.Y.; Fan, P.; Wang, W.Z. Thermodynamic calculation for alloy systems. Metall. Mater. Trans. B 1999, 30, 271–277. [Google Scholar] [CrossRef]
- Ding, X.; Fan, P.; Han, Q. Models of activity and activity interaction parameter in ternary metallic melt. Acta Metall. Sin. 1994, 30, 49–60. [Google Scholar]
- Ueno, S.; Waseda, Y.; Jacob, K.T.; Tamaki, S. Theoretical treatment of interaction parameters in multicomponent metallic solutions. Process Metall. 1988, 59, 474–483. [Google Scholar] [CrossRef]
- Fan, P.; Chou, K.C. A self-consistent model for predicting interaction parameters in multicomponent alloys. Metall. Mater. Trans. A 1999, 30, 3099–3102. [Google Scholar] [CrossRef]
- Wang, F.M.; Li, X.P.; Han, Q.Y.; Zhang, N.X. A model for calculating interaction coefficients between elements in liquid and iron-base alloy. Metall. Mater. Trans. B 1997, 28, 109–113. [Google Scholar] [CrossRef]
- Zhang, N.; Chen, W.; Chen, X.; Ding, X.; Zhou, G. Modeling Activity and Interaction Coefficients of Components of Multicomponent Alloy Melts: An Example of Iron Melt. High Temp. Mater. Process. 2013, 32, 215. [Google Scholar] [CrossRef]
- Ding, X.; Fan, P.; Luo, L. Alloy Melts Thermodynamic Model: Prediction and Software Development; Northeastern University Press: Shenyang, China, 1998. (In Chinese) [Google Scholar]
- Neuhausen, J.; Eichler, B. Extension of Mediema’s Macroscopic Atom Model to the Elements of Group 16 (O, S, Se, Te, Po); Paul Scherrer Inst.: Villigen, Switzerland, 2003. [Google Scholar]
- Hino, M.; Ito, K. Thermodynamic Data for Steelmaking; Tohoku University Press: Sendai, Japan, 2010. [Google Scholar]
- Inoue, R.; Suito, H. Calcium desulfurization equilibrium in liquid iron. Steel Res. 1994, 65, 403–409. [Google Scholar] [CrossRef]
- Hillert, M. Empirical methods of predicting and representing thermodynamic properties of ternary solution phases. Calphad 1980, 4, 1–12. [Google Scholar] [CrossRef]
- Tanaka, T.; Morita, Z.-I.; Gokcen, N.A.; Iida, T. Thermodynamic relationship between enthalpy of mixing and excess entropy in liquid binary alloys. Zeitschrift für Metallkunde 1993, 84, 192–200. [Google Scholar]
- Miedema, A.; De Chatel, P.; De Boer, F. Cohesion in alloys—Fundamentals of a semi-empirical model. Physica B+C 1980, 100, 1–28. [Google Scholar] [CrossRef]
- Niessen, A.; Miedema, A.; De Boer, F.; Boom, R. Enthalpies of formation of liquid and solid binary alloys based on 3d metals: IV. Alloys of cobalt. Physica B+C 1988, 151, 401–432. [Google Scholar] [CrossRef]
- Niessen, A.K.; de Boer, F.R.; Boom, R.; De Châtel, P.F.; Mattens, W.C.M.; Miedema, A.R. Model predictions for the enthalpy of formation of transition metal alloys II. Calphad 1983, 7, 51–70. [Google Scholar] [CrossRef]
- Wu, X.S.; Yan, X.H.; Ma, B.K.; Lin, Z.J.; Yang, X.Z. Calculation of the Heat of Formation of Ternary Compounds—P-Ga-as, N-Ga-as, N-Ga-P. Acta Phys. Sin. 1995, 4, 62–70. [Google Scholar]
- Taguchi, K.; Ono-Nakazato, H.; Nakai, D.; Usui, T.; Marukawa, K. Deoxidation and desulfurization equilibria of liquid iron by calcium. ISIJ Int. 2003, 43, 1705–1709. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, L.; Du, T. Thermodynamics of rare earth elements in liquid iron. J. Less-Common Met. 1985, 110, 187–193. [Google Scholar] [CrossRef]
- Zhang, R.F.; Liu, B.X. Proposed model for calculating the standard formation enthalpy of binary transition-metal systems. Appl. Phys. Lett. 2002, 81, 1219–1221. [Google Scholar] [CrossRef]
- Sun, S.P.; Yi, D.Q.; Jiang, Y.; Zang, B.; Xu, C.H.; Li, Y. An improved atomic size factor used in Miedema’s model for binary transition metal systems. Chem. Phys. Lett. 2011, 513, 149–153. [Google Scholar] [CrossRef]
- Chen, X.-Q.; Podloucky, R. Miedema’s model revisited: The parameter for Ti, Zr, and Hf. Calphad 2006, 30, 266–269. [Google Scholar] [CrossRef]
- Chartrand, P.; Pelton, A.D. On the choice of “Geometric” thermodynamic models. J. Phase Equilib. 2000, 21, 141–147. [Google Scholar] [CrossRef]
Figure 1.
The activity interaction parameter of the 2nd periodic elements j on S.

Figure 2.
The activity interaction parameter of the 3rd periodic elements j on S.

Figure 3.
The activity interaction parameter of the 4th periodic elements j on S.

Figure 4.
The activity interaction parameter of the 5th periodic elements j on S.

Figure 5.
The activity interaction parameter of the 6th periodic elements j on S.

Figure 6.
The distribution locations of elements j in periodic table together with the relative deviations of the corresponding activity interaction parameter between calculated and experimental values (the elements with red color represent that the experimental value of is available, the others represent that the experimental value is not available).
Figure 6.
The distribution locations of elements j in periodic table together with the relative deviations of the corresponding activity interaction parameter between calculated and experimental values (the elements with red color represent that the experimental value of is available, the others represent that the experimental value is not available).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Miedema parameters [19] of the elements O, S, Se, Te, and Po.
Table 1.
Miedema parameters [19] of the elements O, S, Se, Te, and Po.
| Element | μ | |||
|---|---|---|---|---|
| O | 6.97 | 2.66 | 1.70 | 0.04 |
| S | 5.60 | 4.38 | 1.46 | 0.04 |
| Se | 5.17 | 5.17 | 1.40 | 0.04 |
| Te | 4.72 | 6.44 | 1.31 | 0.04 |
| Po | 4.44 | 7.04 | 1.15 | 0.04 |
Table 2.
Comparison of the calculation activity interaction parameter with experimental values recommended by JSPS [20] in Fe-based alloys at 1873 K.
Table 2.
Comparison of the calculation activity interaction parameter with experimental values recommended by JSPS [20] in Fe-based alloys at 1873 K.
| j | Current Model | Ding Model [12] | JSPS | j | Current Model | Ding Model [12] | JSPS | ||
|---|---|---|---|---|---|---|---|---|---|
| H | 4.0396 | 4.5582 | 1.3551 | 0.41 | Pd | 0.0089 | 0.0077 | −0.0192 | |
| Li | −0.8793 | −0.3694 | −0.7415 | Ag | −0.0403 | −0.0310 | −0.0107 | ||
| Be | 0.0297 | 0.0334 | −0.0046 | Cd | −0.0242 | −0.0131 | −0.0138 | ||
| B | 0.4360 | 0.2808 | 0.3909 | 0.134 | In | −0.0332 | −0.0147 | −0.0044 | |
| C | 0.5800 | 0.3849 | 0.3770 | 0.111 | Sn | −0.0166 | −0.0072 | 0.0009 | −0.0044 |
| N | 0.5449 | −0.1025 | 0.3709 | 0.01 | Sb | −0.0117 | −0.0090 | 0.0101 | 0.0037 |
| O | 0.3075 | −0.3524 | 0.3574 | −0.27 | Te | 0.0062 | 0.0042 | −0.0738 | |
| Na | −0.4944 | −0.2318 | −0.2903 | Cs | −0.1929 | −0.0822 | −0.0567 | ||
| Mg | −0.2058 | −0.1314 | −0.1475 | Ba | −0.1029 | −0.0707 | −0.0222 | ||
| Al | 0.0426 | 0.0405 | 0.0112 | 0.041 | Hf | −0.0103 | −0.0047 | −0.0116 | −0.045 |
| Si | 0.0621 | 0.0471 | 0.0941 | 0.075 | Ta | −0.0044 | 0.0016 | 0.0032 | −0.019 |
| P | 0.2013 | 0.0995 | 0.1907 | 0.035 | W | 0.0032 | 0.0028 | 0.0094 | 0.011 |
| S | 0.1488 | 0.0852 | 0.1496 | −0.0461 | Re | 0.0096 | 0.0068 | 0.0117 | |
| K | −0.4969 | −0.2329 | −0.2276 | Os | 0.0133 | 0.0099 | 0.0127 | ||
| Ca | −0.2508 | −0.1668 | −0.1753 | −110.0 | Ir | 0.0164 | 0.0094 | 0.0121 | |
| Sc | −0.1013 | −0.0477 | −0.1258 | Pt | 0.0179 | 0.0086 | 0.0056 | 0.0089 | |
| Ti | −0.0532 | −0.0174 | −0.0820 | −0.18 | Au | 0.0018 | 0.0137 | −0.0039 | 0.0028 |
| V | −0.0367 | −0.0174 | −0.0463 | −0.019 | Hg | −0.0153 | 0.0011 | −0.0091 | |
| Cr | −0.0156 | −0.0089 | −0.0173 | −0.0103 | Tl | −0.0286 | −0.0174 | −0.0068 | |
| Mn | −0.0395 | −0.0221 | −0.0393 | −0.026 | Pb | −0.0284 | −0.0182 | −0.0051 | −0.046 |
| Co | 0.0042 | 0.0029 | 0.0036 | 0.0026 | Bi | −0.0254 | −0.0154 | −0.0042 | |
| Ni | 0.0087 | 0.0061 | 0.0071 | 0 | Po | −0.0302 | −0.0194 | −0.0443 | |
| Cu | −0.0329 | −0.0238 | −0.0214 | −0.0084 | La | −0.0491 | −0.0270 | −0.0320 | −18.3 |
| Zn | −0.0130 | −0.0040 | −0.0095 | Ce | −0.0346 | −0.0176 | −0.0309 | −9.10 | |
| Ga | −0.0009 | 0.0119 | −0.0031 | Pr | −0.0316 | −0.0158 | −0.0301 | ||
| Ge | 0.0181 | 0.0133 | 0.0142 | 0.014 | Nd | −0.0307 | −0.0156 | −0.0290 | −0.76 |
| As | 0.0313 | 0.0241 | 0.0431 | 0.0041 | Pm | −0.0271 | −0.0122 | −0.0279 | |
| Se | 0.0423 | 0.0274 | 0.0456 | Sm | −0.0269 | −0.0130 | −0.0497 | ||
| Rb | −0.4041 | −0.1789 | −0.1711 | Eu | −0.0800 | −0.0500 | −0.0265 | ||
| Sr | −0.1478 | −0.1015 | −0.0872 | Gd | −0.0255 | −0.0128 | −0.0254 | ||
| Y | −0.0657 | −0.0368 | −0.0674 | −0.275 | Tb | −0.0231 | −0.0109 | −0.0248 | |
| Zr | −0.0234 | −0.0157 | −0.0521 | −0.210 | Dy | −0.0226 | −0.0107 | −0.0244 | |
| Nb | −0.0112 | 0.0005 | −0.0259 | −0.013 | Ho | −0.0229 | −0.0113 | −0.0233 | |
| Mo | −0.0024 | −0.0005 | −0.0041 | 0.0027 | Er | −0.0200 | −0.0089 | −0.0230 | |
| Tc | 0.0155 | 0.0109 | 0.0131 | Tm | −0.0198 | −0.0089 | −0.0400 | ||
| Ru | 0.0201 | 0.0142 | 0.0166 | Yb | −0.0600 | −0.04 | −0.0215 | ||
| Rh | 0.0152 | 0.0116 | 0.0111 | Lu | −0.0168 | −0.0066 | −0.0192 | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ju, T.; Ding, X.; Zhang, Y.; Chen, W.; Cheng, X.; Wang, B.; Dai, J.; Yan, X. Estimation of Activity Interaction Parameters in Fe-S-j Systems. Entropy 2018, 20, 808. https://0-doi-org.brum.beds.ac.uk/10.3390/e20100808
AMA Style
Ju T, Ding X, Zhang Y, Chen W, Cheng X, Wang B, Dai J, Yan X. Estimation of Activity Interaction Parameters in Fe-S-j Systems. Entropy. 2018; 20(10):808. https://0-doi-org.brum.beds.ac.uk/10.3390/e20100808
Chicago/Turabian StyleJu, Tianhua, Xueyong Ding, Yingyi Zhang, Weiliang Chen, Xiangkui Cheng, Bo Wang, Jingxin Dai, and Xinlin Yan. 2018. "Estimation of Activity Interaction Parameters in Fe-S-j Systems" Entropy 20, no. 10: 808. https://0-doi-org.brum.beds.ac.uk/10.3390/e20100808
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.