
Enhanced Directed Random Walk for the Identification of Breast Cancer Prognostic Markers from Multiclass Expression Data

 , , ,
, , ,
Abstract
:1. Introduction
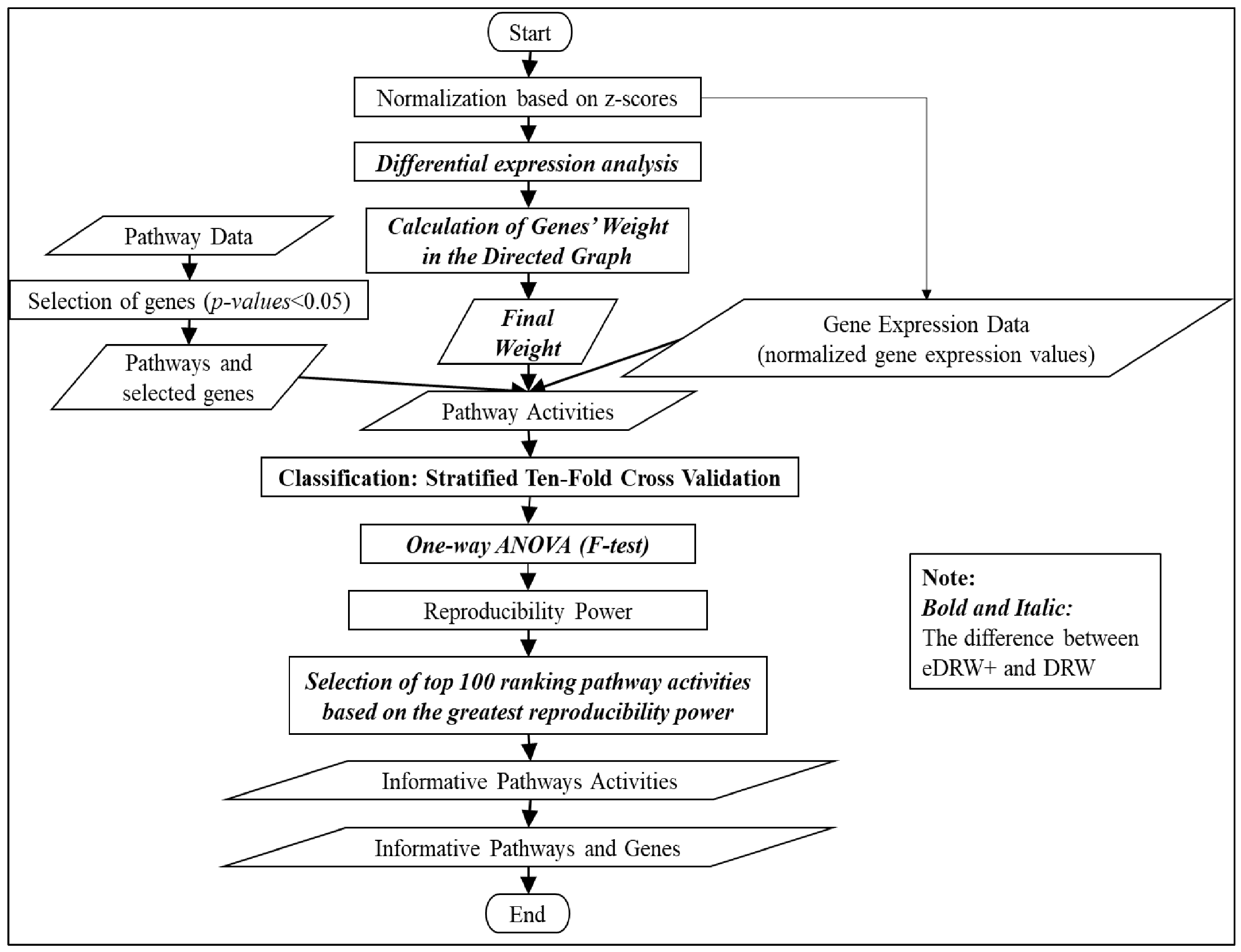
2. Materials and Methods
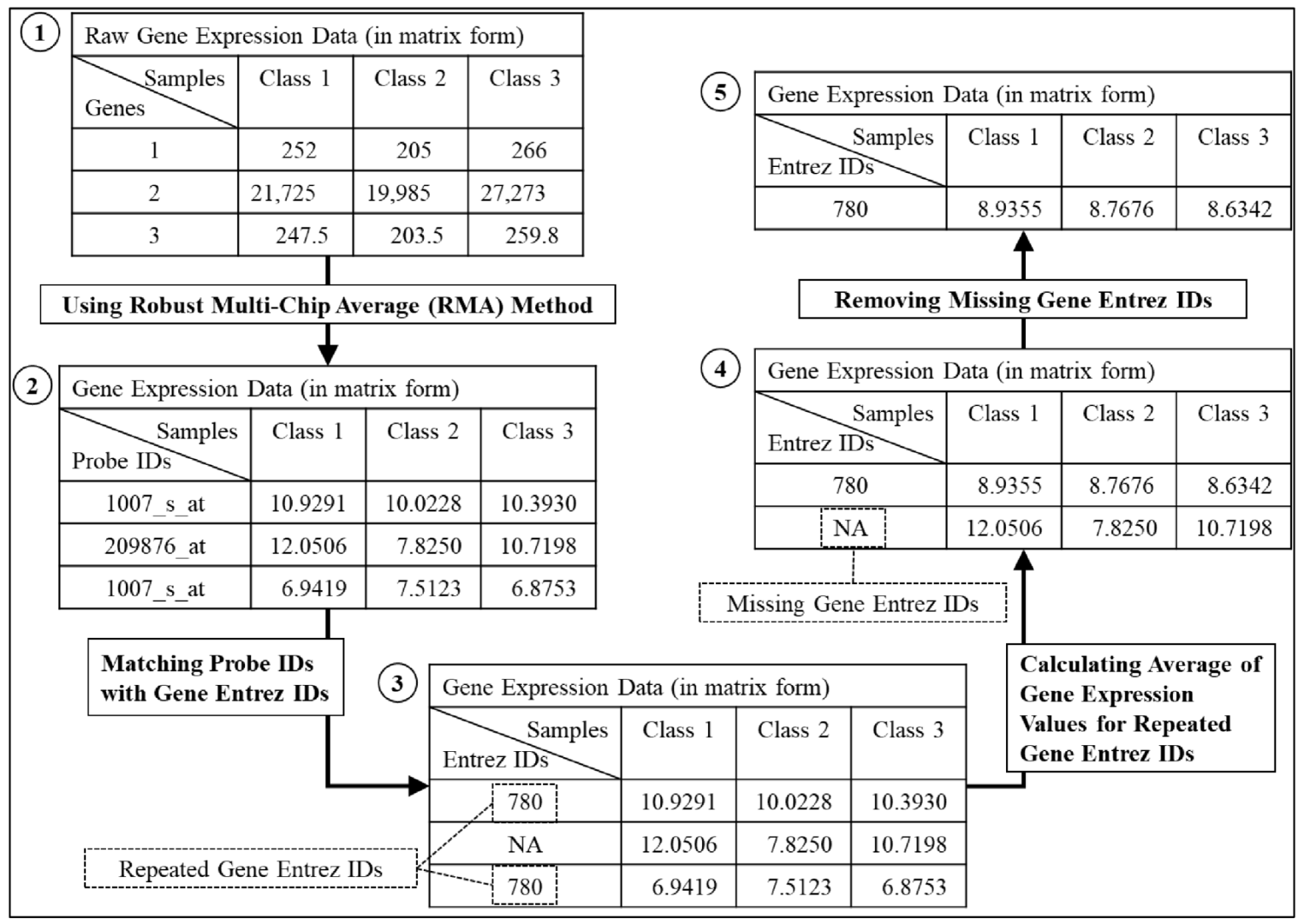
2.1. Data Collection and Pre-Processing
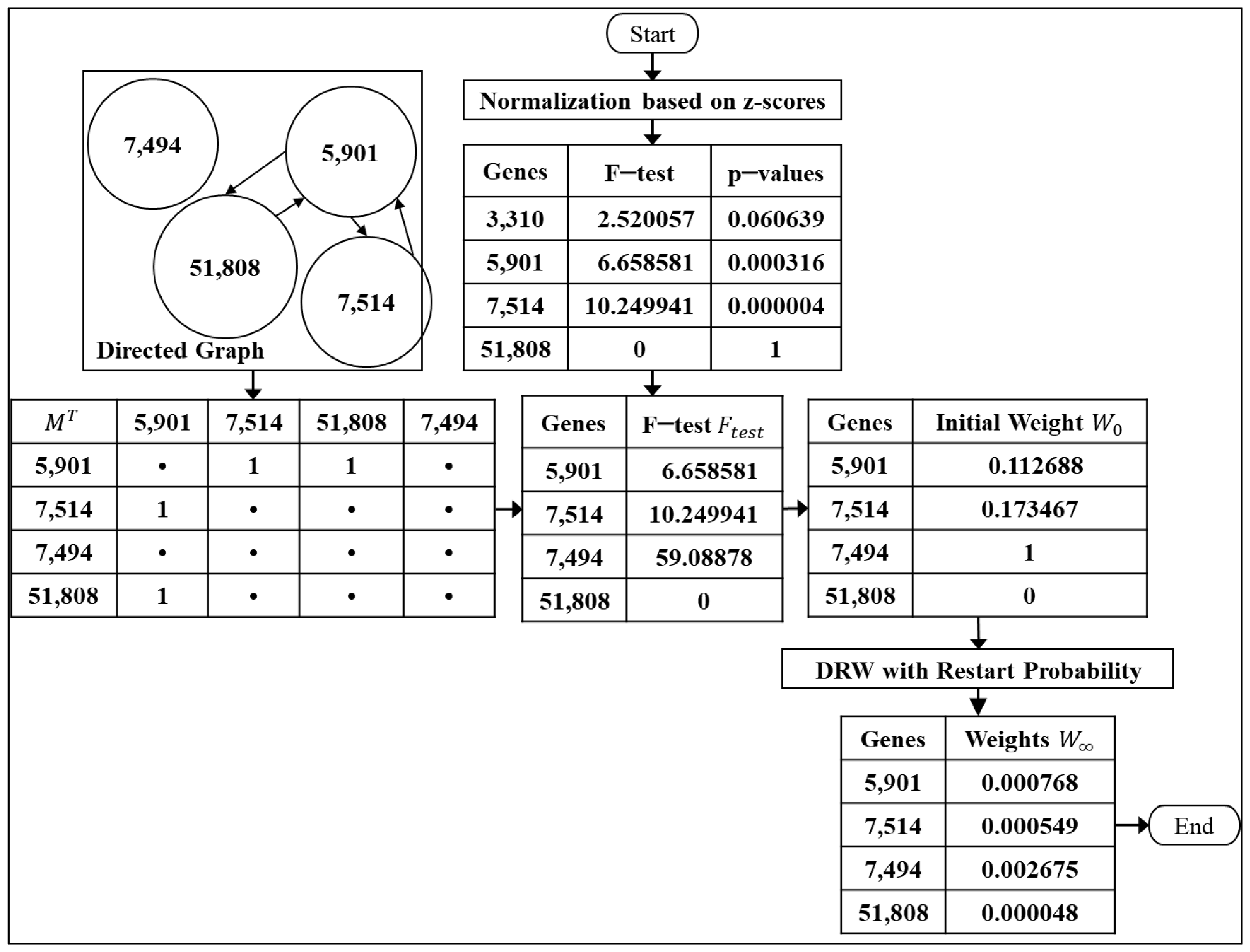
2.2. Step 1: Normalization Based on Z-Scores
2.3. Step 2: Differential Expression Analysis
2.4. Step 3: Calculation of the Genes’ Weight in the Directed Graph
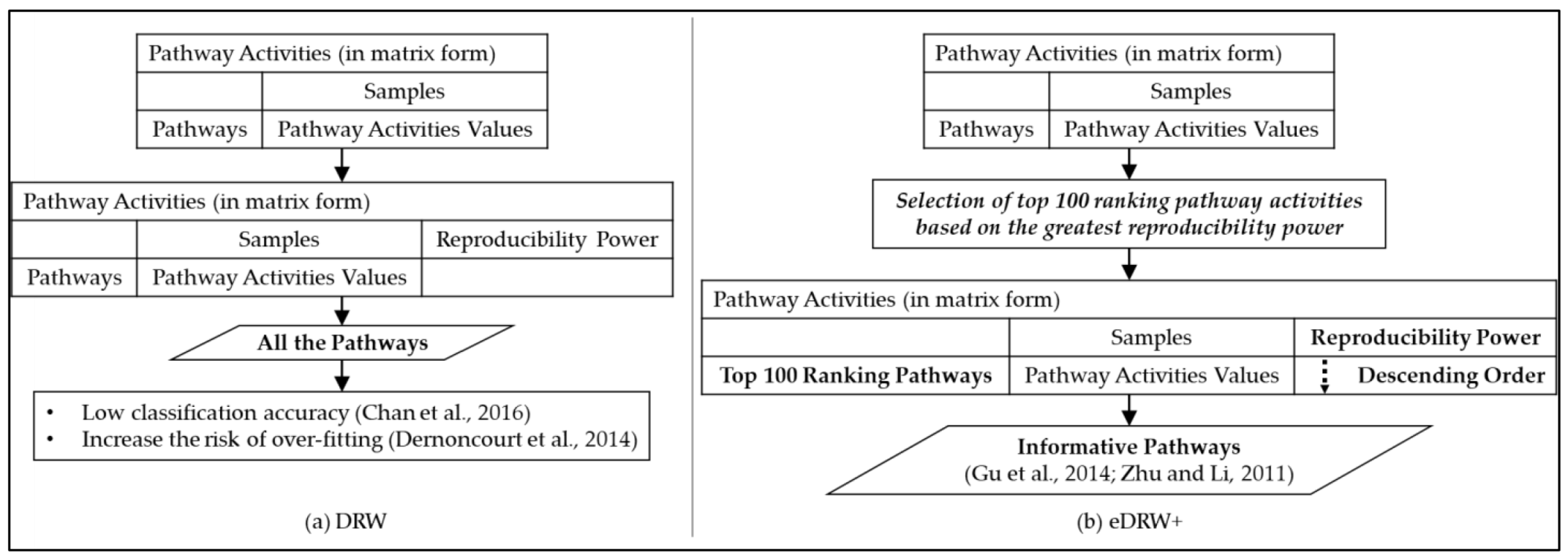
2.5. Step 4: Inferring Pathway Activities
2.6. Step 5: Classification
3. Results
3.1. Multicollinearity Analysis
3.2. Stratified Ten-Fold Cross-Validation within the Datasets
3.3. Stratified Ten-Fold Cross-Validation between the Datasets
3.4. Biological Context Verification and Validation of the Identified Pathways and Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vaske, C.; Benz, S.; Sanborn, J.Z.; Earl, D.; Szeto, C.; Zhu, J.; Haussler, D.; Stuart, J.M. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics 2010, 26, i237–i245. [Google Scholar] [CrossRef]
- Nies, Y.H.; Islahudin, F.; Chong, W.W.; Abdullah, N.; Ismail, F.; Bustamam, R.S.A.; Wong, Y.F.; Jaszle, S.; Shah, N.M. Treatment decision-making among breast cancer patients in Malaysia. Patient Prefer. Adherence 2017, 11, 1767–1777. [Google Scholar] [CrossRef] [Green Version]
- Mohapatra, P.; Chakravarty, S.; Dash, P. Microarray medical data classification using kernel ridge regression and modified cat swarm optimization based gene selection system. Swarm Evol. Comput. 2016, 28, 144–160. [Google Scholar] [CrossRef]
- Liu, W.; Li, C.; Xu, Y.; Yang, H.; Yao, Q.; Han, J.; Shang, D.; Zhang, C.; Su, F.; Li, X.; et al. Topologically inferring risk-active pathways toward precise cancer classification by directed random walk. Bioinformatics 2013, 29, 2169–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macher, J.-P.; Crocq, M.-A. Treatment goals: Response and nonresponse. Dialogues Clin. Neurosci. 2004, 6, 83–91. [Google Scholar]
- AlAjlan, A.; Badr, G. Data Mining in Pathway Analysis for Gene Expression. In Industrial Conference on Data Mining; Springer: Cham, Switzerland, 2015; Volume 9165, pp. 69–77. [Google Scholar]
- Yasrebi, H.; Sperisen, P.; Praz, V.; Bucher, P. Can Survival Prediction Be Improved by Merging Gene Expression Data Sets? PLoS ONE 2009, 4, e7431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karn, T.; Metzler, D.; Ruckhäberle, E.; Hanker, L.; Gätje, R.; Solbach, C.; Ahr, A.; Schmidt, M.; Holtrich, U.; Kaufmann, M.; et al. Data driven derivation of cutoffs from a pool of 3030 Affymetrix arrays to stratify distinct clinical types of breast cancer. Breast Cancer Res. Treat. 2010, 120, 567–579. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. Analysing biological pathways in genome-wide association studies. Nat. Rev. Genet. 2010, 11, 843–854. [Google Scholar] [CrossRef]
- Tian, S.; Chang, H.H.; Wang, C. Weighted-SAMGSR: Combining significance analysis of microarray-gene set reduction algorithm with pathway topology-based weights to select relevant genes. Biol. Direct 2016, 11, 50. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Ainali, C.; Tsoka, S.; Papageorgiou, L.G. Pathway activity inference for multiclass disease classification through a mathematical programming optimisation framework. BMC Bioinform. 2014, 15, 390. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.H.; Sootanan, P.; Larpeampaisarl, P. Feature selection of pathway markers for microarray-based disease classification using negatively correlated feature sets. In Proceedings of the 2011 International Joint Conference on Neural Networks (IJCNN), San Jose, CA, USA, 31 July–5 August 2011; pp. 3293–3299. [Google Scholar]
- Sootanan, P.; Meechai, A.; Prom-on, S.; Chan, J.H. Pathway-based microarray analysis with negatively correlated feature sets for disease classification. In International Conference on Neural Information Processing; Springer: Berlin/Heidelberg, Germany, 2011; Volume 7062, pp. 676–683. [Google Scholar]
- Engchuan, W.; Chan, J.H. Apriori Gene Set-based Microarray Analysis for Disease Classification Using Unlabeled Data. Procedia Comput. Sci. 2013, 23, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Ross, A.; Willson, V.L. Basic and Advanced Statistical Tests; Sense Publishers: Rotterdam, The Netherlands, 2017; pp. 21–24. [Google Scholar]
- Chandra, B.; Gupta, M. An efficient statistical feature selection approach for classification of gene expression data. J. Biomed. Inform. 2011, 44, 529–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engchuan, W.; Chan, J.H. Pathway-Based Multi-class Classification of Lung Cancer. In International Conference on Neural Information Processing; Springer: Berlin/Heidelberg, Germany, 2012; pp. 697–702. [Google Scholar]
- Kar, S.; Das Sharma, K.; Maitra, M. A particle swarm optimization based gene identification technique for classification of cancer subgroups. In Proceedings of the 2016 2nd International Conference on Control, Instrumentation, Energy & Communication (CIEC), Kolkata, India, 28–30 January 2016; pp. 130–134. [Google Scholar]
- Ortiz-Ramón, R.; Larroza, A.; Ruiz-España, S.; Arana, E.; Moratal, D. Classifying brain metastases by their primary site of origin using a radiomics approach based on texture analysis: A feasibility study. Eur. Radiol. 2018, 28, 4514–4523. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Liu, Z.; Ma, X.; Hua, D. Selecting Genes by Test Statistics. J. Biomed. Biotechnol. 2005, 2005, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Engchuan, W.; Meechai, A.; Tongsima, S.; Doungpan, N.; Chan, J.H. Gene-set activity toolbox (GAT): A platform for microarray-based cancer diagnosis using an integrative gene-set analysis approach. J. Bioinform. Comput. Biol. 2016, 14, 1650015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engchuan, W.; Chan, J.H. Pathway activity transformation for multi-class classification of lung cancer datasets. Neurocomputing 2015, 165, 81–89. [Google Scholar] [CrossRef]
- Li, T.; Zhang, C.; Ogihara, M. A comparative study of feature selection and multiclass classification methods for tissue classification based on gene expression. Bioinformatics 2004, 20, 2429–2437. [Google Scholar] [CrossRef]
- Ferdowsi, S.; Voloshynovskiy, S.; Gabryel, M.; Korytkowski, M. Multi-class Classification: A Coding Based Space Partitioning. In International Conference on Artificial Intelligence and Soft Computing; Springer: Cham, Switzerland, 2014; pp. 593–604. [Google Scholar]
- Gu, J.-L.; Lu, Y.; Liu, C.; Lu, H. Multiclass classification of sarcomas using pathway based feature selection method. J. Theor. Biol. 2014, 362, 3–8. [Google Scholar] [CrossRef]
- Yang, S.; Naiman, D.Q. Multiclass cancer classification based on gene expression comparison. Stat. Appl. Genet. Mol. Biol. 2014, 13, 477–496. [Google Scholar] [CrossRef]
- Hung, F.H.; Chiu, H.W. Cancer subtype prediction from a pathway-level perspective by using a support vector machine based on integrated gene expression and protein network. Comput. Methods Programs Biomed. 2017, 141, 27–34. [Google Scholar] [CrossRef]
- Zhe, S.; Naqvi, S.A.Z.; Yang, Y.; Qi, Y. Joint network and node selection for pathway-based genomic data analysis. Bioinformatics 2013, 29, 1987–1996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Yu, M.; Wang, S. A statistical framework for pathway and gene identification from integrative analysis. J. Multivar. Anal. 2017, 156, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creixell, P.; Reimand, J.; Haider, S.; Wu, G.; Shibata, T.; Vazquez, M.; Mustonen, V.; Gonzalez-Perez, A.; Pearson, J.; Sander, C.; et al. Pathway and network analysis of cancer genomes. Nat. Methods 2015, 12, 615–621. [Google Scholar] [CrossRef]
- Evangeline, D.P.; Sandhiya, C.; Anandhakumar, P.; Raj, G.D.; Rajendran, T. Feature subset selection for irrelevant data removal using Decision Tree Algorithm. In Proceedings of the 2013 Fifth International Conference on Advanced Computing (ICoAC), Chennai, India, 18–20 December 2013; pp. 268–274. [Google Scholar]
- Farmer, P.; Bonnefoi, H.; Becette, V.; Tubiana-Hulin, M.; Fumoleau, P.; Larsimont, D.; MacGrogan, G.; Bergh, J.; Cameron, D.; Goldstein, D.; et al. Identification of molecular apocrine breast tumours by microarray analysis. Breast Cancer Res. 2005, 7, P2.11. [Google Scholar] [CrossRef] [Green Version]
- Pawitan, Y.; Bjöhle, J.; Amler, L.; Borg, A.-L.; Egyhazi, S.; Hall, P.; Han, X.; Holmberg, L.; Huang, F.; Klaar, S.; et al. Gene expression profiling spares early breast cancer patients from adjuvant therapy: Derived and validated in two population-based cohorts. Breast Cancer Res. 2005, 7, R953. [Google Scholar] [CrossRef]
- Nur, U.; Shack, L.G.; Rachet, B.; Carpenter, J.R.; Coleman, M.P. Modelling relative survival in the presence of incomplete data: A tutorial. Int. J. Epidemiol. 2009, 39, 118–128. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Wang, W.; Tian, G.; Xie, W.; Lei, L.; Liu, J.; Huang, W.; Xu, L.; Li, E. Topologically inferring pathway activity for precise survival outcome prediction: Breast cancer as a case. Mol. Biosyst. 2017, 13, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.; Biegert, G.; Adamec, J.; Helikar, T. Identification of potential tissue-specific cancer biomarkers and development of cancer versus normal genomic classifiers. Oncotarget 2017, 8, 85692–85715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hochreiter, S.; Clevert, D.-A.; Obermayer, K. A new summarization method for affymetrix probe level data. Bioinformatics 2006, 22, 943–949. [Google Scholar] [CrossRef] [Green Version]
- Usoskin, D.; Furlan, A.; Islam, S.; Abdo, H.; Lönnerberg, P.; Lou, D.; Hjerling-Leffler, J.; Haeggstrom, J.Z.; Kharchenko, O.; Kharchenko, P.V.; et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci. 2015, 18, 145–153. [Google Scholar] [CrossRef]
- An, J.; Kim, K.; Chae, H.; Kim, S. Degpack: A web package using a non-parametric and information theoretic algorithm to identify differentially expressed genes in multiclass RNA-seq samples. Methods 2014, 69, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.; Russo, R. A Student’s Guide to Analysis of Variance; Routledge: London, UK, 2014. [Google Scholar]
- Bernhardson, C.S. 375: Type I Error Rates When Multiple Comparison Procedures Follow a Significant F Test of ANOVA. Biometrics 1975, 31, 229–232. [Google Scholar] [CrossRef]
- Lix, L.M.; Keselman, J.C.; Keselman, H.J. Consequences of assumption violations revisited: A quantitative review of alternatives to the one-way analysis of variance F test. Rev. Educ. Res. 1996, 66, 579–619. [Google Scholar] [CrossRef]
- Yang, R.; Daigle, B.J.; Petzold, L.R.; Doyle, F.J. Core module biomarker identification with network exploration for breast cancer metastasis. BMC Bioinform. 2012, 13, 12. [Google Scholar] [CrossRef] [Green Version]
- Chan, W.H.; Mohamad, M.S.; Deris, S.; Zaki, N.; Kasim, S.; Omatu, S.; Corchado, J.M.; Alashwal, H. Identification of informative genes and pathways using an improved penalized support vector machine with a weighting scheme. Comput. Biol. Med. 2016, 77, 102–115. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Li, T.; Liang, D. Incorporating logistic regression to decision-theoretic rough sets for classifications. Int. J. Approx. Reason. 2014, 55, 197–210. [Google Scholar] [CrossRef]
- Schoenborn, N.L.; Xue, Q.-L.; Pollack, C.E.; Janssen, E.M.; Bridges, J.F.; Wolff, A.C.; Boyd, C.M. Demographic, health, and attitudinal factors predictive of cancer screening decisions in older adults. Prev. Med. Rep. 2019, 13, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Carson, M.B.; Lu, H. Network-based prediction and knowledge mining of disease genes. BMC Med. Genom. 2015, 8, S9. [Google Scholar] [CrossRef] [Green Version]
- Huan, J.; Wang, L.; Xing, L.; Qin, X.; Feng, L.; Pan, X.; Zhu, L. Insights into significant pathways and gene interaction networks underlying breast cancer cell line MCF-7 treated with 17β-Estradiol (E2). Gene 2014, 533, 346–355. [Google Scholar] [CrossRef]
- Zhou, J.; Fu, B.-Q. The research on gene-disease association based on text-mining of PubMed. BMC Bioinform. 2018, 19, 37. [Google Scholar] [CrossRef]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003, 4, R60. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2008, 37, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44. [Google Scholar] [CrossRef] [PubMed]
- Zou, R.; Zhang, D.; Lv, L.; Shi, W.; Song, Z.; Yi, B.; Lai, B.; Chen, Q.; Yang, S.; Hua, P. Bioinformatic gene analysis for potential biomarkers and therapeutic targets of atrial fibrillation-related stroke. J. Transl. Med. 2019, 17, 45. [Google Scholar] [CrossRef]
- Imdadullah, M.; Aslam, M.; Altaf, S. mctest: An R Package for Detection of Collinearity among Regressors. R J. 2016, 8, 495–505. [Google Scholar] [CrossRef]
- Enerly, E.; Steinfeld, I.; Kleivi, K.; Leivonen, S.K.; Aure, M.R.; Russnes, H.G.; Rønneberg, J.A.; Johnsen, H.; Navon, R.; Rødland, E.; et al. miRNA-mRNA integrated analysis reveals roles for miRNAs in primary breast tumors. PLoS ONE 2011, 6, e16915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haakensen, V.D.; Steinfeld, I.; Saldova, R.F.; Shehni, A.A.; Kifer, I.; Naume, B.; Rudd, P.M.; Børresen-Dale, A.-L.; Yakhini, Z. Serum N-glycan analysis in breast cancer patients—Relation to tumour biology and clinical outcome. Mol. Oncol. 2016, 10, 59–72. [Google Scholar] [CrossRef]
- Bischoff, P.; Kornhuber, M.; Dunst, S.; Zell, J.; Fauler, B.; Mielke, T.; Taubenberger, A.V.; Guck, J.; Oelgeschläger, M.; Schönfelder, G. Estrogens determine adherens junction organization and E-Cadherin clustering in breast cancer cells via Amphiregulin. iScience 2020, 23, 101683. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, M.; Xu, F.; Jiang, S. Wnt signaling in breast cancer: Biological mechanisms, challenges and opportunities. Mol. Cancer 2020, 19, 1–35. [Google Scholar] [CrossRef]
- Phongwattana, T.; Engchuan, W.; Chan, J.H. Clustering-based multi-class classification of complex disease. In Proceedings of the 2015 7th International Conference on Knowledge and Smart Technology (KST), Chonburi, Thailand, 28–31 January 2015; pp. 25–29. [Google Scholar]
- Obuchowski, N.A.; Bullen, J.A. Receiver operating characteristic (ROC) curves: Review of methods with applications in diagnostic medicine. Phys. Med. Biol. 2018, 63, 07TR01. [Google Scholar] [CrossRef]
- Zhao, L.; Lee, V.; Ng, M.K.; Yan, H.; Bijlsma, M.F. Molecular subtyping of cancer: Current status and moving toward clinical applications. Brief. Bioinform. 2018, 20, 572–584. [Google Scholar] [CrossRef]
- Sáez, A.; Sánchez-Monedero, J.; Gutiérrez, P.A.; Hervás-Martínez, C. Machine learning methods for binary and multiclass classification of melanoma thickness from dermoscopic images. IEEE Trans. Med Imaging 2016, 35, 1036–1045. [Google Scholar] [CrossRef]
- Wang, J.; Zuo, Y.; Man, Y.-G.; Avital, I.; Stojadinovic, A.; Liu, M.; Yang, X.; Varghese, R.S.; Tadesse, M.G.; Ressom, H.W. Pathway and Network Approaches for Identification of Cancer Signature Markers from Omics Data. J. Cancer 2015, 6, 54–65. [Google Scholar] [CrossRef] [Green Version]
- Breitkreutz, D.; Hlatky, L.; Rietman, E.; Tuszynski, J. Molecular signaling network complexity is correlated with cancer patient survivability. Proc. Natl. Acad. Sci. USA 2012, 109, 9209–9212. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.C.; Ma, F.; Shen, Y.; Hu, Y.Q.; Pan, S. Overexpression of SMAR1 enhances radio-sensitivity in human breast cancer cell line MCF7 via activation of p53 signaling pathway. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2015, 22, 293–300. [Google Scholar]
- Sun, G.; Shan, M.H.; Ma, B.L.; Geng, Z.L.; Alibiyati, A.; Zhong, H.; Wang, J.; Ren, G.H.; Li, H.T.; Dong, C. Identifying crosstalk of mTOR signaling pathway of lobular breast carcinomas. Eur. Rev. Med. Pharmacol. Sci. 2012, 16, 1355–1361. [Google Scholar] [PubMed]
- Pécuchet, N.; Popova, T.; Manié, E.; Lucchesi, C.; Battistella, A.; Vincent-Salomon, A.; Caux-Moncoutier, V.; Bollet, M.; Sigal-Zafrani, B.; Sastre-Garau, X.; et al. Loss of heterozygosity at 13q13 and 14q32 predicts BRCA2 inactivation in luminal breast carcinomas. Int. J. Cancer 2013, 133, 2834–2842. [Google Scholar] [PubMed]
- Quintela-Fandino, M.; Arpaia, E.; Brenner, D.; Goh, T.; Yeung, F.A.; Blaser, H.; Alexandrova, R.; Lind, E.F.; Tusche, M.W.; Wakeham, A.; et al. HUNK suppresses metastasis of basal type breast cancers by disrupting the interaction between PP2A and cofilin-1. Proc. Natl. Acad. Sci. USA 2010, 107, 2622–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Yan, M.; Patra, J.; Natrajan, R.; Yan, Y.; Swagemakers, S.; Tomaszewski, J.M.; Verschoor, S.; Millar, E.K.; Van der Spek, P.; et al. Enhanced RAD21 cohesin expression confers poor prognosis and resistance to chemo-therapy in high grade luminal, basal and HER2 breast cancers. Breast Cancer Res. 2011, 13, R9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, F.; Esnakula, A.; Ricks-Santi, L.J.; Zafar, R.; Kanaan, Y.; Naab, T. Loss of PTEN in high grade advanced stage triple negative breast ductal cancers in African American women. Pathol. Res. Pract. 2018, 214, 673–678. [Google Scholar] [CrossRef]
- Koni, M.; Pinnarò, V.; Brizzi, M.F. The Wnt Signalling Pathway: A Tailored Target in Cancer. Int. J. Mol. Sci. 2020, 21, 7697. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, J.; Li, L.; Xia, H.; Lin, Z.; Zhong, T. AMPH-1 is critical for breast cancer progression. J. Cancer 2018, 9, 2175–2182. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GEO Accession Numbers | Number of Genes (After Pre-Processed) | Number of Samples with Classes | Links |
|---|---|---|---|
| GSE1561 [32] | 12,437 | Luminal: 27 Basal: 16 Apocrine: 6 | www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE1561 (accessed on 15 February 2021) |
| GSE1456 [33] | 12,437 | Luminal: 62 Basal: 25 ERBB2 1: 15 Normal: 37 | www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE1456 (accessed on 15 February 2021) |
| Comparisons | DRW [4] | eDRW+ |
|---|---|---|
| Gene expression data | Binary class | Multiclass |
| Pathways | Metabolic and Non-metabolic Pathways | |
| Number of genes in the directed graph | 4113 genes | |
| Repeated and missing genes | No | Yes (required for datasets pre-processing) |
| Normalization based on z-scores | Yes | |
| Differential expression analysis | t-test (equal variances) | One-way ANOVA (F-test) |
| Restart probability (r) | 0.7 | |
| Interaction between genes | Interaction between two genes (directed edges) | |
| Position of genes | Yes | |
| Weight of the genes | Yes | |
| Pathway selection | No | Yes |
| Main Variable | Other Variables | VIF 1 | TOL 2 | Leamer 3 | Red Indicator 4 | R25 |
|---|---|---|---|---|---|---|
| Subtype | SURV_RELAPSE | 12.68 | 0.08 | 0.28 | 0.63 | 0.24 |
| SURV_DEATH | 9.16 | 0.11 | 0.33 |
| Method | eDRW+ | DRW (Re-Run) | Mean-Centering & Magnitude-Normalization | PCA & AHC | NCFS-CORG | NCFS-i | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Classifier | LR | SVM | NB | LR | CPR | CR | LR | NB | LR | LR (GS) | NB | SVM |
| GSE1561 [32] | 0.95 | 0.92 | 0.94 | 0.97 | 0.80 | NA | NA | NA | 0.95 | NA | 0.95 | NA |
| GSE1456 [33] | 0.91 | 0.80 | 0.92 | 0.91 | NA | 0.55 | 0.75 | 0.83 | 0.75 | 0.86 | 0.84 | 0.90 |
| eDRW+ (LR) vs. Compared Methods | Mean-Centering & Magnitude-Normalization | NCFS-i |
|---|---|---|
| p-values | 0.0678 | |
| 95% Confidence Interval | (0.1546, 0.1783) | (−0.0336, 0.0013) |
| Difference (%) | 16.65 | 0.65 |
| eDRW+ (LR) vs Compared Methods | PCA & AHC | NCFS-CORG | NCFS-i (Genetic Search) |
|---|---|---|---|
| p-values | |||
| 95% Confidence Interval | (0.3566, 0.3671) | (0.1586, 0.1691) | (0.0446, 0.0551) |
| Difference (%) | 36.18 | 16.38 | 4.98 |
| Training Set | GSE1456 | GSE1456 | GSE1561 |
|---|---|---|---|
| Test Set | GSE1561 | GSE19536 | GSE1456 |
| eDRW+ (Logistic regression model) | 0.82 | 0.80 | 0.68 |
| eDRW+ (Support vector machine) | 0.54 | 0.53 | 0.49 |
| eDRW+ (Naïve Bayes) | 0.76 | 0.81 | 0.69 |
| Datasets | Number of Identified Pathways | Number of Cancer Pathway Markers | Number of Identified Genes | Number of Cancer Gene Markers | ||
|---|---|---|---|---|---|---|
| PubMed | DAVID | PubMed | DAVID | |||
| GSE1561 | 24 | 18 | 7 | 536 | 144 | 4 |
| GSE1456 | 52 | 39 | 13 | 953 | 315 | 9 |
| KEGG Pathway IDs | Pathway Names | DRW | eDRW+ |
|---|---|---|---|
| HSA04010 | MAPK signaling pathway | Yes | No |
| HSA04020 | Calcium signaling pathway | Yes | No |
| HSA04144 | Endocytosis | Yes | Yes |
| HSA04310 | Wnt signaling pathway | Yes | Yes |
| HSA04520 | Adherens junction | No | Yes |
| HSA04810 | Regulation of actin cytoskeleton | Yes | No |
| Gene Entrez ID | KEGG Pathway IDs | OMIM_DISEASE |
|---|---|---|
| 207 (AKT1) | HSA04150: mTOR signalling pathway, HSA04620: Toll-like receptor signalling pathway, HSA04630: Jak-STAT signalling pathway, HSA04914: Progesterone-mediated oocyte maturation, HSA04920: Adipocytokine signalling pathway | Breast cancer, Colorectal cancer, Ovarian cancer |
| 7251 (TSG101) | HSA04144: Endocytosis | Breast cancer |
| 367 (AR) | HSA04114: Oocyte meiosis | Prostate cancer, Breast cancer |
| 675 (BRCA2) | HSA03440: Homologous recombination | Breast cancer, Prostate cancer, Pancreatic cancer |
| 841 (CASP8) | HSA04115: p53 signalling pathway, HSA04620: Toll-like receptor signalling pathway, HSA04622: RIG-I-like receptor signalling pathway | Breast cancer, Hepatocellular carcinoma, Lung cancer |
| 999 (CDH1) | HSA04520: Adherens junction | Breast cancer, Gastric cancer, Ovarian cancer, Prostate cancer |
| 5888 (RAD51) | HSA03440: Homologous recombination | Breast cancer |
| 7157 (TP53) | HSA04110: Cell cycle, HSA04115: p53 signalling pathway, HSA04310: Wnt signalling pathway | Breast cancer, Colorectal cancer, Hepatocellular carcinoma, Pancreatic cancer, Nasopharyngeal carcinoma, Basal cell carcinoma |
| 11,200 (CHEK2) | HSA04110: Cell cycle, HSA04115: p53 signalling pathway | Breast cancer, Prostate cancer, Colorectal cancer |
| 5290 (PIK3CA) | HSA04070: Phosphatidylinositol signalling system, HSA04150: mTOR signalling pathway, HSA04660: T cell receptor signalling pathway, HSA04910: Insulin signalling pathway | Breast cancer, Colorectal cancer, Ovarian cancer, Non-small cell lung cancer, Hepatocellular cancer, Gastric cancer |
| 8493 (PPM1D) | HSA04115: p53 signalling pathway | Breast cancer |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nies, H.W.; Mohamad, M.S.; Zakaria, Z.; Chan, W.H.; Remli, M.A.; Nies, Y.H. Enhanced Directed Random Walk for the Identification of Breast Cancer Prognostic Markers from Multiclass Expression Data. Entropy 2021, 23, 1232. https://0-doi-org.brum.beds.ac.uk/10.3390/e23091232
Nies HW, Mohamad MS, Zakaria Z, Chan WH, Remli MA, Nies YH. Enhanced Directed Random Walk for the Identification of Breast Cancer Prognostic Markers from Multiclass Expression Data. Entropy. 2021; 23(9):1232. https://0-doi-org.brum.beds.ac.uk/10.3390/e23091232
Chicago/Turabian StyleNies, Hui Wen, Mohd Saberi Mohamad, Zalmiyah Zakaria, Weng Howe Chan, Muhammad Akmal Remli, and Yong Hui Nies. 2021. "Enhanced Directed Random Walk for the Identification of Breast Cancer Prognostic Markers from Multiclass Expression Data" Entropy 23, no. 9: 1232. https://0-doi-org.brum.beds.ac.uk/10.3390/e23091232