Introduction
Organic compounds obtained from radical coupling of phenylpropenoidic phenols have an important biological role. In fact, they constitute organic polymers such as lignin [
1], lignans [
2], suberin [
3] and algal cell walls [
4].
The lignans represent a structurally very diverse class of vascular plant products. They are typically dimers and their primary physiological role in plants is in plant defense [
5]. Some lignans have found application in medicine, such as podophyllotoxin in venereal wart treatment [
6], or its semisynthetic derivatives etoposide, and teniposide in cancer therapies [
7]. Stereoselective total syntheses of lignans can involve multistep procedures [
8]. In particular Charlton’s group has synthesized important podophyllotoxin analogs with applications in medicine. [
9].
A bimolecular phenoxy radical coupling could be an alternative way to perform stereoselective syntheses of lignans, but, unlike most biological oxidations, the bimolecular phenoxy radical coupling reaction is not under a strictly regio- and stereospecific control [
10]. This is due to the fact that phenoxy radicals are very persistent and the dimerization reaction is slow. Hence the stereogenic carbons formed in the oxidative phenol coupling reaction in vitro are racemic [
11]. Sarkanenen has studied the radical phenol coupling since 1973 and he has demonstrated that the β-β coupling of phenols such as (
E)-isoeugenol is remarkably stereospecific and produces exclusively
threo-compounds, whereas the (
Z)-isoeugenol gives
threo- and
erythro-coupling products in equal amounts. He has proposed that the differences in the probabilities of coupling modes in the oxidations of (
E) and (
Z)-isoeugenol must be considered to be due to the characteristics of these intermediate complexes rather than to the differences in spin densities [
12]. A similar enzymatic oxidative coupling of ferulic acid derivatives was observed by us for the diastereoselective synthesis of benzo[b]phenylcoumarans [
13].
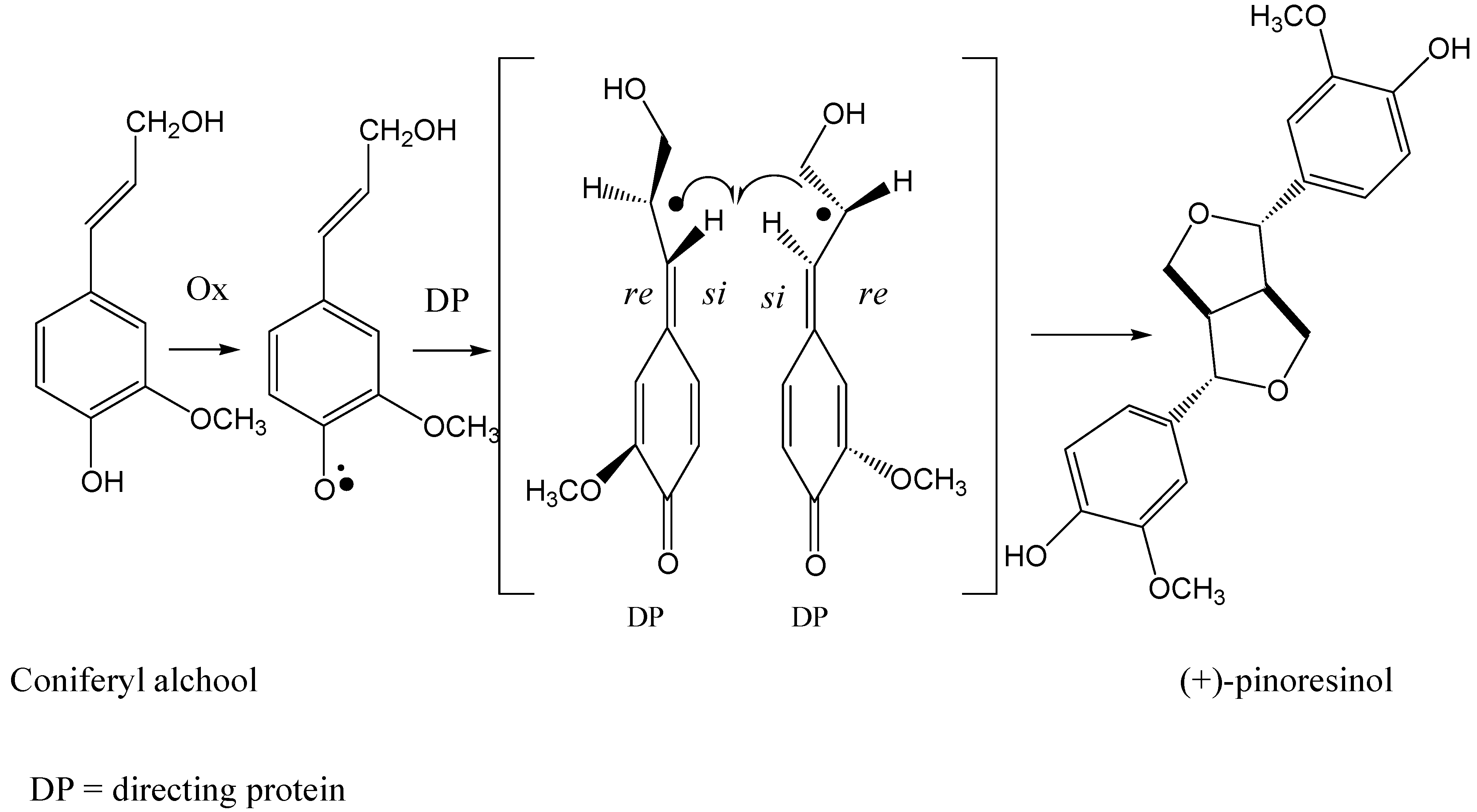
Scheme 1.
The formation of (+) pinoresinol by directing protein mediated coupling of coniferyl alcohol.
Scheme 1.
The formation of (+) pinoresinol by directing protein mediated coupling of coniferyl alcohol.
Recently Lewis has proposed a new biosynthetic pathway to enantiopure lignans. A protein isolated from
Forsythia species is suggested to be responsible for the formation of enantiomeric pure pinoresinol from coniferyl alcohol [
14]. In this case, the protein acts as a chiral inducer. The mechanism of steroselective coupling of coniferyl phenoxy radical mediated by a directing protein proposed by Lewis is shown in
Scheme 1 [
15]. The role of the directing protein is supposed to be at the level of β-β coupling of phenols. Stereocontrol in enzymatic oxidative coupling of ferulic acid derivatives to enantioselectively give benzo[b]phenylcoumarans [
16] has been recently shown by us to be possible in the oxidative phenol coupling reaction [
17] using a chiral inducer and the horseradish peroxidase (HRP)-catalyzed oxidative coupling in presence of hydrogen peroxide as the oxidant. The chiral inducers were aminoacid ethyl esters, camphorsultam and aryloxazolidinones[
18].
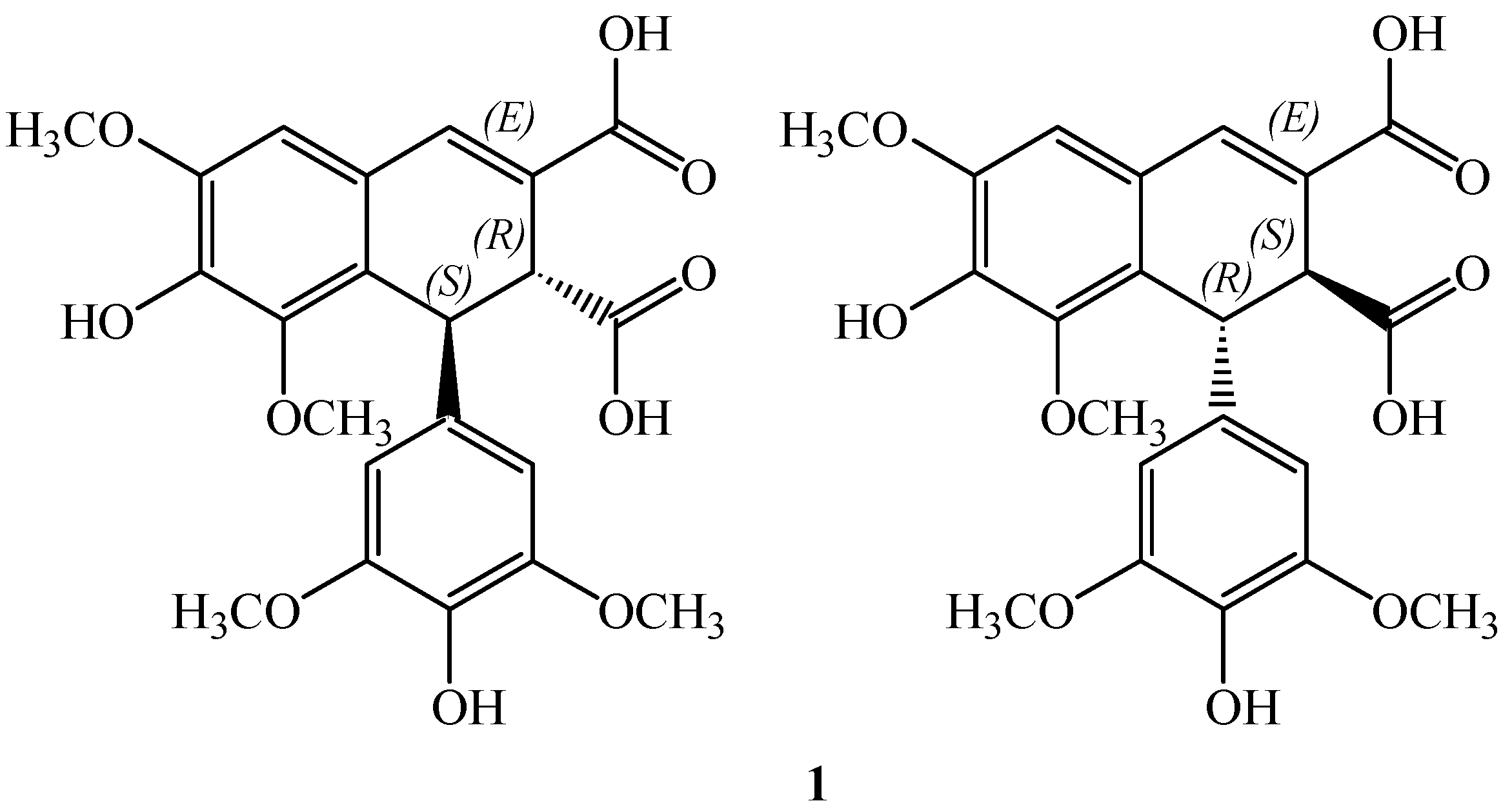
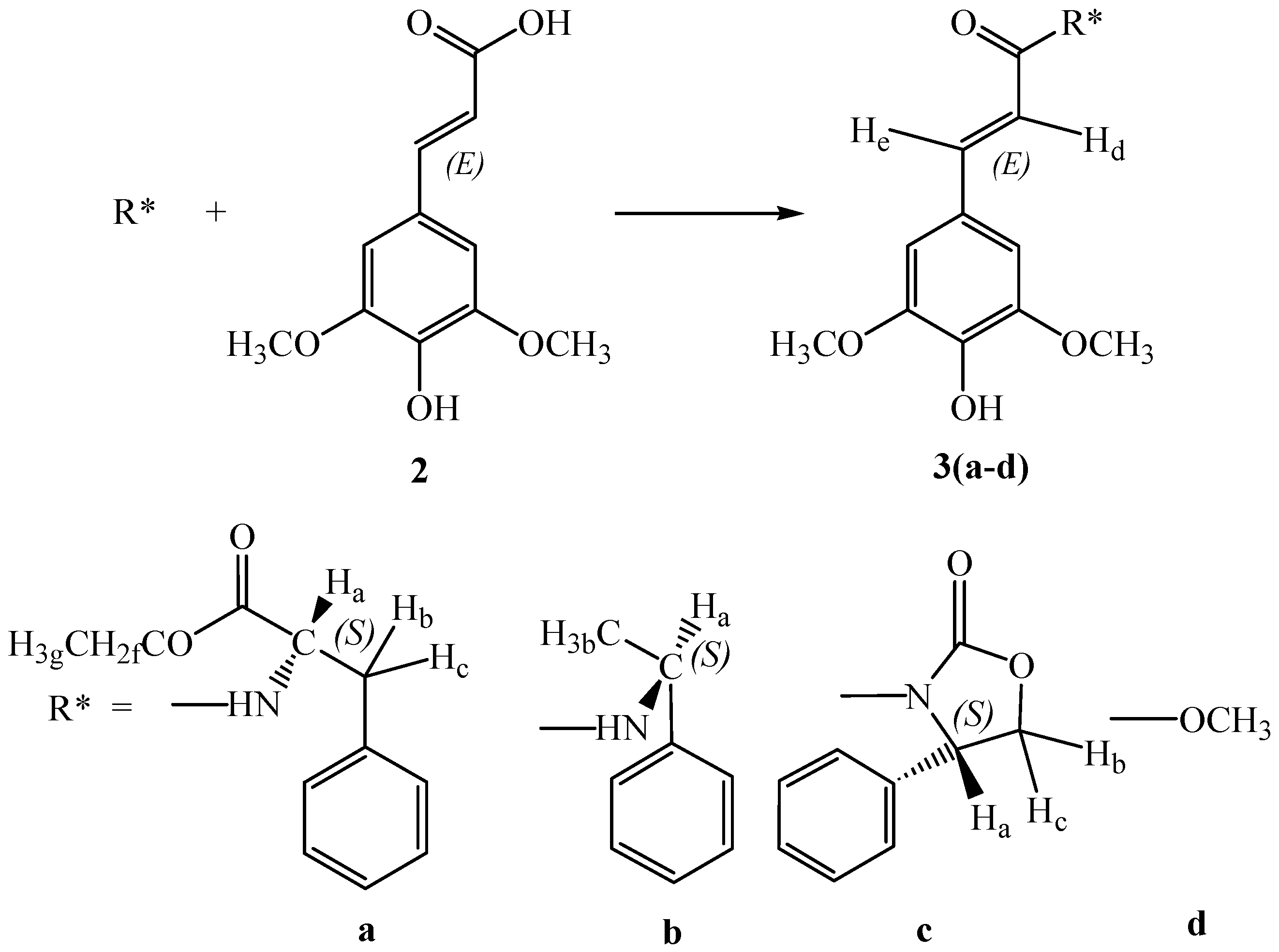
On these bases we focused our attention to thomasidioic acid (
1,
Figure 1). This lignan in the
trans configuration was isolated for the first time from
Ulmus thomasii in 1969 and shown to be cytotoxic [
19]. Recently this acid has been shown to prevent the peroxynitrite-mediated protein nitration [
20].
Several synthetic methods have been developed to prepare racemic
trans thomasidioic acid: Stobbe condensation of 3,4,5-trimethoxybenzaldehyde with dimethylsuccinate [
21]; alkaline treatment of sinapic acid
2 [
22], oxidative coupling of sinapic acid
2 [
23] or of sinapic acid methyl ester
3d [
24] with ferric chloride in aqueous acetone (
Scheme 2). Higher yields were obtained using hydrogen peroxide as the oxidant and horseradish peroxidase as the catalyst [
25].
Scheme 2.
Synthesis of amides from sinapic acid.
Scheme 2.
Synthesis of amides from sinapic acid.
These reactions are important in food processing because sinapic acid
2 is a major constituent of canola oil [
26]. This finding stimulated the question whether thomasidioic acid 1 is really a natural product. Charlton has demonstrated that
trans thomasidioic acid can be formed also by air oxidation of sinapic acid at pH 13. On this basis and on the basis that “natural”
trans thomasidioic acid is optically inactive, he suggested that this acid is not a real natural product of plant metabolism, but it is generated during the extraction procedure from basic aqueous extracts of
Ulmus thomasii [
27]. Chiral induction was obtained in a preliminary study showing that Me (
R)-mandelyl sinapate could be dimerized diastereoselectively to give a 1,2-
trans thomasidioate diester [
28].
Results and Discussion
Here we report an enantioselective biomimetic approach to synthesize thomasidioic acid (1). In order to achieve this result we performed a preliminary oxidation of methyl-(E)-sinapate 3d catalyzed by horseradish peroxidase (HRP, EC 1.11.1.7), using hydrogen peroxide as the reactant. The best results of oxidative coupling were obtained in a dioxane 30%-buffer pH 4, 70% solution.
The yield of racemic mixture of
trans-thomasidioic acid dimethylesters
4d,
5d was 70%. In agreement with literature data it was no amounts of
cis-thomasidioic acid dimethylesther were observed. The enantioselective oxidative phenol coupling of sinapic acid derivatives
3(a-c) having an amide bond with
(S)-phenylalanine ethyl ester,
(S)-methylbenzylamine,
(S)-2-phenyloxazolidinone as chiral auxiliaries was performed using the same oxidation system. These sinapamides with the chiral auxiliaries were obtained in high yields in a one pot reaction from sinapic acid
2 (
Scheme 2). The mixture of the two
trans diastereoisomers
4,
5 (
Figure 2) was obtained with the diastereoisomeric excesses reported in
Table 1.
Table 1.
The enantioselective oxidative phenol coupling with chiral sinapamides.
Table 1.
The enantioselective oxidative phenol coupling with chiral sinapamides.
| Chiral auxiliary R* | Yields in 4 + 5 (%) | Yield in peak 1 (%) | Yield in peak 2 (%) | Diastereoisomeric excess (%) |
|---|
| (S)-phenylalanine ethyl ester | 60 | 45 | 15 | 50 |
| (S)-methylbenzylamine | 50 | 35 | 15 | 40 |
| (S)-2-phenyloxazolidinone | 40 | 34 | 6 | 70 |
The diastereoisomeric excess was evaluated by RP-HPLC analysis of the crude reaction mixture. Separation by preparative RP-HPLC allowed to obtain the individual diastereoisomers which were characterized by 1H-NMR.
The hydrolysis of the individual
trans-thomasidioic acid amides
4 and
5 (
Scheme 2) was performed with LiOOH in tetrahydrofuran (
Table 2).
Table 2.
The hydrolysis of thomasidioic acid derivatives.
Table 2.
The hydrolysis of thomasidioic acid derivatives.
| Chiral auxiliary R* | Reactant | Solvent | Time (hours) | Yield of 1 (%) |
|---|
| (S)-Phenylalanine ethyl ester | LiOOH | THF | 5 | 20 |
| (S)-Methylbenzylamine | LiOOH | THF | 4 | 10 |
| (S)-2-Phenyloxazolidinone | LiOOH | THF | 5 | 60 |
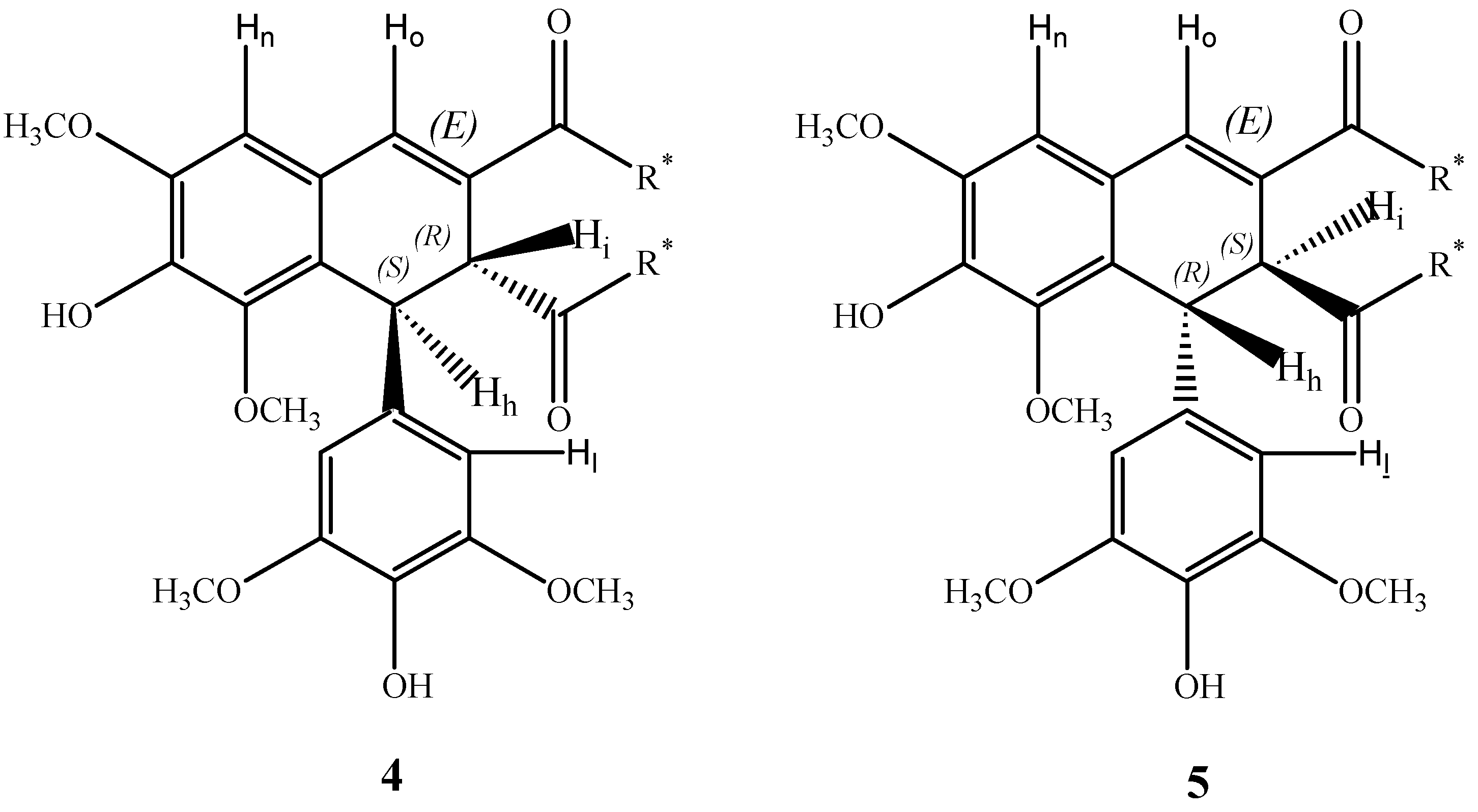
In order to account for the origin of the observed enantioselection and diasteroselection in the formation of thomasidioic acid derivatives
4,
5, the reaction mechanism has been investigated in detail by quantum-mechanical methods. As shown in
Table 1 the chiral auxiliaries
(S)-2-phenyl-oxazolidinone gave considerably higher selectivities (e.e. 70 %) compared to the
(S)-phenylalanine ethyl ester and
(S)-methylbenzylamine. Therefore, the compounds having
(S)-2-phenyloxazolidinone as the chiral auxiliary were chosen for the theoretical study.
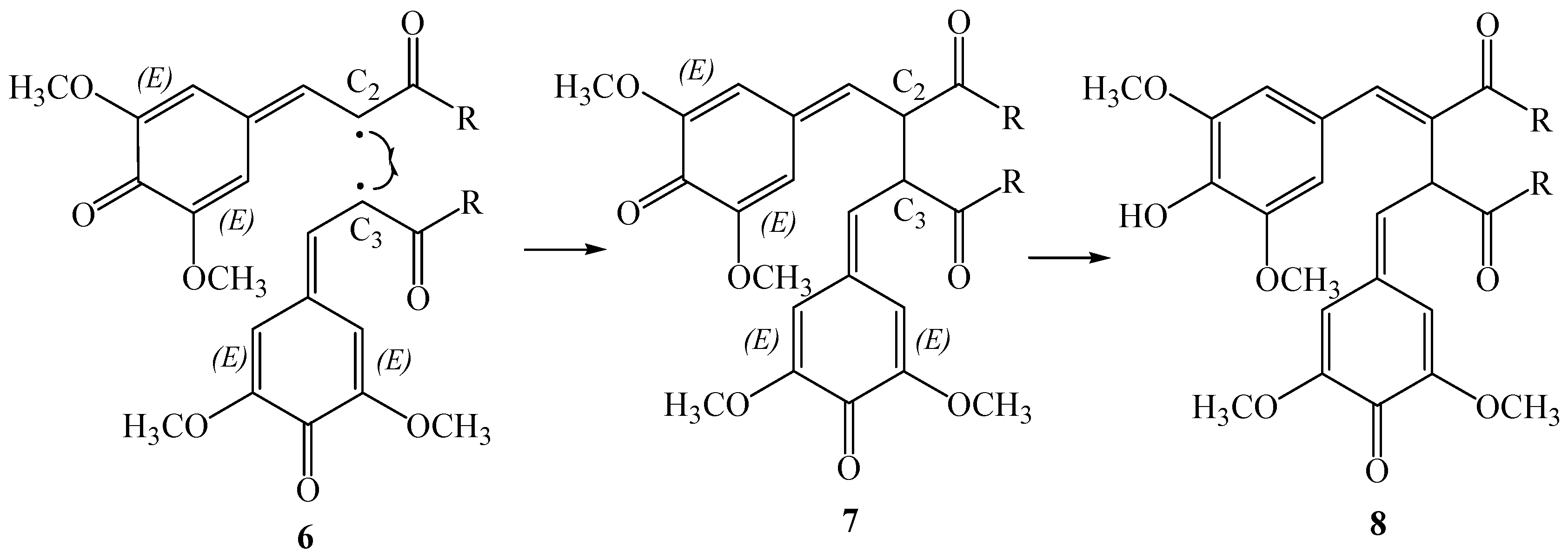
The proposed reaction pathway involves two steps; the first step is the β-β oxidative coupling of two quinomethide radicals
6 to give the bisquinomethide
7, and the second step is the ring closure of
7 to give the final product (see
Scheme 3). For this mechanism the absolute configuration of thomasidioic acid should be determined by the absolute configuration of the two stereogenic centers of the bisquinomethide
7 formed in the β-β oxidative coupling, and by the
trans or
cis ring closure of the bisquinomethide
7.
The relative orientation of the two quinomethide radicals 6 in the β-β oxidative coupling determines the configuration of the stereogenic centres in 7. In fact, coupling at the re-re and si-si face lead to the formation of R,R- and S, S-bisquinomethide 7, while coupling at the re-si face results in the corresponding R,S/S,R mesoform. In this context, the preferred absolute configuration of 7 should be controlled by the different stabilities of the pre-reactive van der Waals complexes of dimers of 6, and the corresponding transition states along the reaction path to give the bisquinomethide 7.
Scheme 3.
Radical coupling to form bisquinone methide 7 followed by aromatisation of one ring.
Scheme 3.
Radical coupling to form bisquinone methide 7 followed by aromatisation of one ring.
It should be noted that the computational study of the β-β oxidative coupling is complicated by the diradical nature of the molecular system along the reaction path, which can only be modelled accurately by using the Configuration Interaction approach (CI). For this reason the semiempirical CI-PM3 level of theory has been adopted to investigate the β-β oxidative coupling of quinomethide radicals 6. Due to the large size of the molecular system only the frontier orbitals were included in the active space for the CI vector.
Initially, a conformational analysis of the quinomethide radical 6 has been performed in order to find the most stable conformations involved in the β-β coupling. In this analysis two energy minimum conformers has been characterized, one in which the amide bond is trans (6a) and the other one in which the amide bond is cis (6b). The former is more stable than the latter by about 2 kcal∙mol-1. Therefore, the conformer 6a with trans amide bond was chosen for the study of the β-β oxidative coupling.
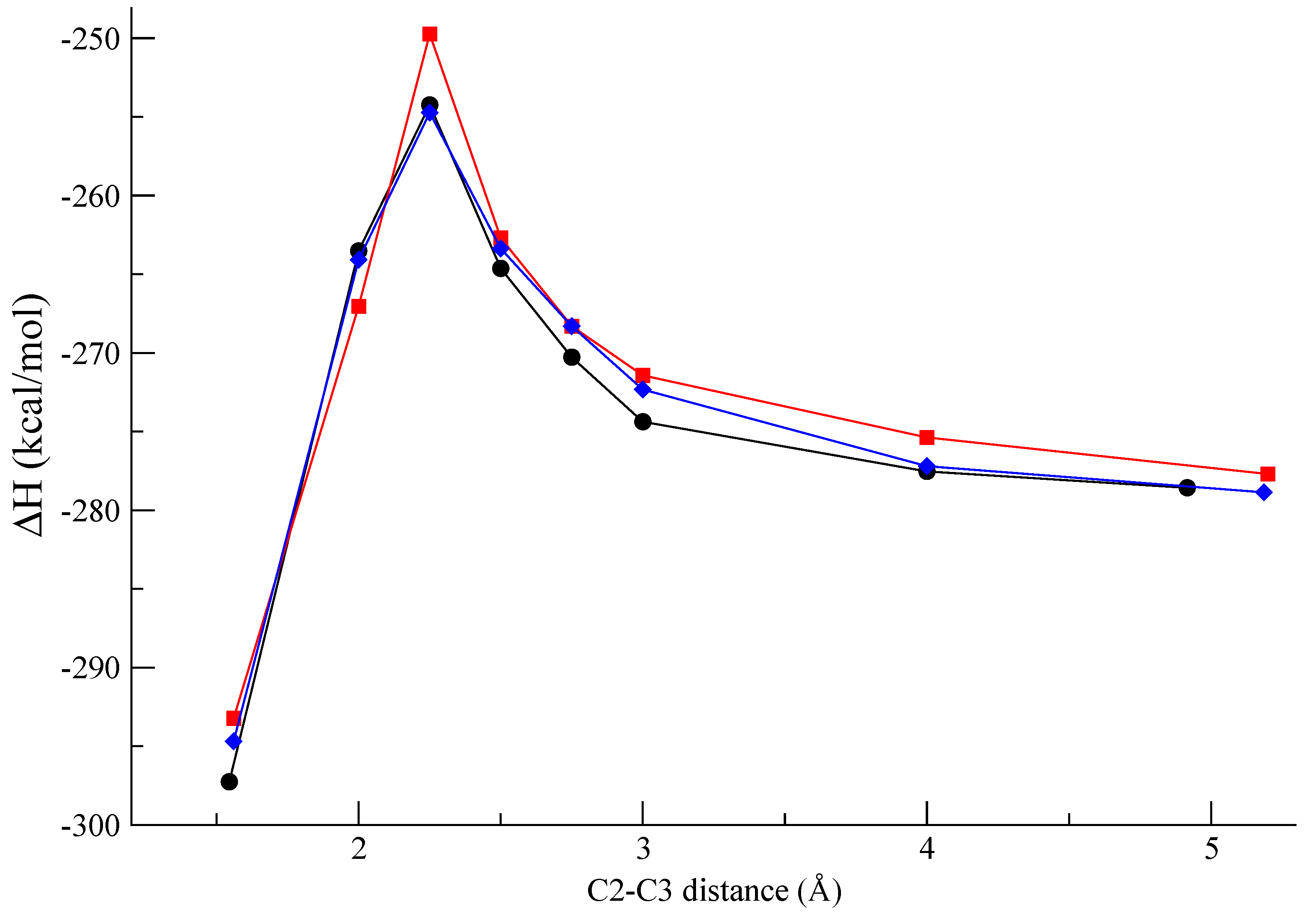
Figure 3 shows the computed profile along the reaction pathway for the β-β coupling at the
re-re,
si-si, and
re-si faces. In
Figure 4 the molecular geometries of the pre-reactive van der Waals complexes and the corresponding bisquinomethide products in the first conformation after the formation of the C-C in the radical coupling are also shown. It is interesting to note that the energy barrier calculated for the
si-si coupling is higher by more than 4 kcal∙mol
-1 than that calculated for the
re-re and
re-si coupling. This result suggests that the formation of the
R,R (from
re-re coupling), and
R,S/
S,R (from
re-si coupling) bisquinomethide
7 is preferred with respect to the formation of the
S,S (from
si-si coupling) bisquinomethide
7.
Sarkanen
et al. suggested that the excess of the
threo β-β coupling products over the
erithreo ones, observed for some quinomethide derivatives such as syringylresinol and 2-propenylphenol [
12] is due to the different stability of the
re-re,
si-si and
re-si pre-reactive complexes. However, as shown in
Figure 1, the pre-reactive complexes calculated in this work are almost isoenergetic, suggesting that for the bisquinomethide
7 the stereochemistry is not controlled by the formation of this pre-reactive complex.
As discussed above, the second step of the reaction mechanism is the ring closure of the bisquinomethide
7 (see
Scheme 2). The cyclization step determines the observed diasteroselection to the
trans isomer. The diasteroselection should be controlled by the relative orientation of the two quinomethide rings which should feature a preferred conformation in order to give the “correct” ring closure, as well as to the energy of the transition state along the reaction path to give the final product.
Figure 3.
Reaction profile of the β-β oxidative coupling of two quinometide radicals
6 at the re-re (●-●),
si-si (
![Molecules 13 00129 i001]()
), and
re-si (
![Molecules 13 00129 i002]()
) faces.
Figure 3.
Reaction profile of the β-β oxidative coupling of two quinometide radicals
6 at the re-re (●-●),
si-si (
![Molecules 13 00129 i001]()
), and
re-si (
![Molecules 13 00129 i002]()
) faces.
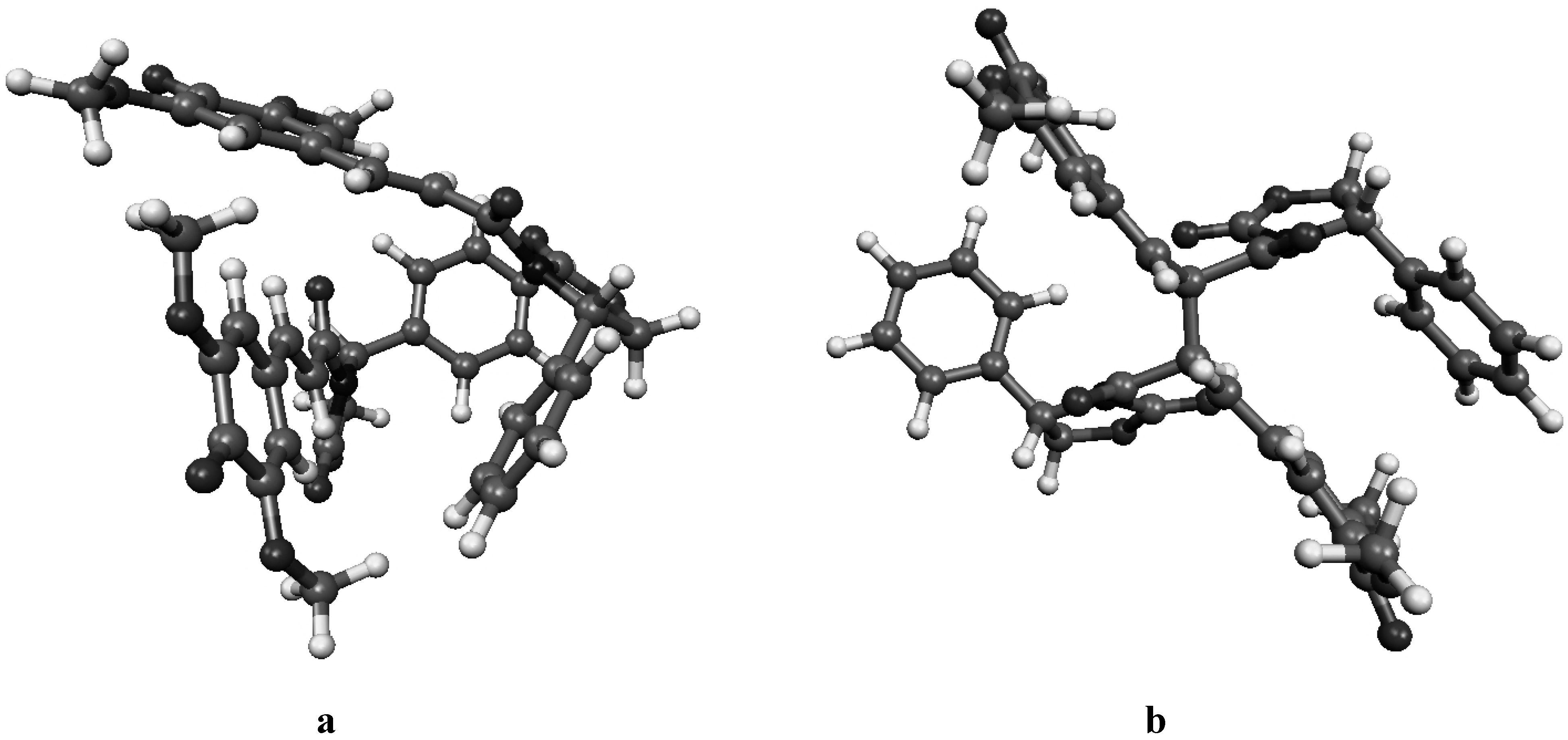
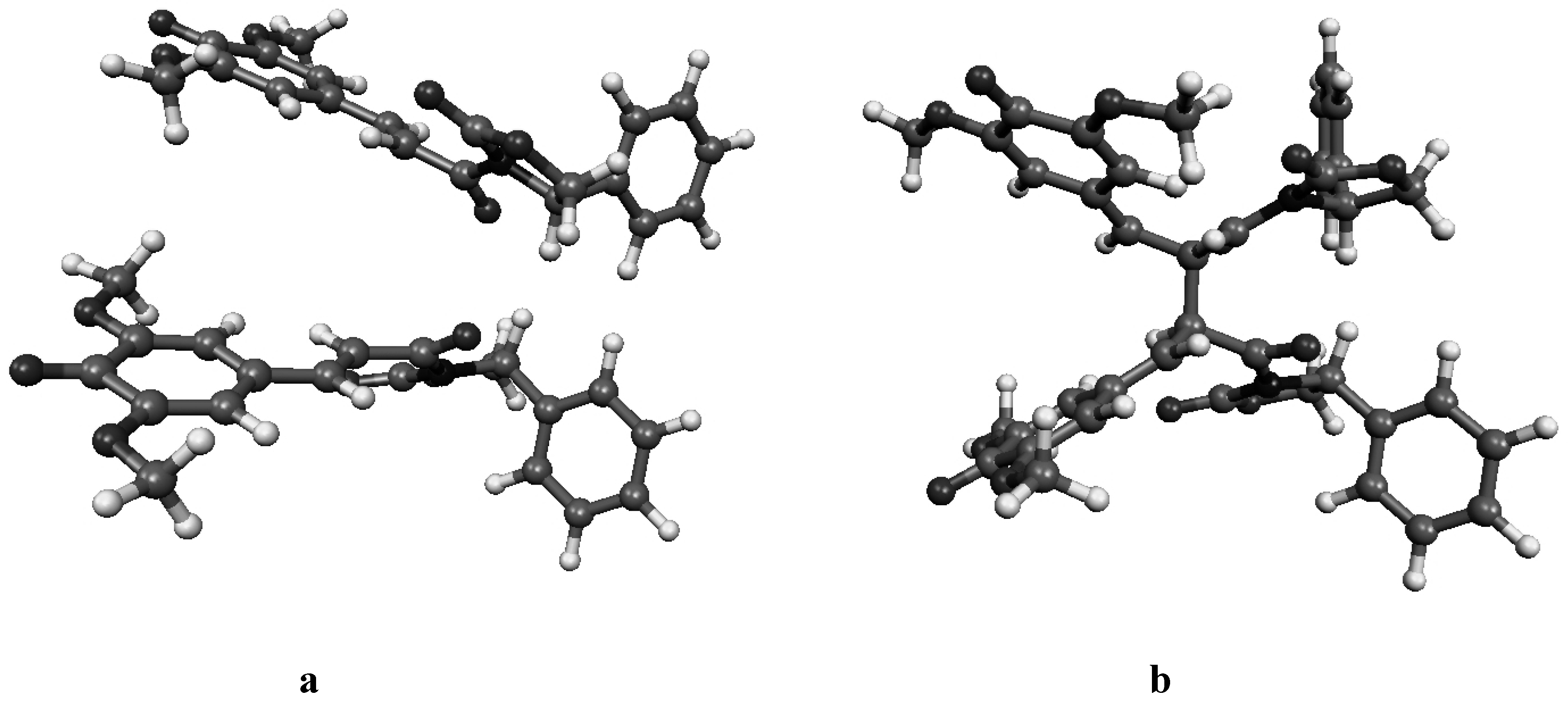
Figure 4a.
Molecular geometry of the van der Waals pre-reactive complex with the monomers at the re-re face (a), and the corresponding bisquinomethide product (b).
Figure 4a.
Molecular geometry of the van der Waals pre-reactive complex with the monomers at the re-re face (a), and the corresponding bisquinomethide product (b).
It should be noted that the cyclization should occur after the aromatization of one of the rings of bisquinomethide
7 with loss of one stereogenic center (
Scheme 3, compound
8). However, the carbon backbone of the intermediate
8 is much more rigid than in bisquinomethide
7, possibly preventing rearrangements to conformations with the rings in a different orientation. On the basis of this observation, the diasteroselection to the
trans isomer should be controlled by the relative orientation of the two quinomethide rings in the most stable conformations of bisquinomethide
7. The same assumption has been used by us to show that the observed
trans-diastereoselectivity in the cyclization of ferulic acid derivatives to dihydrobenzo[b]furans [
29] derives from thermodynamic control in the formation of diastereoisomeric quinomethide intermediates. These considerations prompted us to carry out a theoretical investigation of the potential energy surface (PES) of the quinomethide intermediate in the
R,R enantiomer and
R,S mesoforms.
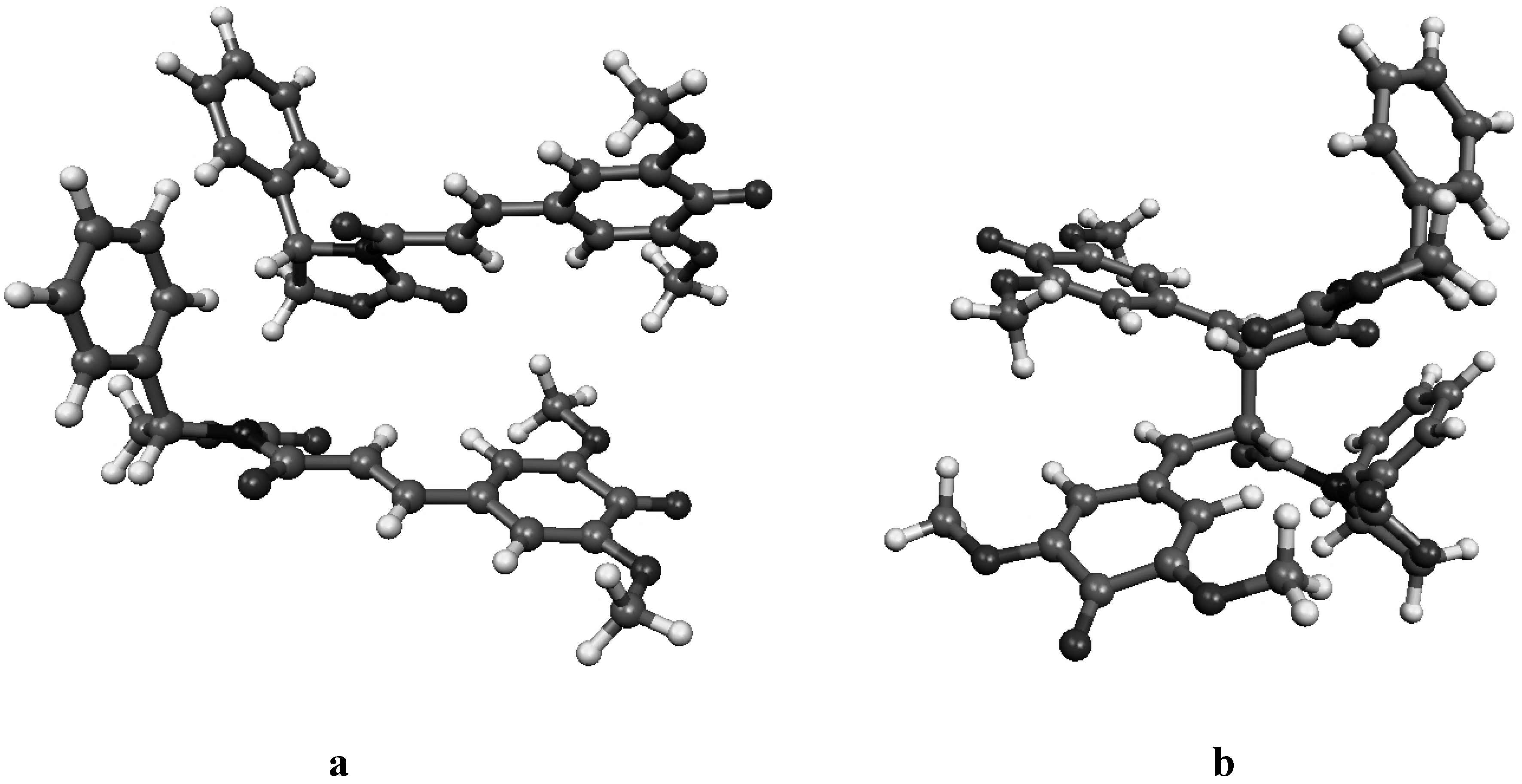
Figure 4b.
Molecular geometry of the van der Waals pre-reactive complex with the monomers at the si-si face (a), and the corresponding bisquinomethide product (b).
Figure 4b.
Molecular geometry of the van der Waals pre-reactive complex with the monomers at the si-si face (a), and the corresponding bisquinomethide product (b).
Figure 4c.
Molecular geometry of the van der Waals pre-reactive complex with the monomers at the re-si face (a), and the corresponding bisquinomethide product (b).
Figure 4c.
Molecular geometry of the van der Waals pre-reactive complex with the monomers at the re-si face (a), and the corresponding bisquinomethide product (b).
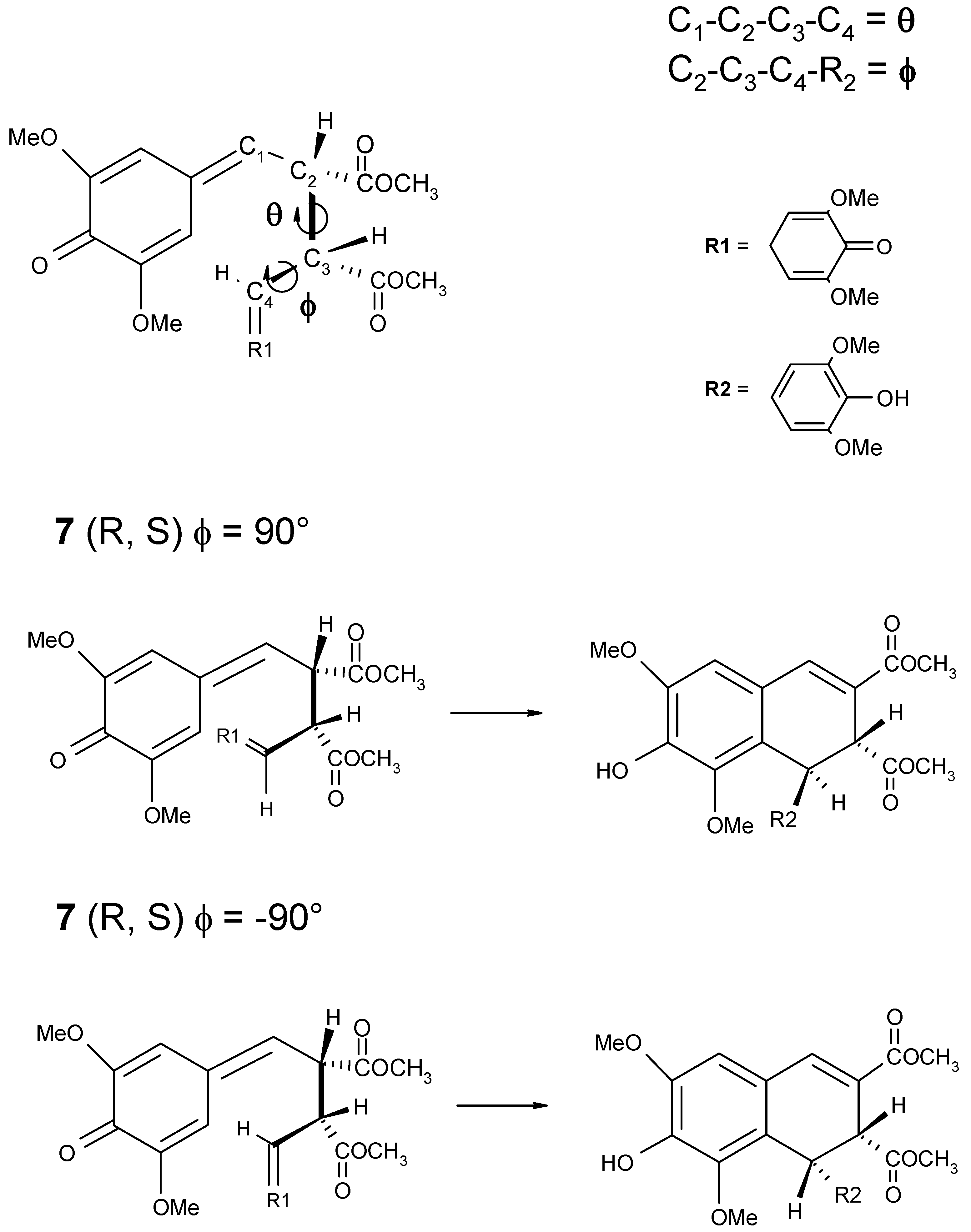
Scheme 4.
Schematic representation of the θ and ϕ dihedral angles (on the top). For the mesoform 2R,3S positive values of the ϕ angle should lead to the formation of trans-thomasidioic acid 1, while negative values the ϕ angle should lead to the formation of cis isomer (on the bottom).
Scheme 4.
Schematic representation of the θ and ϕ dihedral angles (on the top). For the mesoform 2R,3S positive values of the ϕ angle should lead to the formation of trans-thomasidioic acid 1, while negative values the ϕ angle should lead to the formation of cis isomer (on the bottom).
Because of the large number of degrees of freedom in the bisquinomethide
7 with the two chiral auxiliaries, we preliminarly performed an analysis of the PES for the bisquinomethide in which the chiral auxiliaries are replaced by -OCH
3 groups
7d, and for which, diasteroselection to the
trans isomer is also observed. The PES of the quinomethide
7d has been calculated as a function of the ϕ and θ dihedral angles as shown in
Scheme 4.
An inspection of the molecular structure of the quinomethide
7 shows that for the
R,S mesoform, conformations with
positive dihedral angle ϕ are expected to lead to the formation of thomasidioic acid
1 in the
trans configuration, while conformations with
negative dihedral angle ϕ should lead to the formation of compound
1 in the
cis configuration. The contrary holds true for the
R,R enantiomer, in which
negative and
positive values of the dihedral angle ϕ lead to the formation of
trans- and
cis-compound
1, respectively (see
Table 3).
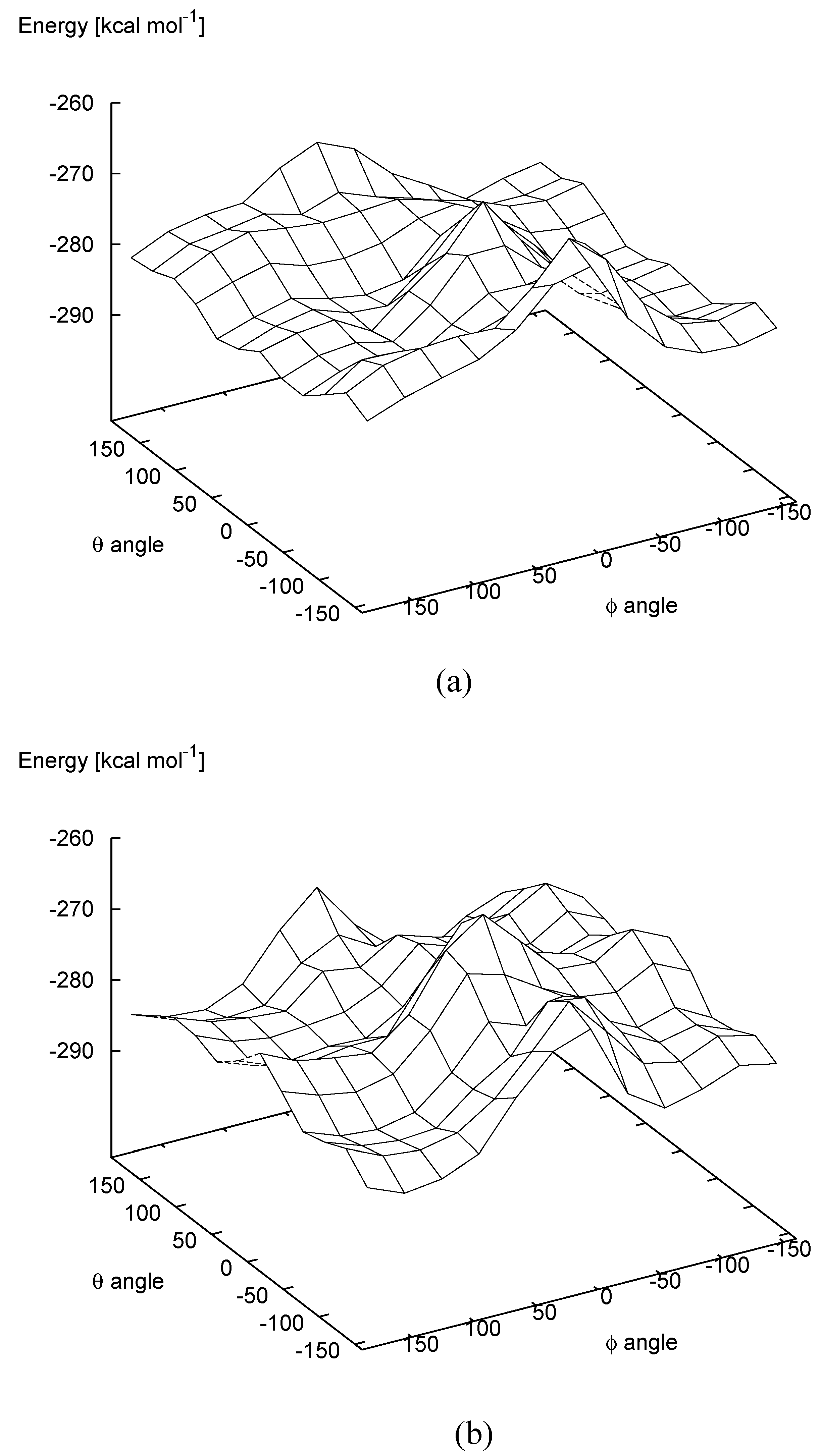
In addition, the two carbon atoms involved in the ring closure approach to each other for values of the θ angle around 0°, while they are at the maximum distance for a value of θ angle equal to about 180°. The PES computed at the semiempirical PM3 level of theory for the quinomethide
7 R,S mesoform and
R,R enantiomer are shown in
Figure 5a and
Figure 5b, respectively. In
Table 3 the values of the heat of formation corresponding to the local minima identified on the PES are reported.
Table 3.
Energy minima on the PES of the quinomethides
7 calculated as a function of theθ and ϕ dihedral angles (see
Scheme 3) at the semiempirical PM3 level of theory.
Table 3.
Energy minima on the PES of the quinomethides 7 calculated as a function of theθ and ϕ dihedral angles (see Scheme 3) at the semiempirical PM3 level of theory.
| Quinomethide 7 | θ (Degrees) | ϕ (Degrees) | Heat of formation
(kcal∙mol-1) | Thomasidioic acid (1) (predicted) |
|---|
| R,S | 180 | 120 | -286.6 | 1R,2S/1S,2R trans |
| R,S | 60 | 120 | -284.8 | 1R,2S/1S,2R trans |
| R,S | -60 | 120 | -284.3 | 1R,2S/1S,2R trans |
| R,S | 180 | -60 | -286.1 | 1R,2R/1S,2S cis |
| R,S | 60 | -90 | -279.9 | 1R,2R/1S,2S cis |
| R,S | -60 | -90 | -279.4 | 1R,2R/1S,2S cis |
| R,R | 180 | -90 | -285.3 | 1R,2S trans |
| R,R | 60 | -120 | -286.0 | 1R,2R trans |
| R,R | -60 | -120 | -284.6 | 1R,2S trans |
| R,R | 60 | 30 | -282.1 | 1R,2R cis |
In the case of the R,S mesoform a total of six local minima have been located on the PES; three, with positive values, and three with negative values of ϕ. Two almost isoenergetic minima are characterized by a value of θ of about 180° and values of ϕ of about 120° (-286.5 kcal∙mol-1) and -60° (-286.1 kcal∙mol-1). However, as noted above, in these conformations the two carbon atoms involved in the ring closure are at their maximum distance. The other local minima are located along the reaction coordinate corresponding to the rotation of the θ angle from 180° to 0°, for which the two carbon atoms are close to each other. It is interesting to note that the two minima characterized by a positive value of ϕ (θ = 60°, ϕ = 120° ΔH = -284.8; θ = −60°, ϕ = 120° ΔH = -284.3) are lower in energy by about 5 kcal∙mol-1 with respect to those characterized by negative values of ϕ (θ = 60°, ϕ = −90° ΔH = -279.9; θ = −60°, ϕ = −90° ΔH = -279.4). This clearly indicates that the most stable conformation in the region of the PES where the two carbon atoms are close enough to give rise ring closure predicts for the formation of trans-(1S,2R, or 1R,2S) thomasidioic acid 1.
In the case of the R,R enantiomer only four local minima have been located on the PES; three with negative values, and one with a positive value of ϕ. The lowest energy minimum is characterized by a value of θ of about 60° and a value of ϕ of about -120° (-286.0 kcal∙mol-1). This minimum is located along the reaction coordinate which leads to the ring closure with the formation of trans-thomasidioic acid 1. The only local energy minimum which should lead to the formation of cis-thomasidioic acid (1) θ = 60°, ϕ = 30° ΔH = -282.1) is about 4 kcal∙mol-1 higher in energy. Again, the most stable conformation in the region of the PES where the two carbon atoms are close enough to give rise to ring closure predicts for the formation of trans-(1S,2R, or 1R,2S) thomasidioic acid (1).
Figure 5.
Potential energy surface as a function of the θ and ϕ angles of quinomethide 7 with the R,R (a) and R,S (b) absolute configurations.
Figure 5.
Potential energy surface as a function of the θ and ϕ angles of quinomethide 7 with the R,R (a) and R,S (b) absolute configurations.
The same conformational analysis is than performed in order to predict the enantioselectivity in the formation of
trans-thomasidioic acid
1 from quinomethides having a (
S)-2-phenyloxazolidinone chiral auxiliary group.
Table 1 shows the enantioselectivity obtained experimentally. Three diastereoisomers are possible for this intermediate; a)
R,
R,
SS; b)
S,
S,
SS; c)
S,
R,
SS/
R,
S,
SS (mesoform). As for the case of the compound free of auxiliary groups, the conformational analysis was performed at the PM3 level by rotating the θ and ϕ angles (see
Scheme 3). Conformations corresponding to energy minima on the PES was then fully optimized. For the three isomers the energies of the most stable conformations, and the corresponding θ and ϕ angles are reported in
Table 4.
The relative stabilities of the different conformations allows to predict the amount of each quinomethide
7 isomer at the equilibrium. In this respect, we note that all energy minimum conformations calculated for the 2
S,3
S,
SS isomer are significantly less stable than that calculated for the
R,
R,
SS and
R,
S,
SS isomers (see
Table 4). The low stability observed for the
S,
S,
SS isomer is due to a larger steric hindrance between the two phenyl groups in the chiral auxiliary group. On the other hand, the energies of the
R,
R,
SS and
R,
S, SS isomers are very similar, both for the conformation with the θ angle near to 180° (in which the two carbon atoms involved in the ring closure are at the maximum distance), and for the conformation with the θ angle near to 0° (in which the two carbon atoms involved in the ring closure are at the minimum distance). As discussed for the compounds free of auxiliary groups, in the case of the
R,
R,
SS isomer conformations with
positive dihedral angle ϕ are expected to lead to the formation of thomasidioic acid
1 in the
cis configuration, while conformations with
negative dihedral angle ϕ should lead to the formation of compound
1 in the
trans configuration (see
Table 4).
Table 4.
Energy minima on the PES of the quinomethides
7 calculated as a function of the θ and ϕ dihedral angles (see
Scheme 3) at the semiempirical PM3 level of theory.
Table 4.
Energy minima on the PES of the quinomethides 7 calculated as a function of the θ and ϕ dihedral angles (see Scheme 3) at the semiempirical PM3 level of theory.
| Quinomethide 7 | R* | θ (Degrees) | ϕ (Degrees) | Heat of formation
(kcal∙mol-1) | Thomasidioic acid (1) (predicted) |
|---|
R,R
(isomer 1) | (S)-2-phenyloxazolidinone | 67 | -117 | -294.6 | 1S, 2R trans |
| 50 | 66 | -290.1 | 1R, 2R cis |
| 180 | 91 | -286.4 | --- |
| -175 | -95 | -295.5 | --- |
R,S/S,R
(mesoform) | (S)-2-phenyloxazolidinone | 47 | 73 | -294.1 | 1R, 2S / 1S, 2R trans |
| -61 | -88 | -287.6 | 1R, 2S / 1S, 2R cis |
| -84 | -113 | -293.2 | --- |
| -178 | 94 | -288.2 | --- |
S,S
(isomer 2) | (S)-2-phenyloxazolidinone | -80 | -118 | -291.5 | 1R, 2S trans |
| -59 | 128 | -286.3 | 1S, 2S cis |
| 161 | 117 | -281.9 | --- |
| 172 | -94 | -276.2 | --- |
Notably, the conformation with a negative value of ϕ is about 4 kcal∙mol-1 more stable than that with a positive value of ϕ, indicating that the ring closure to give the trans 1S,2R absolute configuration thomasidioic acid 1 should be preferred to the ring closure to the cis 1R,2R absolute configuration. In the case of the R,S mesoform, the ring closure to give the trans configuration is still significantly preferred with respect to that which give the cis configuration. However, in this case the reaction of the two non equivalent prochiral carbon atoms present at the quinomethide double bonds with the two ortho carbons of the phenyl ring can give both the trans 1S,2R and, trans 1R,2S absolute configurations.
In
Table 5 are also reported the stabilities of conformers of the aromatic intermediate
8 for two different values of the ϕ angle (see
Scheme 3). The conformations have been obtained by the geometry optimization of stable conformers of bisquinomethide
7 in which the two rings approach to each other. As discussed above, the conformational analysis of
8 is simpler than that of
7, due to the loss of one stereogenic centre and to the rigidity of the carbon backbone. For both the
R,
SS and
S,
SS isomers the two conformers are characterized by a negative and positive value of ϕ, respectively. Interestingly the trend discussed above for the bisquinomethide
7 is fully confirmed. In fact, for the
R,
SS isomer, the conformer with a negative ϕ angle is more than 10 kcal∙mol
-1 stable than the conformer with a positive ϕ angle, while the opposite is true in the case of the
S,
SS isomer. The ring closure starting from the most stable conformations of the
R,
SS and
S,
SS isomers leads in both cases to the thomasidioic acid
1 in the
trans configuration, with the 1
S, 2
R and 1
R, 2
S absolute configuration, respectively.
Table 5.
Heat of formation of conformers of the aromatic intermediate
8, obtained for two different values of ϕ angle (see
Scheme 3), and of the corresponding Thomasidioic acid amide obtained after the ring closure computed at the semiempirical PM3 level of theory.
Table 5.
Heat of formation of conformers of the aromatic intermediate 8, obtained for two different values of ϕ angle (see Scheme 3), and of the corresponding Thomasidioic acid amide obtained after the ring closure computed at the semiempirical PM3 level of theory.
| Aromatic intermediate 8 | ϕ
(Degrees) | Heat of formation
(kcal∙mol-1) | | Thomasidioic acid amide | Heat of formation
(kcal∙mol-1) |
|---|
| R | -121 | -300.2 | → | 1S, 2R trans | -326.6 |
| R | 16 | -287.3 | → | 1R, 2R cis | -321.2 |
| S | 130 | -299.3 | → | 1R, 2S trans | -320.8 |
| S | -106 | -291.3 | → | 1S, 2S cis | -329.4 |
The results reported above, show that the diasteroselection to the
trans thomasidioic acid
1 begin at the level of bisquinomethide
7 where the conformers that leads to the ‘correct’ ring closure are more stable with respect to the conformers that leads to the
cis isomer. The difference in stability is even larger after the aromatization of
7 to give the intermediate
8. Finally, it should be noted that
trans-thomasidioic acid amides
4,
5 are more stable than the corresponding
cis isomers by about 5 kcal∙mol
-1 (see
Table 5). If the assumption is made that the energy of activation for the ring closure of
8 is correlated to the stability of the products, we can predict that the energy barrier of the cyclization to the
trans thomasidioic acid is also lower than the energy barrier to the
cis isomer. The larger stabilities of
7 and
8 conformers which can give ring closure to the
trans isomer, and the lower energies of the corresponding transition states along the cyclization pathway clearly explain the total diasteroselectivity to the
trans thomasidioic acid
1.
Experimental
General
All chemicals were from Sigma-Aldrich. IR spectra were recorded with a FT-IR Jasco spectrophotometer in KBr pellets. Mass spectra were determined by the direct injection system mode with positive electron impact using a VG 7070 EQ instrument. 1H-NMR spectra were recorded at 300 MHz with a Bruker AMX 300 instrument (in CDCl3 solutions). Chemical shifts are given as ppm from tetramethylsilane and J values are given in Hz. HPLC analyses were performed using a Waters 600 E instrument equipped with a HP 1040 Diode Array Detector and a column Agilent XDB-C18 (5 µm, 4.6 x 250 mm) was used. Water-acetonitrile (1:1) was used as a mobile phase. The total flow was 1 mL/min and the injection volume was 20 µL.
Preparation of compound 3a
To a solution of (S)-phenylalanine ethyl ester hydrochloride (1.00 g, 4.35 mmol) in anhydrous tetrahydrofuran (100 mL), triethylamine (0.6 mL, 4.35 mmol) were added dropwise. After solubilisation of the salt, sinapic acid 2 (0.97 g, 4.35 mmol) and dicyclohexylcarbodiimide (DCC, 0.90 g, 4.35 mmol) were added under stirring and the mixture was stirred for further 4 h at room temperature. A few drops of acetic acid were then added, the urea was filtered off and the resulting solution was evaporated under reduced pressure to give a residue which was dissolved in ethyl acetate (50 mL), washed with water saturated with NaCl (60 mL) and dried over Na2SO4. The solvent was evaporated under reduced pressure, and the residue was purified by silica gel flash chromatography (eluent ethyl acetate-petroleum ether 7:3). Compound 3a was obtained in 50% yield. Anal. calc. for C22H25NO6 (399.44): C 66.15%, H 6.31%, N 3.51%, O 24.03%; found: C 66.17%, H 6.29%, N 3.49%; MS: 399.4 (M+); 1H-NMR: δH 1.12 (3 H, t, J=8.0, Hg), 2.95 (1 H, dd, J=15.0-8.0, Hb), 3.20 (1 H, dd, J=15.0-4.0, Hc), 3.60 (2 H, q, J=8.0, Hf), 3.75 (6 H, s, OCH3), 4.55 (1 H, ddd, J=12.0-8.0-4.0, Ha), 5.62 (1 H, s, O-H), 5.78 (1 H, d, J=12.0 N-H) 6.55 (1 H, d, J=15.0, He), 7.20 (2 H, s, Ar-H), 7.30 (1 H, d, J=15.0, Hd), 7.35-7.50 (5 H, m, Ar-H); FT-IR (KBr, νmax, cm-1): 3347 (N-H), 3161 (O-H), 1661 (C=O), 1603 (C=C), 1109 (O-CH3).
Preparation of compound 3b
To a solution of sinapic acid (2, 1.00 g, 4.46 mmol) in anhydrous dichloromethane (100 mL), N-methylmorpholine (0.50 mL, 4.46 mmol) and isobutylchloroformate (0.80 mL, 4.46 mmol) were added. The mixture was stirred at 0° C for 1 h, then N-methylmorpholine (0.50 mL) and (S)-methyl-benzylamine (0.60 mL) were added. The mixture was stirred at room temperature for 2 h. The resulting solution was washed with a pH 4 solution of aqueous HCl (60 mL), with a 10% aqueous NaHCO3 solution (60 mL), with saturated brine (60 mL) and finally dried over Na2SO4. The solvent was evaporated under reduced pressure, and the residue was purified by silica gel flash chromatography, (eluent ethyl acetate and petroleum ether 7:3). The yield was 40% to give 3b. Anal. calc. for C19H21NO4 (327.34): C 69.71%, H 6.47%, N 4.28%, O 19.55%; found: C 69.65%, H 6.50%, N 4.31%; MS: 327.3 (M+); 1H-NMR: δH 1.50 (3 H, d, J=6.8, Hb), 3.75 (6 H, s, OCH3), 5.20 (1 H, m, J=6.8-12.0, Ha), 5.62 (1 H, s, O-H), 5.78 (1 H, d, J=12.0, N-H), 6.55 (1 H, d, J=15.5, He), 6.80 (2 H, s, Ar-H), 7.30 (1 H, d, J=15.5, Hd), 7.20-7.40 (5 H, m, Ar-H); FT-IR (KBr, νmax, cm-1): 3345 (N-H), 3155 (O-H), 1665 (C=O), 1602 (C=C), 1111 (O-CH3).
Preparation of compound 3c
Sinapic acid 2 (1 g, 4.46 mmol), 2-chloro-1-methylpyridine iodide (0.52 g, 4.46 mmol) and (S)-2-phenyloxazolidinone (0.35 g, 4.46 mmol) was dissolved in 20 ml of dry CH2Cl2 under nitrogen atmosphere. A solution of triethylamine (0.7 mL, 5.6 mmol), dissolved in dry CH2Cl2 (4 mL) was added dropwise for 15 min. The mixture was stirred at room temperature for 120 hours, then a saturated aqueous NaCl solution (20 mL) was added. The organic phase was separated, then washed with a pH 4 solution of aqueous HCl (20 mL), with a 10% aqueous NaHCO3 solution (20 mL), with water saturated with NaCl (20 mL) and finally dried over Na2SO4. The solvent was evaporated under reduced pressure, and the residue was purified by silica gel flash chromatography, (eluent CH2Cl2-acetone 7:3). Compound 3c was obtained in 40% yield. Anal. calc. for C20H19NO6 (369.37): C 65.03%, H 5.18%, N 3.79%, O 25.99%; found: C 65.10%, H 5.15%, N 3.80%; MS: 369.4 (M+); 1H-NMR: δH 3.75 (6 H, s, OCH3), 4.20 (1 H, dd, J=9.0-9.0, Ha), 4.73 (1 H, dd, J=9.0-9.0 Hb), 4.97 (1 H, dd, J=9.0-9.0, Hc), 5.62 (1 H, s, O-H), 6.55 (1 H, d, J=15.0, He), 7.20 (2 H, s, Ar-H), 7.30 (1 H, d, J=15.0, Hd), 7.20-7.40 (5 H, m, Ar-H); FT-IR (KBr, νmax, cm-1): 3155 (O-H), 1750 (C=O), 1659 (C=O), 1605 (C=C), 1110 (O-CH3).
Horseradish Peroxidase (HRP)-catalyzed oxidative phenol coupling methyl synapate 3d
A solution of 3d (1.0 mmol) in dioxane (14 mL) and 0.02 M phosphate/citric acid buffer pH 3.5 (4.0 mL) was added of a 0.86 M aqueous hydrogen peroxide solution (0.60 mL, 0.5 mmol) and aqueous HRP (0.93 mL, 837 U) at 0 °C in small portions over 15 minutes. The mixture was then stirred at 0 °C. After 4 hours a saturated aqueous NaCl solution (20 mL) was added. The organic phase was then evaporated under reduced pressure and the resulting solution was extracted with ethyl acetate (4 x 20 mL). The combined organic extracts were washed with 10% aqueous NaHCO3 (25 mL), then with water saturated with NaCl (25 mL), and dried over Na2SO4. The solvent was evaporated under reduced pressure, and the residue was chromatographed on a silica gel flash column with hexane-ethyl acetate (gradient mode, from 4:1 to 1:1) yielding a racemic mixture of trans thomasidioic metyl ester (4-5d), yield = 70%. Racemic mixture of 4-5d had: m.p. 203 °C; 1H-NMR: δH 3.69 (6 H, s, OCH3), 3,70 (3 H, s, OCH3) , 3.75 (6 H, s, OCH3), 3.90 (3 H, s, OCH3), 3,99 (1 H, s, Hi) 4.99 (1 H, s, Hh), 5.00 (1 H, s, OH), 5.40 (1 H, s, OH), 6.21 (2 H, s, Hl), 6.58 (1 H, s, Hn), 7.60 (1 H, s, Ho); MS: 474 (M+); FT-IR (KBr, νmax, cm-1): 3300 (O-H), 1730 (C=O), 1700 (C=O), 1110 (O-CH3)
Horseradish Peroxidase (HRP)-catalyzed oxidative phenol coupling with compounds 3(a-c)
A solution of
3(a-c) (1.0 mmol) in dioxane (14 mL) and 0.02 M phosphate/citric acid buffer pH 3.5 (4.0 mL) was added of a 0.86 M aqueous hydrogen peroxide solution (0.60 mL, 0.5 mmol) and aqueous HRP (0.93 mL, 837 U) at 0 °C in small portions over 15 minutes. The mixture was then stirred at 0 °C. After 4 hours a saturated aqueous NaCl solution (20 mL) was added. The organic phase was then evaporated under reduced pressure, and the resulting solution was extracted with ethyl acetate (4 x 20 mL). The combined organic extracts were washed with 10% aqueous NaHCO
3 (25 mL), then with water saturated with NaCl (25 mL), and dried over Na
2SO
4. The solvent was evaporated under reduced pressure, and the residue was purified with preparative RP-HPLC to obtain the individual diastereoisomers, named peak1 and peak 2 in eluative order. The diastereoisomeric excess was calculated by HPLC (
Table 1). Compounds
4a,
5a had:
Peak 1: Anal. calc. for C
44H
48N
2O
12 (796.86): C 66.32%, H 6.07%, N 3.52%, O 24.09%; found: C 66.30%, H 6.05%, N 3.55%; MS: 796.9 (M
+); FT-IR (KBr, ν
max, cm
-1): 3155 (O-H), 1750 (C=O), 1659 (C=O), 1605 (C=C), 1110 (O-CH
3); [α]
D25 = -160.27°;
1H-NMR: δ
H 1.12 (3 H, t,
J=8.0, H
g), 1.30 (3 H, t,
J=8.0, H
g), 3.08 (2 H, d-d,
J=6.0-15.0, H
b), 3.15 (2 H, d-d,
J=8.0-15.0 H
c), 3.69 (3 H, s, OCH
3), 3,70 (1 H, s, H
i) , 3.75 (6 H, s, OCH
3), 3.90 (3 H, s, OCH
3), 4.05 (2 H, q,
J=8.0, H
f), 4.20 (2 H, q,
J=8.0, H
f), 4.69 (1 H, d-d,
J=6.0-8.0 , H
a), 4.87 (1 H, d-d,
J=6.0-8.0, H
a), 4.99 (1 H, s, H
h), 5.00 (1 H, s, OH), 5.40 (1 H, s, OH), 6.21 (2 H, s, H
l), 6.58 (1 H, s, H
n), 6.75 (1 H, d,
J=8.0 Hz, N-H), 7.00-7.20 (10 H, m, Ar-H), 7.60 (1 H, d,
J=8.0 Hz, N-H), H
o is in the aromatic zone.
Peak 2: Anal. calc. for C
44H
48N
2O
12 (796.86): C 66.32%, H 6.07%, N 3.52%, O 24.09%; found: C 66.30%, H 6.05%, N 3.55%; MS: 796.9 (M
+); FT-IR (KBr, ν
max, cm
-1): 3155 (O-H), 1750 (C=O), 1659 (C=O), 1605 (C=C), 1110 (O-CH
3); [α]
D25 = +127.88°;
1H-NMR: δ
H 1.15 (3 H, t,
J=8.0, H
g), 1.20 (3 H, t,
J=8.0, H
g), 3.08 (2 H, d-d,
J=6.0-15.0, H
b), 3.15 (2 H, d-d,
J=8.0-15.0 H
c), 3.69 (3 H, s, OCH
3), 3,70 (1 H, s, H
i) , 3.75 (6 H, s, OCH
3), 3.90 (3 H, s, OCH
3), 4.10 (2 H, q,
J=8.0, H
f), 4.15 (2 H, q,
J=8.0, H
f), 4.70 (1 H, d-d,
J=6.0-8.0 , H
a), 4.80 (1 H, d-d,
J=6.0-8.0, H
a), 5.02 (1 H, s, H
h), 5.00 (1 H, s, OH), 5.40 (1 H, s, OH), 6.21 (2 H, s, H
l), 6.58 (1 H, s, H
n), 6.75 (1 H, d,
J=8.0 Hz, N-H), 7.00-7.20 (10 H, m, Ar-H), 7.60 (1 H, d,
J=8.0 Hz, N-H), H
o is in the aromatic zone. Compounds
4b,
5b had:
Peak 1: Anal. calc. for C
38H
40N
2O
8 (652.73): C 69.92%, H 6.18%, N 4.29%, O 19.61%; found: C 69.88%, H 6.21%, N 4.34%; MS: 652.7 (M
+); FT-IR (KBr, ν
max, cm
-1): 3155 (O-H), 1750 (C=O), 1659 (C=O), 1110 (O-CH
3); [a]
D25 = -80.21°;
1H-NMR: δ
H 1.15 (3 H, d,
J=5.0 Hz, H
b), 1.40 (3 H, d,
J=5.0 Hz, H
b), 3.45 (6 H, s, OCH3), 3,65 (1 H, s, H
i), 3.67 (3 H, s, OCH
3), 3.90 (3 H, s, OCH
3), 4.72 (1 H, m,
J=5.0 Hz, H
a), 4.92 (1 H, s, H
h), 5.15 (1 H, m,
J=5.0 Hz,H
a), 5.30 (1 H, s, OH), 5.73 (1 H, s, OH), 6.21 (2 H, s, H
l), 6.52 (1 H, d,
J=8.0 Hz, N-H), 6.58 (1 H, s, H
n), 7.02-7.20 (10 H, m, Ar-H), 7.55 (1 H, d,
J=8.0 Hz, N-H), H
o is in the aromatic zone.
Peak 2: Anal. calc. for C
38H
40N
2O
8 (652.73): C 69.92%, H 6.18%, N 4.29%, O 19.61%; found: C 69.88%, H 6.21%, N 4.34%; MS: 652.7 (M
+); FT-IR (KBr, ν
max, cm
-1): 3155 (O-H), 1750 (C=O), 1659 (C=O), 1605 (C=C), 1110 (O-CH
3); [α]
D25 = +51.63°;
1H-NMR: δ
H 1.35 (3 H, d,
J=5.0 Hz, H
b), 1.45 (3 H, d,
J=5.0 Hz, H
b), 3.65 (6 H, s, OCH3), 3.72 (1 H, s, H
i), 3.75 (3 H, s, OCH
3), 3.81 (3 H, s, OCH
3), 4.88 (1 H, m,
J=5.0 Hz, H
a), 4.92 (1 H, s, H
h), 5.08 (1 H, m,
J=5.0 Hz, H
a), 5.30 (1 H, s, OH), 5.73 (1 H, s, OH), 6.21 (2 H, s, H
l), 6.52 (1 H, d,
J=8.0 Hz, N-H), 6.58 (1 H, s, H
n), 7.02-7.20 (10 H, m, Ar-H), 7.55 (1 H, d,
J=8.0 Hz, N-H), H
o is in the aromatic zone. Compounds
4c,
5c had:
Peak 1: Anal. calc. for C
40H
36N
2O
12 (736.72): C 65.21%, H 4.93%, N 3.80%, O 26.06%; found: C 65.25%, H 4.90%, N 3.77%; MS: 736.7 (M
+); FT-IR (KBr, ν
max, cm
-1): 3155 (O-H), 1750 (C=O), 1659 (C=O), 1605 (C=C), 1110 (O-CH
3);
1H-NMR: 3.65 (3 H, s, OCH
3), 3,69 (1 H, s, H
i) , 3.75 (6 H, s, OCH
3), 3.90 (3 H, s, OCH
3), 4.10 (1 H, dd,
J=9.0-9.0, H
a), 4.20 (1 H, dd,
J=9.0-9.0, H
a), 4.61 (1 H, dd,
J=9.0-9.0 H
b), 4.73 (1 H, dd,
J=9.0-9.0 H
b), 4.85 (1 H, dd,
J=9.0-9.0, H
c), 4.97 (1 H, dd,
J=9.0-9.0, H
c), 4.99 (1 H, s, H
h), 5.00 (1 H, s, O-H), 5.40 (1 H, s, O-H), 6.21 (2 H, s, H
l), 6.58 (1 H, s, H
n), 7.00-7.20 (10 H, m, Ar-H), H
o is in the aromatic zone.
Peak 2: Anal. calc. for C
40H
36N
2O
12 (736.72): C 65.21%, H 4.93%, N 3.80%, O 26.06%; found: C 65.25%, H 4.90%, N 3.77%; MS: 736.7 (M
+); FT-IR (KBr, ν
max, cm
-1): 3155 (O-H), 1750 (C=O), 1659 (C=O), 1605 (C=C), 1110 (O-CH
3);
1H-NMR: 3.65 (3 H, s, OCH
3), 3,69 (1 H, s, H
i) , 3.75 (6 H, s, OCH
3), 3.90 (3 H, s, OCH
3), 4.10 (1 H, dd,
J=9.0-9.0, H
a), 4.20 (1 H, dd,
J=9.0-9.0, H
a), 4.61 (1 H, dd,
J=9.0-9.0 H
b), 4.73 (1 H, dd,
J=9.0-9.0 H
b), 4.85 (1 H, dd,
J=9.0-9.0, H
c), 4.97 (1 H, dd,
J=9.0-9.0, H
c), 4.99 (1 H, s, H
h), 5.00 (1 H, s, O-H), 5.40 (1 H, s, O-H), 6.21 (2 H, s, H
l), 6.58 (1 H, s, H
n), 7.00-7.20 (10 H, m, Ar-H), H
o is in the aromatic zone.
LiOOH hydrolysis of aryltetralines
To a solution of aryltetralin 4 peak 1 (0.171 mmol) in tetrahydrofuran (20 mL) containing 30% hydrogen peroxide (1 mL), LiOH (69 mg, 2.87 mmol) dissolved in water (3 mL) was added and the solution was stirred at room temperature for 18 h. The resulting suspension was then cooled at 0 °C and a saturated solution of sodium bisulphite was added until peroxides were completely reduced, then concentrated under reduced pressure, acidified with a HCl solution pH 5 and extracted with ethyl acetate. The organic phase was dried over anhydrous sodium sulphate, then evaporated at reduced pressure to give thomasidioic acid 1. Anal. calc. for C22H22O10 (446.40): C 59.19%, H 4.97%, O 35.84%; found: C 59.22%, H 5.00%; MS: 446.4 (M+); FT-IR (KBr, νmax, cm-1): 3155 (O-H), 1750 (C=O), 1659 (C=O), 1605 (C=C), 1110 (O-CH3); 1H-NMR: δH 3.65 (6 H, s, OCH3), 3.70 (3 H, s, OCH3), 3.81 (3 H, s, OCH3), 4.03 (1 H, s, Hi), 4.95 (1 H, s, Hh), 5.30 (1 H, s, O-H), 5.75 (1 H, s, O-H), 6.20 (2 H, s, Hl), 6.62 (1 H, s, Hn), 7.57 (1 H, s, Ho); [α]D25 = -90.27°
Computational details
All calculations have been performed at the semiempirical PM3 level [
30] of theory using Gaussian 03 [
31] and Mopac2000 [
32] suite of programs.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

 ), and re-si (
), and re-si (  ) faces.
) faces.





