Bioactive Pentacyclic Triterpenes from the Stems of Combretum laxum

Abstract
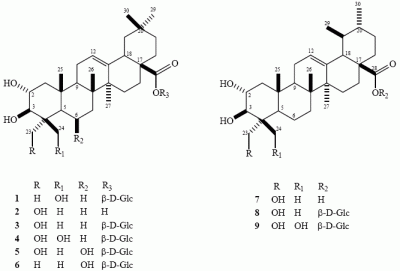
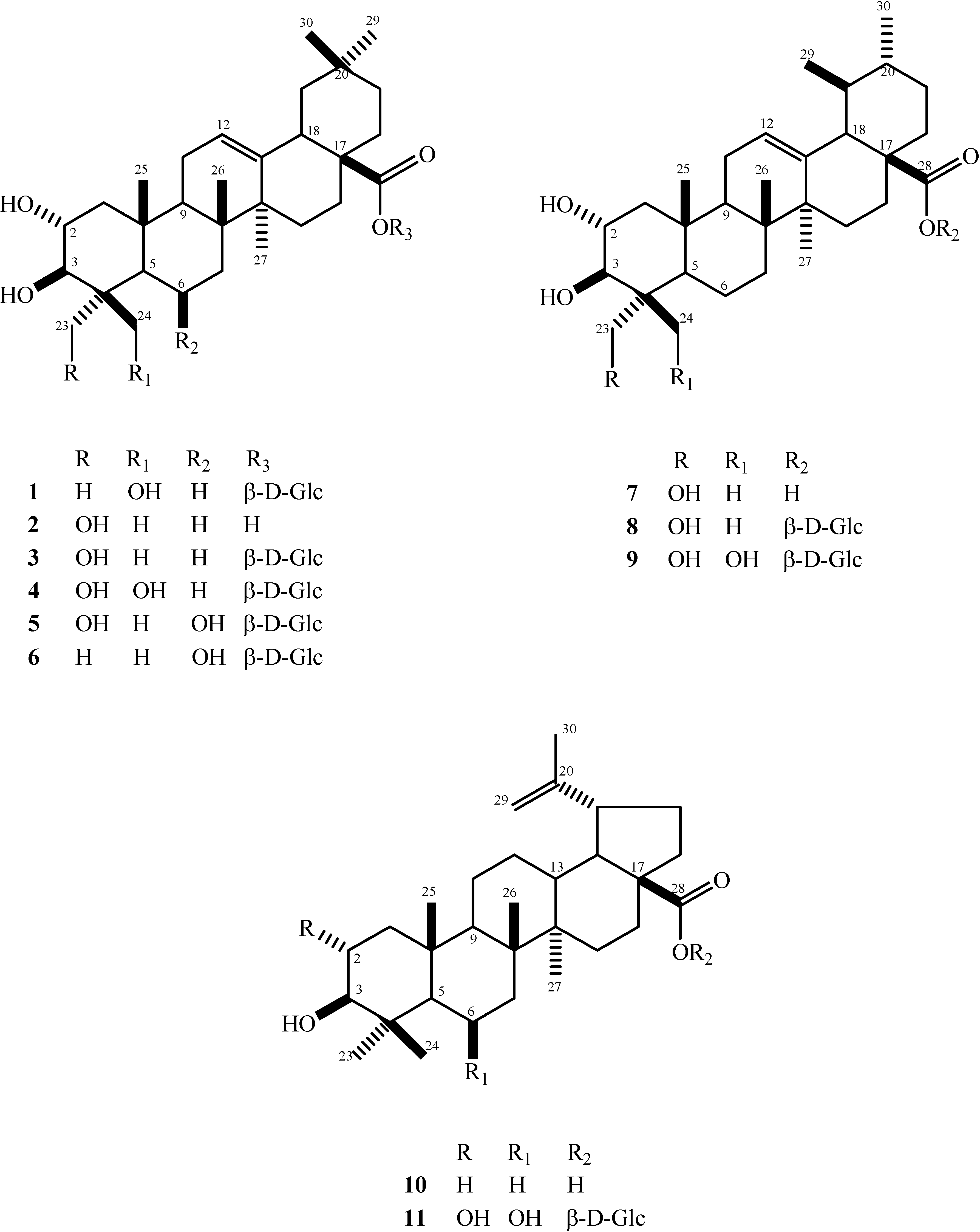
:
Introduction
Results and Discussion


{kind=link}
{kind=link}
| Compounds | Candida albicans | Candida krusei | Cryptococcus neoformans | |
|---|---|---|---|---|
| ATCC 90028 | ATCC 6258 | ATCC 32045 | ||
| 1 | 200 | 200 | 200 | |
| 2 | 50 | 50 | 50 | |
| 5 | 200 | 200 | 100 | |
| 6 | 200 | 200 | 200 | |
| 10 | 100 | 200 | 100 | |
| 11 | 200 | 200 | 200 | |
| 2/7 | 200 | 200 | 200 | |
| 3/8 | 200 | 200 | 200 | |
| 4/9 | 200 | 200 | 200 | |
| amphotericin B | 0.25 | 0.50 | 0.50 | |
Experimental
General
Plant Material
Extraction and Isolation
 : + 22.79o (c 0.17, MeOH); HRESIMS m/z 685.3755 [M+Cl]- (calcd. for C36H58O10Cl 685.3720); 1H-NMR: δ 4.33 (1H, m, H-2); 3.54 (1H, d, J = 9.3 Hz, H-3); 5.38 (1H, brs, H-12); 3.16 (1H, dd, J = 14.3, 3.8 Hz, H-18); 3.69 (1H, d, J = 11.0 Hz, H-24a); 4.42 (1H, d, J = 11.0 Hz, H-24b); 1.54 (3H, s, H-23); 0.97 (3H, s, H-25); 1.06 (3H, s, H-26); 1.18 (3H, s, H-27); 0.87 (3H, s, H-29); 0.85 (3H, s, H-30); sugar signals: δ 6.29 (1H, d, J = 7.9 Hz, H-1’); 4.00 (1H, m, H-5’); 4.18-4.45* (5H, m H-2’, H-3’, H-4’, H-6’) *overlapping signals; 13C-NMR: δ 47.8 (C-1); 68.6 (C-2); 85.7 (C-3); 44.0 (C-4); 56.5 (C-5); 19.3 (C-6); 33.4 (C-7); 40.0 (C-8); 48.3 (C-9); 38.3 (C-10); 24.1 (C-11); 122.7 (C-12); 144.1 (C-13); 42.1 (C-14); 28.2 (C-15); 23.4 (C-16); 47.0 (C-17); 41.7 (C-18); 46.2 (C-19); 30.7 (C-20); 34.0 (C-21); 32.5 (C-22); 24.1 (C-23); 65.7 (C-24); 17.3 (C-25); 17.4 (C-26); 26.0 (C-27); 176.4 (C-28); 33.1 (C-29); 23.6 (C-30); sugar signals: δ 95.8 (C-1’); 74.1 (C-2’); 78.9 (C-3’); 71.1 (C-4’); 79.3 (C-5’); 62.3 (C-6’).
: + 22.79o (c 0.17, MeOH); HRESIMS m/z 685.3755 [M+Cl]- (calcd. for C36H58O10Cl 685.3720); 1H-NMR: δ 4.33 (1H, m, H-2); 3.54 (1H, d, J = 9.3 Hz, H-3); 5.38 (1H, brs, H-12); 3.16 (1H, dd, J = 14.3, 3.8 Hz, H-18); 3.69 (1H, d, J = 11.0 Hz, H-24a); 4.42 (1H, d, J = 11.0 Hz, H-24b); 1.54 (3H, s, H-23); 0.97 (3H, s, H-25); 1.06 (3H, s, H-26); 1.18 (3H, s, H-27); 0.87 (3H, s, H-29); 0.85 (3H, s, H-30); sugar signals: δ 6.29 (1H, d, J = 7.9 Hz, H-1’); 4.00 (1H, m, H-5’); 4.18-4.45* (5H, m H-2’, H-3’, H-4’, H-6’) *overlapping signals; 13C-NMR: δ 47.8 (C-1); 68.6 (C-2); 85.7 (C-3); 44.0 (C-4); 56.5 (C-5); 19.3 (C-6); 33.4 (C-7); 40.0 (C-8); 48.3 (C-9); 38.3 (C-10); 24.1 (C-11); 122.7 (C-12); 144.1 (C-13); 42.1 (C-14); 28.2 (C-15); 23.4 (C-16); 47.0 (C-17); 41.7 (C-18); 46.2 (C-19); 30.7 (C-20); 34.0 (C-21); 32.5 (C-22); 24.1 (C-23); 65.7 (C-24); 17.3 (C-25); 17.4 (C-26); 26.0 (C-27); 176.4 (C-28); 33.1 (C-29); 23.6 (C-30); sugar signals: δ 95.8 (C-1’); 74.1 (C-2’); 78.9 (C-3’); 71.1 (C-4’); 79.3 (C-5’); 62.3 (C-6’).Antifungal assay
Acknowledgements
References
- Available online: http://www.ibge.gov.br/home/presidencia/noticias/21052004biomashtml/shtm accessed in September 2008.
- Garcez, F.R.; Garcez, W.S.; Martins, M.M.; Lopes, F.A. Triterpenoids, lignan and flavans from Terminalia argentea (Combretaceae). Biochem Syst. Ecol. 2003, 31, 229–231. [Google Scholar] [CrossRef]
- Garcez, F.R.; Garcez, W.S.; Miguel, D.L.S.; Serea, A.A.T.; Prado, F.C. Chemical constituents from Terminalia glabrescens. J. Braz. Chem. Soc. 2003, 14, 461–465. [Google Scholar] [CrossRef]
- Garcez, F.R.; Garcez, W.S.; Santana, A.L.B.D.; Alves, M.M.; Matos, M.F.C.; Scaliante, A.M. Bioactive flavonoids and triterpenes from Terminalia fagifolia (Combretaceae). J. Braz. Chem. Soc. 2006, 17, 1223–1228. [Google Scholar]
- Pott, A.; Pott, V.J. Plantas do Pantanal; Empresa Brasileira de Pesquisa Agropecuária: Brasília, 1994; pp. 82–84. [Google Scholar] and references cited therein.
- Facundo, V.A.; Rios, K.A.; Medeiros, C.M.; Militão, J.S.L.T.; Miranda, A.L.P.; Epifanio, R.A.; Carvalho, M.P.; Andrade, A.T.; Pinto, A.C.; Rezende, C.M. Arjunolic acid in the ethanolic extract of Combretum leprosum root and its use as a potential multi-functional phytomedicine and drug for neurodegenerative disorders: anti-inflammatory and anticholinesterasic activities. J. Braz. Chem. Soc. 2005, 16, 1309–1312. [Google Scholar] [CrossRef]
- Martini, N.D.; Katerere, D.R.; Eloff, J.N. Seven flavonoids with antibacterial activity isolated from Combretum erythrophyllum. S. Afr. J. Bot. 2004, 70, 310–312. [Google Scholar]
- Cirla, A.; Mann, J. Combretastatins. From natural products to drug discovery. Nat. Prod. Rep. 2003, 20, 558–564. [Google Scholar] [CrossRef]
- Simon, G.; Dewelle, J.; Nacoulma, O.; Guissou, P.; Kiss, R.; Daloze, D.; Braekman, J.C. Cytotoxic triterpenes from Combretum nigricans. Fitoterapia 2003, 74, 339–344. [Google Scholar] [CrossRef]
- Adnyana, I.K.; Tezuka, Y.; Awale, S.; Banskota, A.H.; Tran, K.Q.; Kadota, S. 1-O-galloyl-6-O-(4-hydroxy-3,5-dimethoxy)benzoyl-β-d-glucose, a new hepatoprotective constituent from Combretum quadrangulare. Planta Med. 2001, 67, 370–371. [Google Scholar] [CrossRef]
- Adnyana, I.K.; Tezuka, Y.; Banskota, A.H; Tran, K.Q.; Kadota, S. Three new triterpenes from the seeds of Combretum quadrangulare and their hepatoprotective activity. J. Nat. Prod. 2001, 64, 360–363. [Google Scholar] [CrossRef]
- Adnyana, I.K.; Tezuka, Y.; Awale, S.; Banskota, A.H.; Tran, K.Q.; Kadota, S. Quadranosides VI-XI, six new triterpene glucosides from the seeds of Combretum quadrangulare. Chem. Pharm. Bull. 2000, 48, 1114–1120. [Google Scholar] [CrossRef]
- Adnyana, I.K.; Tezuka, Y.; Awale, S.; Banskota, A.H; Xiong, Q.B.; Tran, K.Q.; Kadota, S. Quadranosides I-V, new triterpene glucosides from the seeds of Combretum quadrangulare. J. Nat. Prod. 2000, 63, 496–500. [Google Scholar] [CrossRef]
- Banskota, A.H; Tezuka, Y.; Tran, K.Q.; Tanaka, K.; Saiki, I.; Kadota, S. Methyl quadrangularates A-D and related triterpenes from Combretum quadrangulare. Chem. Pharm. Bull. 2000, 48, 496–504. [Google Scholar] [CrossRef]
- Malan, E.; Swinny, E. Substituted bibenzyls, phenanthrenes and 9,10-dihydrophenanthrenes from the heartwood of Combretum apiculatum. Phytochemistry 1993, 34, 1139–1142. [Google Scholar] [CrossRef]
- Adrian-Romero, M.; Blunden, G.; Patel, A.V.; Armstrong, N.; Melendez, P.; Cuervo, A.C. Betaines and N-methylprolines from Venezuelan plants. Nat. Prod. Comm. 2007, 2, 863–868. [Google Scholar]
- Ahmad, V.U.; Rahman, A.U. Handbook of Natural Products Data. Volume 2. Pentacyclic Triterpenoids; Elsevier: Amsterdam, the Netherlands, 1994; pp. 1–1200. [Google Scholar]
- Agrawal, P.K. NMR spectroscopy in the structural elucidation of oligosaccharides and glycosides. Phytochemistry 1992, 31, 3307–3330. [Google Scholar] [CrossRef]
- Boyd, J.; Turvey, J.R. Identification by N.M.R.-C-13 spectroscopy of oligosaccharides derived from alginic acid. Carbohydr. Res. 1978, 61, 223–226. [Google Scholar] [CrossRef]
- García-Granados, A.; López, P.E.; Melguizo, E.; Parra, A.; Simeó, Y. Remote hydroxylation of methyl groups by regioselective cyclopalladation. Partial synthesis of hyptatic acid-A. J. Org. Chem. 2007, 72, 3500–3509. [Google Scholar] [CrossRef]
- Romussi, G.; De Tommasi, N. Arjunglucoside II from Quercus ilex L. and Quercus suber L. 16. Contents of Cupuliferae. Pharmazie 1992, 47, 877–878. [Google Scholar]
- Nandy, A.K.; Podder, G.; Sahu, N.P.; Mahato, S.B. Triterpenoids and their glucosides from Terminalia bellerica. Phytochemistry 1989, 28, 2769–2772. [Google Scholar] [CrossRef]
- Kundu, A.P.; Mahato, S.B. Triterpenoids and their glucosides from Terminalia chebula. Phytochemistry 1993, 32, 999–1002. [Google Scholar] [CrossRef]
- Gallo, M.B.C.; Silva, F.C.; Vieira, P.C.; Fernandes, J.B.; Silva, M.F.G.F. New natural products from Siphoneugena densiflora Berg (Myrtaceae) and their chemotaxonomic significance. J. Braz. Chem. Soc. 2006, 17, 279–288. [Google Scholar] [CrossRef]
- Ageta, M.; Nonaka, G.I.; Nishioka, I. Tannins and related compounds. LXVII. Isolation and characterization of castanopsinins A-H, novel ellagitannins containing a triterpenoid glycoside core, from Castanopsis cuspidate var. sieboldii Nakai. Chem. Pharm. Bull. 1988, 36, 1646–1663. [Google Scholar] [CrossRef]
- Weniger, B.; Lobstein, A.; Um, B.H.; Vonthron-Senechau, C.; Anton, R.; Usuga, N.J.; Basaran, H.; Lugnier, C. Bioactive triterpenoids from Vochisia pacifica interact with cyclic nucleotide phosphodiesterase isozyme PDE4. Phytother. Res. 2005, 19, 75–77. [Google Scholar]
- Liu, M.; Seidel, V.; Katerere, D.R.; Gray, A.I. Colorimetric broth microdilution method for the antifungal screening of plant extracts against yeasts. Methods 2007, 42, 325–329. [Google Scholar] [CrossRef]
- Masoko, P.; Picard, J.; Eloff, J.N. The antifungal activity of twenty-four southern African Combretum species (Combretaceae). South Afr. J. Bot. 2007, 73, 173–183. [Google Scholar] [CrossRef]
- Asres, K.; Mazumder, A.; Bucar, F. Antibacterial and antifungal activities of extracts of Combretum molle. Ethiop. Med. J. 2006, 44, 269–277. [Google Scholar]
- Batawila, K.; Kokou, K.; Koumaglo, K.; Gbéassor, M.; de Foucault, B.; Bouchet, Ph.; Akpagana, K. Antifungal activities of five Combretaceae used in Togolese traditional medicine. Fitoterapia 2005, 76, 264–268. [Google Scholar] [CrossRef]
- Fyhrquist, P.; Mwasumbi, L.; Hæggström, C.-A.; Vuorela, H.; Hiltunen, R.; Vuorela, P. Antifungal activity of selected species of Terminalia, Pteleopsis and Combretum (Combretaceae) collected in Tanzania. Pharm. Biol. 2004, 42, 308–317. [Google Scholar] [CrossRef]
- Srivastava, S.K.; Srivastava, S.D.; Chouksey, B.K. New antifungal constituents from Terminalia alata. Fitoterapia 2001, 72, 106–112. [Google Scholar] [CrossRef]
- Chouksey, B.K.; Srivastava, S.K. New constituent from the roots of Terminalia arjuna: antifungal agent. Indian J. Chem. 2001, 40B, 354–356. [Google Scholar]
- Nandy, A.K.; Chakraborty, A.; Podder, G. Antimicrobial activity of Terminalia bellerica triterpenoids. Fitoterapia 1997, 68, 178–180. [Google Scholar]
- Masoko, P.; Mdee, L.K.; Mampuru, L.J.; Eloff, J.N. Biological activity of two related triterpenes isolated from Combretum nelsonii (Combretaceae) leaves. Nat. Prod. Res. 2008, 22, 1074–1084. [Google Scholar] [CrossRef]
- National Committee for Clinical Laboratory Standards (NCCLS). Approved Standards M27-A2. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts, 2nd edNCCLS: Wayne, PA, USA, 2002; Vol. 22, pp. 1–29. [Google Scholar]
- Sample Availability: Contact the authors.
© 2008 by the authors. Licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bisoli, E.; Garcez, W.S.; Hamerski, L.; Tieppo, C.; Garcez, F.R. Bioactive Pentacyclic Triterpenes from the Stems of Combretum laxum. Molecules 2008, 13, 2717-2728. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules13112717
Bisoli E, Garcez WS, Hamerski L, Tieppo C, Garcez FR. Bioactive Pentacyclic Triterpenes from the Stems of Combretum laxum. Molecules. 2008; 13(11):2717-2728. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules13112717
Chicago/Turabian StyleBisoli, Eder, Walmir Silva Garcez, Lidilhone Hamerski, Caroline Tieppo, and Fernanda Rodrigues Garcez. 2008. "Bioactive Pentacyclic Triterpenes from the Stems of Combretum laxum" Molecules 13, no. 11: 2717-2728. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules13112717