Synthesis and Structural Characterization of the 5-(2-Haloethyl)pyrimidines – Hydrogen-Bonded chains in α-(1-Carbamyliminomethylene)-γ-Butyrolactone

Abstract
:
Introduction

Results and Discussion
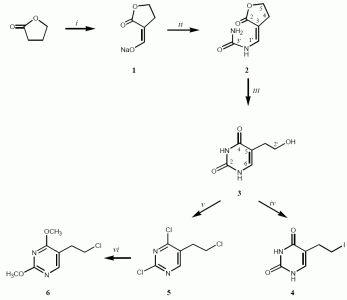
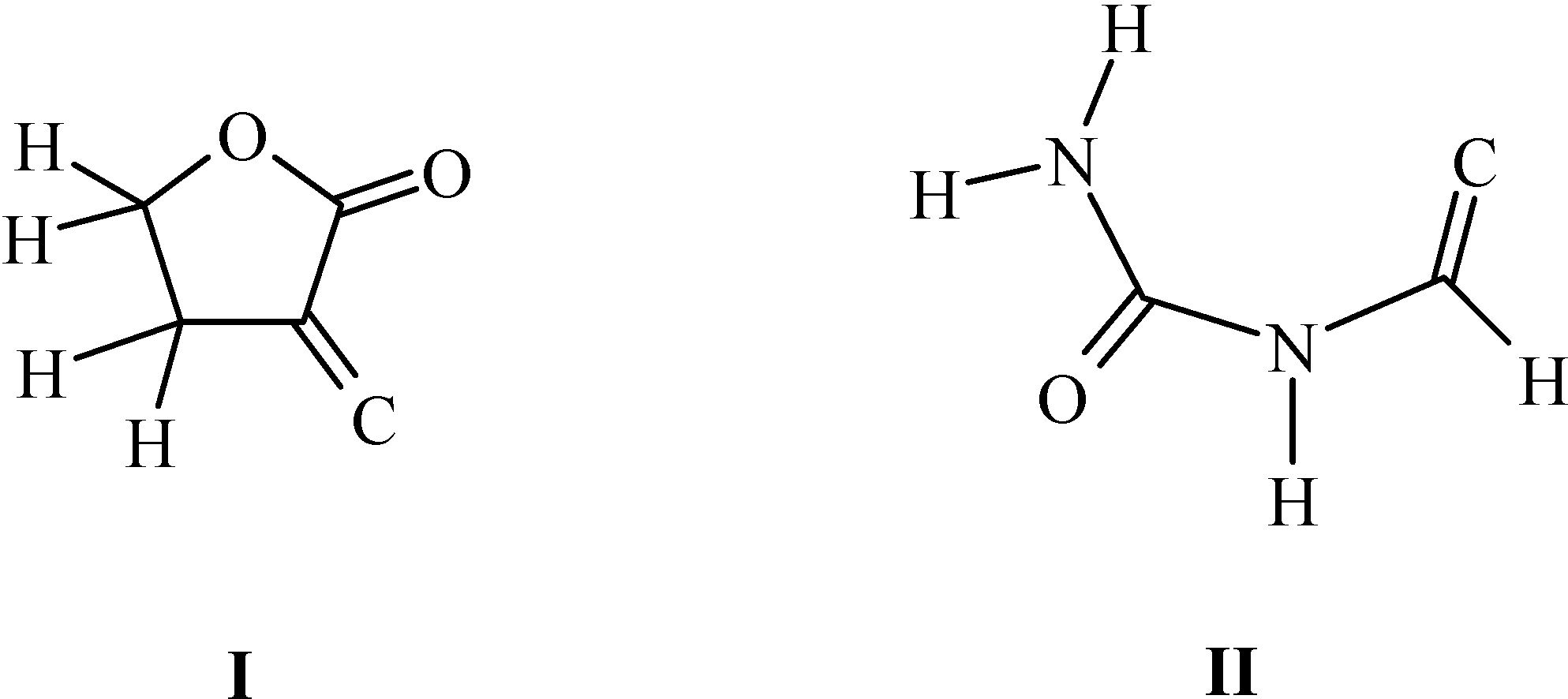
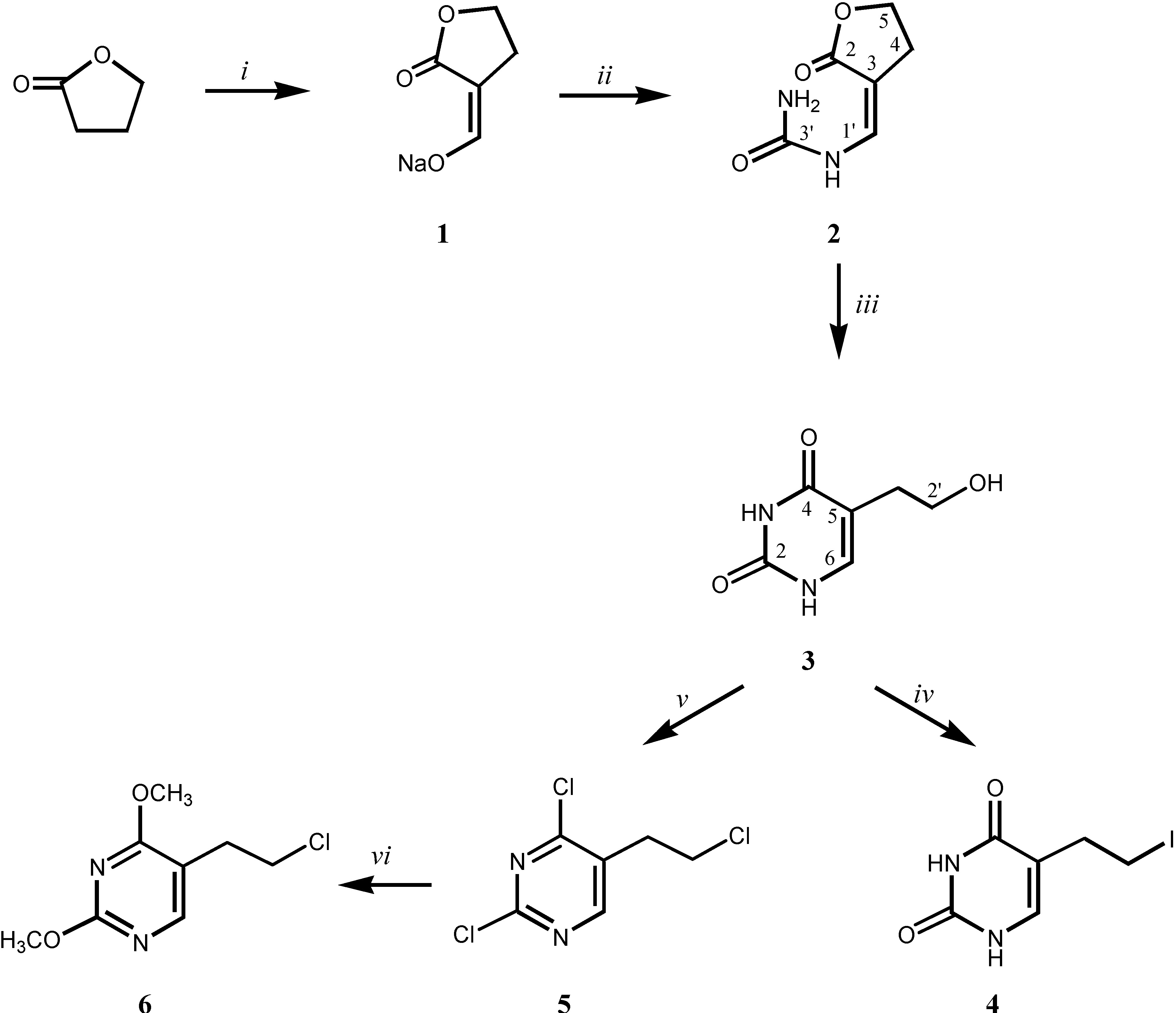
Synthesis
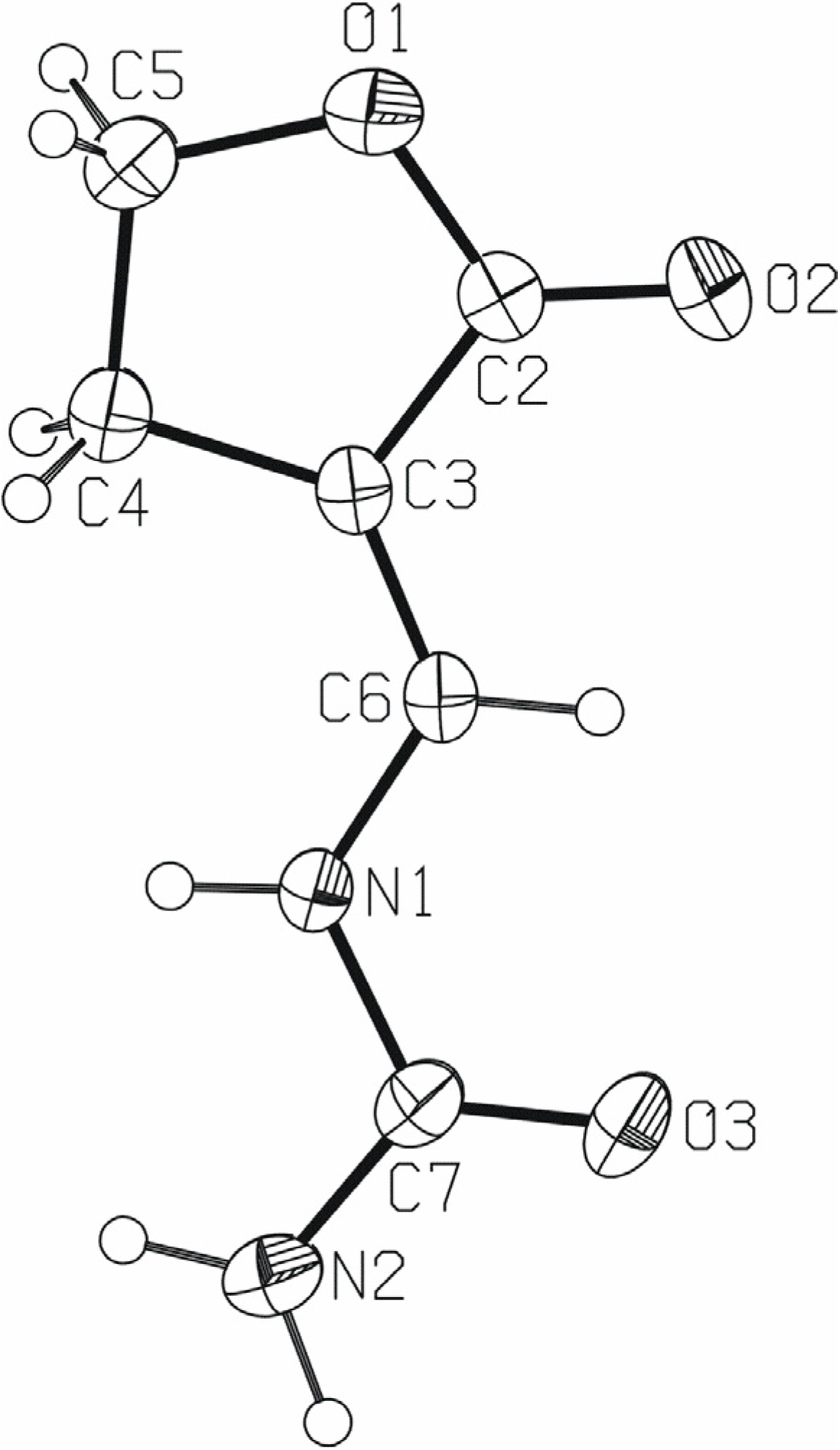
X-ray crystal structure study



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N1−C6 | 1.363(2) | O3−C7 | 1.2251(19) | |
| N1−C7 | 1.3900(18) | C2−C3 | 1.453(2) | |
| N2−C7 | 1.339(2) | C3−C6 | 1.333(2) | |
| O1−C2 | 1.3417(17) | C3−C4 | 1.495(2) | |
| O1−C5 | 1.450(2) | C4−C5 | 1.518(2) | |
| O2−C2 | 1.2173(19) | |||
| C6−N1−C7 | 121.43(13) | C2−C3−C4 | 108.73(12) | |
| C2−O1−C5 | 110.77(12) | C3−C4−C5 | 102.74(13) | |
| O2−C2−O1 | 120.86(14) | O1−C5−C4 | 107.73(12) | |
| O2−C2−C3 | 129.10(15) | C3−C6−N1 | 125.09(13) | |
| O1−C2−C3 | 110.02(12) | O3−C7−N2 | 124.88(15) | |
| C6−C3−C2 | 121.18(13) | O3−C7−N1 | 121.05(15) | |
| C6−C3−C4 | 130.00(14) | N2−C7−N1 | 114.07(13) |
| D−H···A | D−H (Å) | H···A (Å) | D···A (Å) | D−H··· A |
|---|---|---|---|---|
| N1−H1···O2i | 0.85(2) | 2.06(2) | 2.869(2) | 158(2) |
| N2−H2A···O2i | 0.90(2) | 2.16(2) | 2.974(2) | 150(2) |
| N2−H2B···O3ii | 0.93(2) | 1.96(2) | 2.884(2) | 174(2) |
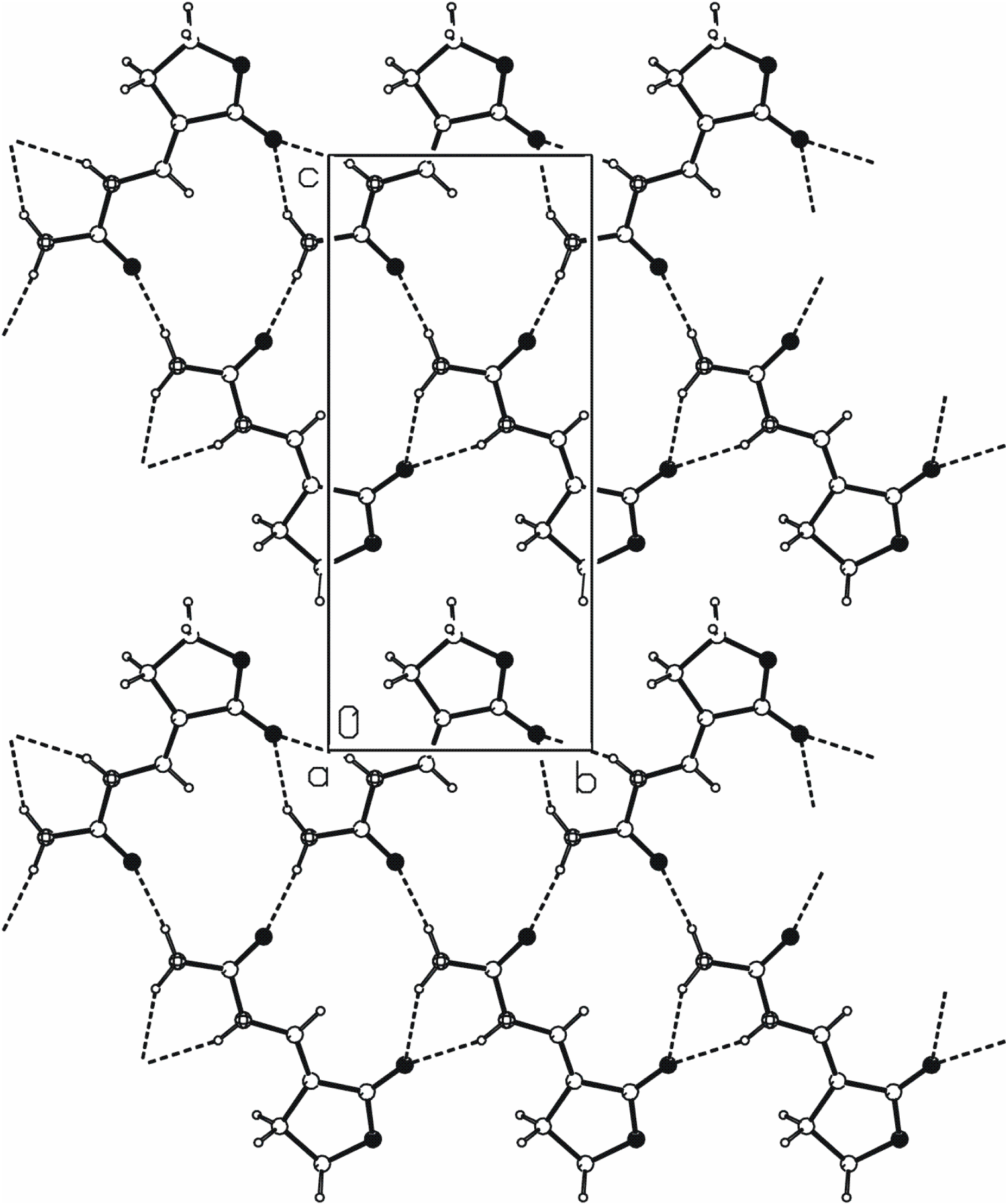
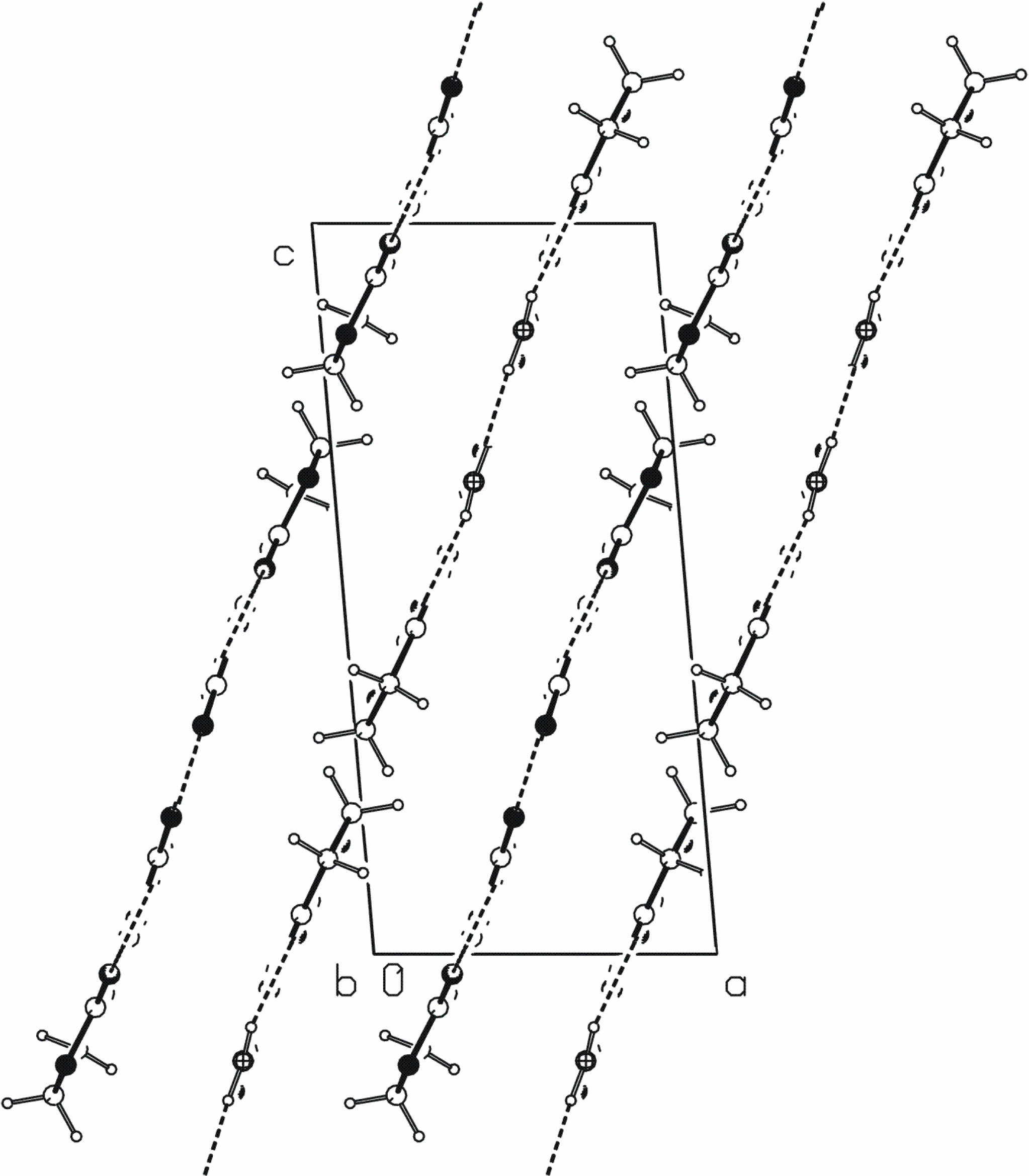
 [20] chain of rings parallel to the b axis (Figure 3). The N2···O3ii hydrogen bond links the molecules into C(4) chains that are also parallel to the b axis. The combination of these two motifs forms new ring of
[20] chain of rings parallel to the b axis (Figure 3). The N2···O3ii hydrogen bond links the molecules into C(4) chains that are also parallel to the b axis. The combination of these two motifs forms new ring of  type. These three N−H···O hydrogen bonds generate mutually parallel one-dimensional chains of edge-fused rings (Figure 3).
type. These three N−H···O hydrogen bonds generate mutually parallel one-dimensional chains of edge-fused rings (Figure 3).

Experimental Section
General
Synthesis
Crystal structure determination of 2
| Formula | C6H8N2O3 |
| Formula weight | 156.14 |
| Crystal system | monoclinic |
| Space group | P 21/c |
| Unit cell dimensions | |
| a (Å) | 7.0522(8) |
| b (Å) | 6.6691(6) |
| c (Å) | 15.1031(16) |
| β (o) | 94.869(9) |
| V (Å3) | 707.76(13) |
| Z | 4 |
| Dcalc. (g cm-3) | 1.465 |
| Absorption coefficient μ (mm-1) | 0.119 |
| F(000) | 328 |
| θ range (°) | 2.71 - 27.99 |
| Index ranges | -9 ≤ h ≤ 9 |
| -8 ≤ k ≤ 8 | |
| -11 ≤ l ≤ 19 | |
| Collected reflections No. | 2941 |
| Independent reflections No. / Rint. | 1702 / 0.0231 |
| Reflections No. I ≥ 2σ(I) | 1063 |
| Refined parameters No. | 112 |
| Goodness-of-fit on F2, S | 1.001 |
| R [I ≥ 2σ(I)] / R [all data] | 0.0398 / 0.0783 |
| wR [I ≥ 2σ(I)] / wR [all data] | 0.1061 / 0.1231 |
| Max./min. electron density (e Å-3) | 0.189 / -0.141 |
Acknowledgements
References
- Balzarini, J.; Bohman, C.; De Clercq, E. Differential mechanism of cytostatic effect of (E)-5-(2-bromovinyl)-2'-deoxyuridine, 9-(1,3-dihydroxy-2-propoxymethyl)guanine, and other antiherpetic drugs on tumor cells transfected by the thymidine kinase gene of herpes simplex virus type 1 or type 2. J. Biol. Chem. 1993, 268, 6332–6337. [Google Scholar]
- Balzarini, J.; De Clercq, E.; Ayusawa, D.; Seno, T. Murine mammary FM3A carcinoma cells transformed with the herpes simplex type 1 thymidine kinase gene are highly sensitive to the growth-inhibitory properties of (E)-5-(2-bromovinyl)-2'-deoxyuridine and related compounds. FEBS Lett. 1985, 185, 95–100. [Google Scholar] [CrossRef]
- De Clercq, E. Biochemical aspects of the selective antiherpes activity of nucleoside analogues. Biochem. Pharmacol. 1984, 33, 2159–2169. [Google Scholar] [CrossRef]
- De Clercq, E.; Descamps, J.; Verhelst, G.; Walker, R.T.; Jones, A.S.; Torrence, P.F.; Shugar, D. Comparative efficacy of antiherpes drugs against different strains of herpes simplex virus. J. Infect. Dis. 1981, 41, 563–574. [Google Scholar]
- Yu, C.-S.; Eisenbarth, J.; Runz, A.; Weber, K.; Zeisler, S.; Oberdorfer, F. Synthesis of 5-(2-radiohaloethyl)- and 5-(2-radiohalovinyl)-2’-deoxyuridines. Novel types of radiotracer for monitoring cancer gene therapy with PET. J. Label. Compd. Radiopharm. 2003, 46, 421–439. [Google Scholar] [CrossRef]
- Prekupec, S.; Makuc, D.; Plavec, J.; Šuman, L.; Kralj, M.; Pavelić, K.; Balzarini, J.; De Clercq, E.; Mintas, M.; Raić-Malić, S. Novel C-6 fluorinated acyclic side chain pyrimidine derivatives: Synthesis, 1H and 13C NMR conformational studies, and antiviral and cytostatic evaluations. J. Med. Chem. 2007, 50, 3037–3045. [Google Scholar] [CrossRef]
- Raić-Malić, S.; Johayem, A.; Ametamey, S.M.; Batinac, S.; De Clercq, E.; Folkers, G.; Scapozza, L. Synthesis, 18F-radiolabelling and biological evaluations of C-6 alkylated pyrimidine nucleoside analogues. Nucleos. Nucleot. Nucleic Acids 2004, 23, 1707–1721. [Google Scholar] [CrossRef]
- Johayem, A.; Raić-Malić, S.; Lazzati, K.; Schubiger, P.A.; Scapozza, L.; Ametamey, S.M. Synthesis and characterization of a C(6) nucleoside analogue for the in vivo imaging of the gene expression of herpes simplex virus type-1 thymidine kinase (HSV1 TK). Chem. Biodiver. 2006, 3, 274–283. [Google Scholar] [CrossRef]
- Gazivoda, T.; Plevnik, M.; Plavec, J.; Kraljević, S.; Kralj, M.; Pavelić, K.; Balzarini, J.; De Clercq, E.; Mintas, M.; Raić-Malić, S. The novel pyrimidine and purine derivatives of l-ascorbic acid: Synthesis, one- and two-dimensional 1H and 13C NMR study, cytostatic and antiviral evaluation. Bioorg. Med. Chem. 2005, 13, 131–139. [Google Scholar] [CrossRef]
- Gazivoda, T.; Šokčević, M.; Kralj, M.; Šuman, L.; Pavelić, K.; De Clercq, E.; Andrei, G.; Snoeck, R.; Balzarini, J.; Mintas, M.; Raić-Malić, S. Synthesis and antiviral and cytostatic evaluations of the new C-5 substituted pyrimidine and furo[2,3-d]pyrimidine 4',5'-didehydro-l-ascorbic acid derivatives. J. Med. Chem. 2007, 50, 4105–4112. [Google Scholar] [CrossRef]
- Gazivoda, T.; Raić-Malić, S.; Marjanović, M.; Kralj, M.; Pavelić, K.; Balzarini, J.; De Clercq, E.; Mintas, M. The novel C-5 aryl, alkenyl and alkynyl substituted uracil derivatives of l-ascorbic acid: Synthesis, cytostatic and antiviral activity evaluations. Bioorg. Med. Chem. 2007, 15, 749–758. [Google Scholar] [CrossRef]
- Sigmond, J.; Peters, G.J. Pyrimidine and purine analogues, effects on cell cycle regulation and the role of cell cycle inhibitors to enhance their cytotoxicity. Nucleos. Nucleot. Nucleic Acids 2005, 24, 1997–2022. [Google Scholar] [CrossRef]
- De Clercq, E. Chemotherapeutic approaches to the treatment of the acquired immunodeficiency syndrome (AIDS). J. Med. Chem. 1986, 29, 1561–1569. [Google Scholar] [CrossRef]
- Herdewijn, P.; Van Aershot, A.; Balzarini, J.; De Clercq, E. 3’-Fluoro- and 3’-azido-substituted 2’,3’-dideoxynucleosides: Structure-activity relationship. Med. Chem. Res. 1991, 1, 9–19. [Google Scholar]
- De Clercq, E. Toward Improved Anti-HIV Chemotherapy: Therapeutic strategies for intervention with HIV infections. J. Med. Chem. 1995, 38, 2491–2517. [Google Scholar] [CrossRef]
- Fissekis, J.D.; Myles, A.; Brown, G.B. Synthesis of 5-hydroxyalkylpyrimidines from lactones. J. Org. Chem. 1964, 29, 2670–2673. [Google Scholar] [CrossRef]
- Fissekis, J.D.; Sweet, F. The chemistry of some 5-(2-hydroxyalkyl)uracil derivatives and a synthesis of 5-vinyluracil. J. Org. Chem. 1973, 38, 264–269. [Google Scholar] [CrossRef]
- Allen, F.H. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Crystallogr. 2002, B58, 380–388, (Version 5.29, January 2008). [Google Scholar] [CrossRef]
- Dinsmore, A.; Doyle, P.M.; Hitchcock, P.B.; Young, D.W. Extension of ring switching strategy to the glutamate antagonist 2-(pyrimidin-2,4-dione-5-yl-methyl)-(2S)-glycine and related compounds with two chiral centres. Tetrahedron Lett. 2000, 41, 10153–10158. [Google Scholar]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.-L. Patterns in hydrogen bonding: functionality and graph set analysis in crystals. Angew. Chem. Int. Ed. Engl. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Stoe & Cie (1995). STADI4. Diffractometer control program, Version 1.05B; Darmstadt, Germany.
- Stoe & Cie (1995). X-RED. Diffractometer reduction program. Version 1.05B; Darmstadt, Germany.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from T. Gazivoda.
© 2008 by the authors. Licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gazivoda, T.; Raić-Malić, S.; Hergold-Brundić, A.; Cetina, M. Synthesis and Structural Characterization of the 5-(2-Haloethyl)pyrimidines – Hydrogen-Bonded chains in α-(1-Carbamyliminomethylene)-γ-Butyrolactone. Molecules 2008, 13, 2786-2795. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules13112786
Gazivoda T, Raić-Malić S, Hergold-Brundić A, Cetina M. Synthesis and Structural Characterization of the 5-(2-Haloethyl)pyrimidines – Hydrogen-Bonded chains in α-(1-Carbamyliminomethylene)-γ-Butyrolactone. Molecules. 2008; 13(11):2786-2795. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules13112786
Chicago/Turabian StyleGazivoda, Tatjana, Silvana Raić-Malić, Antonija Hergold-Brundić, and Mario Cetina. 2008. "Synthesis and Structural Characterization of the 5-(2-Haloethyl)pyrimidines – Hydrogen-Bonded chains in α-(1-Carbamyliminomethylene)-γ-Butyrolactone" Molecules 13, no. 11: 2786-2795. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules13112786