Structure and Catalytic Properties of Carboxylesterase Isozymes Involved in Metabolic Activation of Prodrugs

Laboratory of Drug Metabolism and Biopharmaceutics, Faculty of Pharmaceutical Sciences, Chiba Institute of Science, Shiomi-Cho, Choshi-City, Chiba 288-0025, Japan
Molecules 2008, 13(2), 412-431; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules13020412
Submission received: 14 December 2007
/
Revised: 9 February 2008
/
Accepted: 11 February 2008
/
Published: 18 February 2008
(This article belongs to the Special Issue Prodrugs)
Abstract
:Mammalian carboxylesterases (CESs) comprise a multigene family whose gene products play important roles in biotransformation of ester- or amide-type prodrugs. They are members of an α,β-hydrolase-fold family and are found in various mammals. It has been suggested that CESs can be classified into five major groups denominated CES1-CES5, according to the homology of the amino acid sequence, and the majority of CESs that have been identified belong to the CES1 or CES2 family. The substrate specificities of CES1 and CES2 are significantly different. The CES1 isozyme mainly hydrolyzes a substrate with a small alcohol group and large acyl group, but its wide active pocket sometimes allows it to act on structurally distinct compounds of either a large or small alcohol moiety. In contrast, the CES2 isozyme recognizes a substrate with a large alcohol group and small acyl group, and its substrate specificity may be restricted by the capability of acyl-enzyme conjugate formation due to the presence of conformational interference in the active pocket. Since pharmacokinetic and pharmacological data for prodrugs obtained from preclinical experiments using various animals are generally used as references for human studies, it is important to clarify the biochemical properties of CES isozymes. Further experimentation for an understanding of detailed substrate specificity of prodrugs for CES isozymes and its hydrolysates will help us to design the ideal prodrugs.
Introduction
Mammalian carboxylesterases (CESs, EC 3.1.1.1) comprise a multi-gene family whose gene products are localized in the endoplasmic reticulum (ER) of many tissues. These enzymes efficiently catalyze the hydrolysis of a variety of ester- and amide-containing chemicals as well as drugs (including prodrugs) to the respective free acids. They are involved in detoxification or metabolic activation of various drugs, environmental toxicants and carcinogens. CESs also catalyze the hydrolysis of endogenous compounds such as short- and long-chain acyl-glycerols, long-chain acyl-carnitine, and long-chain acyl-CoA esters [1,2,3,4,5,6,7,8,9]. We have reviewed the characteristics of CESs in relation to the metabolism of xenobiotics [10,11,12]. Multiple isozymes of hepatic microsomal CES exist in various animal species [3,13,14], and some of these isozymes are involved in the metabolic activation of certain carcinogens as well as being associated with hepato-carcinogenesis [6].
Mammalian CES are members of an α,β-hydrolase-fold family and are found in various mammal spp. [3,15,16,17,18,19,20,21,22,23,24,25]. It has been suggested that CESs can be classified into five major groups denominated CES1-CES5, according to the homology of the amino acid sequence [10,11,12], and the majority of CESs that have been identified belong to the CES1 or CES2 family. It has also been shown that striking species differences exist [3,13,26]. For example, Inoue et al. [27] showed that esterase activity in the dog intestine is very weak and produced no appreciable active band in a disc electrophoresis coupled with staining of esterase activity. On the other hand, esterase activities were observed in the intestines of other species (human, rat, mouse, guinea pig and rabbit) [12,26,28,29,30] and found to produce a few active bands in an electrophoretic assay. Since pharmacokinetic and pharmacological data for ester-prodrugs obtained from preclinical experiments using various animals are generally used as references for human studies, it is important to clarify the biochemical properties of each CES isozyme such as substrate specificity, tissue distribution and transcriptional regulation.
CESs show ubiquitous tissue expression profiles with the highest levels of CES activity present in liver microsomes in many mammals [1,6,8,31,32,33,34,35,36,37,38]. Drug-metabolizing enzymes that are present predominantly in the liver are involved in biotransformation of both endogenous and exogenous compounds to polar products to facilitate their elimination. These reactions are categorized into phase I and phase II reactions. CESs are categorized as phase-I drug-metabolizing enzymes that can hydrolyze a variety of ester-containing drugs and prodrugs, such as angiotensin-converting enzyme inhibitors (temocapril, cilazapril, quinapril, and imidapril) [1,39,40,41], anti-tumor drugs (CPT-11 and Capecitabin) [42,43,44,45,46,47,48,49] and narcotics (cocaine, heroin and meperidine) [16,50,51]. In this regard, it is thought that CESs are one of the major determinants for pharmacokinetics and pharmacodynamics of ester-drugs or ester prodrugs (Figure 1). Actually, it has been shown that the dog CES1 isozyme was involved in a pulmonary first-pass effect in the disposition of a propranolol ester prodrug [28,52]. It has also been shown that the expression level of the human CES isozyme was correlated with the conversion ratio of CPT-11 to SN-38, the active metabolite, which is thought to be a key step for the chemotherapeutic action of this anti-tumor drug [47,53,54,55].
This review focuses on the molecular characteristics of CES isozymes, the different structure-activity relationship of substrates with each CES families and genomic structure and regulation of CES genes. This information is important in the design and the development of the prodrugs.
Important role of CES in drug metabolism with other related enzymse or transporters
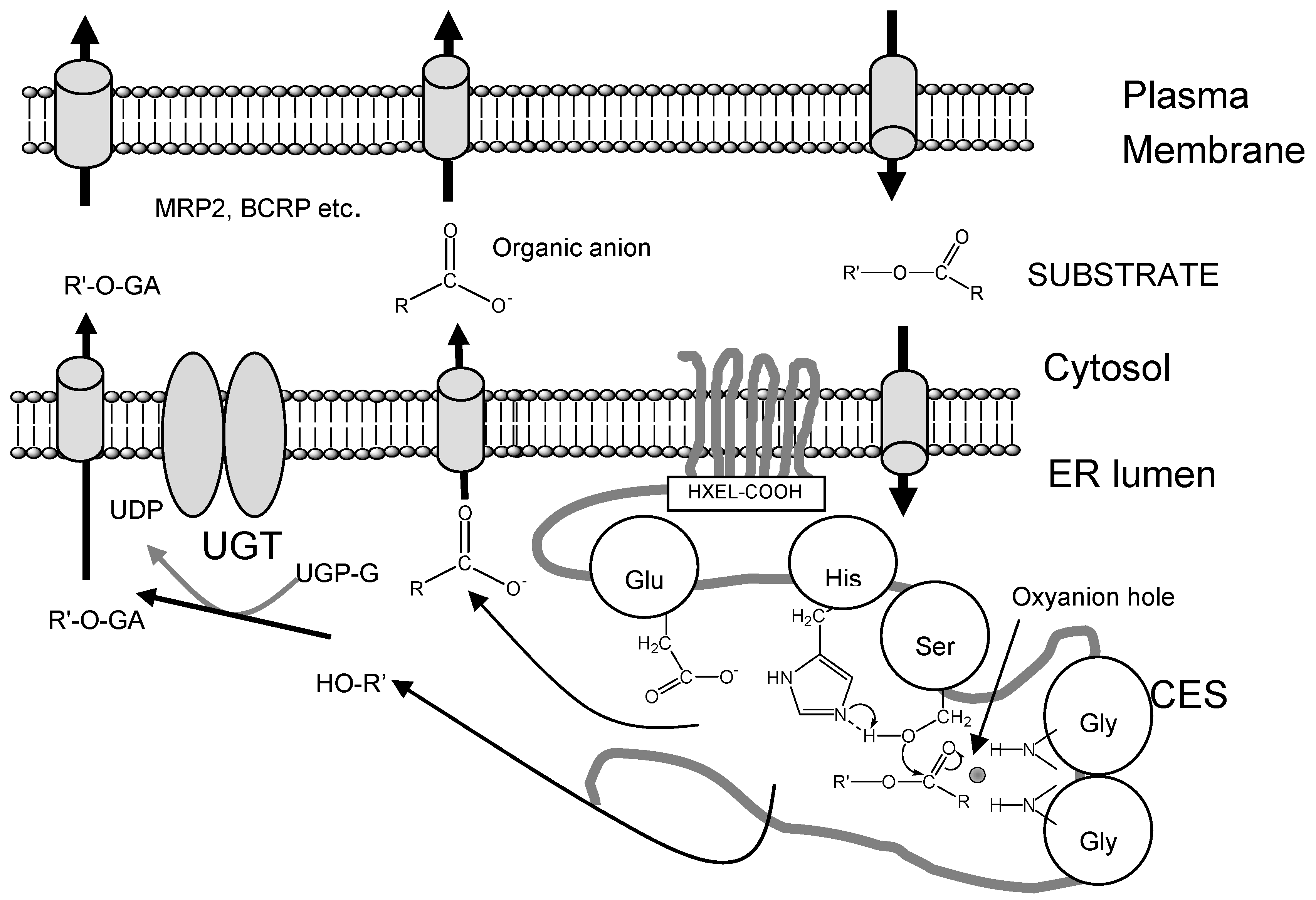
Since many drug metabolizing enzymes, such as cytochrome P450(CYP), CES, UDP-glucuronosyl- ntransferase (UGT) and sulfotransferase, and transporters, such as P-glycoprotein (P-GP), multidrug resistance-associated protein 2 (MRP2) and breast cancer resistance protein (BCRP), were co-expressed in liver and small inteisine, the hydrolase activity in the liver and small intestine is contribute to drug metabolism and drug transport with phase II drug metabolizing enzyme or drug transporter. The CESs and the UGT family, the catalytic domains of which are localized in the luminal sides of the endoplasmic reticulum (ER) membrane, are two major enzyme groups responsible for phase I and II reactions (Figure 2). Products hydrolyzed by CESs, such as SN-38 from CPT-11, are also good substrates for UGT. Thus, we speculated that CES-UGT interaction in the luminal side of the ER membrane is important for drug metabolism. As shown in Figure 1, two hydrolyzed products from ester-substrate are formed by CES: alcohol or phenol, which are substrates for UGT, and organic anions, which are substrates for organic anion transporter such as MRP2 or BCRP. In this regard, we thought that CES is major drug-metabolizing enzymes for enzyme-enzyme interaction or enzyme-transporter interaction.
Figure 1.
CES-UGT interaction in the luminal side of the ER membrane and CES- transporter interaction in the cell. Two hydrolyzed products from ester-substrate are formed by CES; alcohol or phenol, which are substrates for UGT, and organic anions, which are substrates for organic anion transporter such as multidrug resistance-associated protein 2 (MRP2) or breast cancer resistance protein (BCRP).
Figure 1.
CES-UGT interaction in the luminal side of the ER membrane and CES- transporter interaction in the cell. Two hydrolyzed products from ester-substrate are formed by CES; alcohol or phenol, which are substrates for UGT, and organic anions, which are substrates for organic anion transporter such as multidrug resistance-associated protein 2 (MRP2) or breast cancer resistance protein (BCRP).
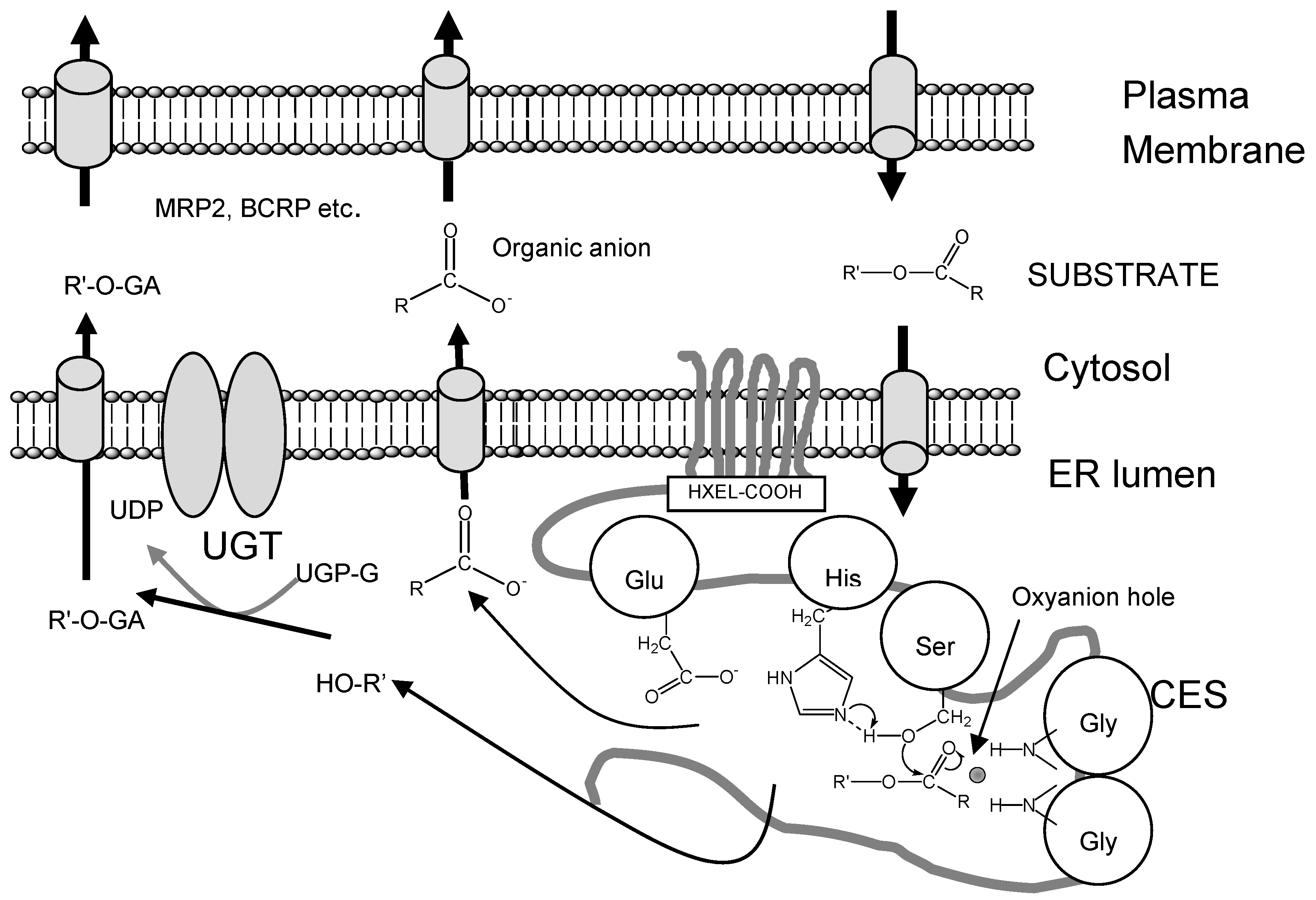
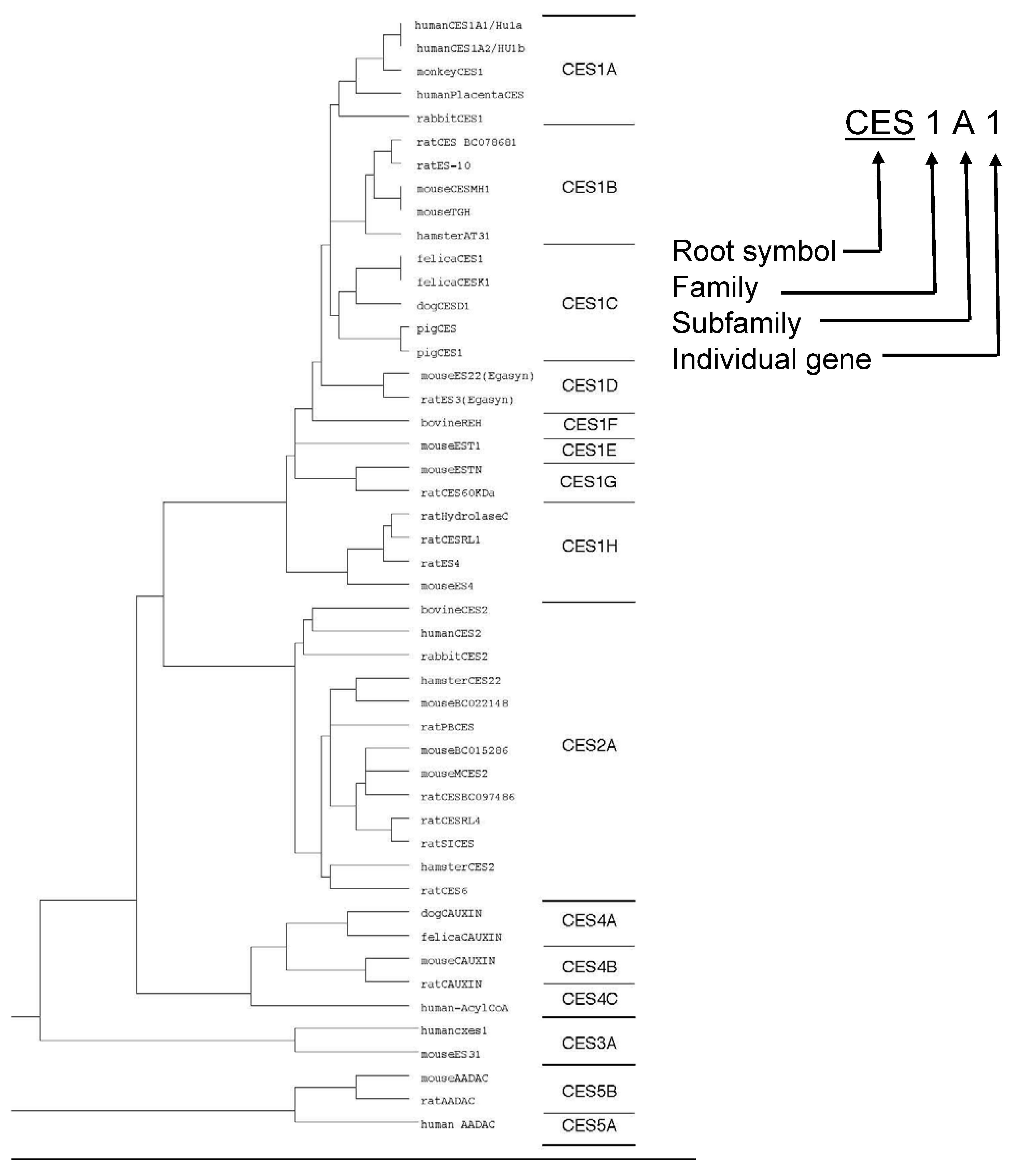
Figure 2.
Phylogenic tree and nomenclature of CES families. CES isozymes are classified into five families, CES1, CES2, CES3, CES4 and CES5. Each families also divided into subfamilies.
Figure 2.
Phylogenic tree and nomenclature of CES families. CES isozymes are classified into five families, CES1, CES2, CES3, CES4 and CES5. Each families also divided into subfamilies.

Classification and Nomenclature of CES Superfamily
According to the classification of esterase by Aldridge [56], the serine superfamily of esterase, i.e., acetylcholine-esterase, butyrylcholine-esterase and CES, fall into the B-esterase group. It is becoming increasingly clear that esterases tend to have a broad and overlapping substrate specificity toward amides and esters. A single esterolytic reaction is frequently mediated by several kinds of enzyme. Recent studies on esterases as well as other enzymes, such as acetylcholine esterase, butyrylcholine esterase, cholesterol esterase, triacylglycerol lipase and CES, involved in xenobiotic metabolism have provided evidence of multiple forms. It seems almost impossible to classify these CES isozymes based on their substrate specificity along the lines of the International Union of Biochemistry (I.U.B.) Classification because the individual hydrolases exhibit properties of CES, lipase or both. Mentlein et al. [37] proposed classification of these hydrolases as "unidentified CESs” (EC 3.1.99.1 to 3.1.99.x). Based on amino acid sequence alignment of the encoding genes, we tried to classify CES isozymes into five families, CES 1, CES 2, CES 3, CES4 and CES 5 (Figure 2) [11,12]. The CES 1 family includes the major forms of CES isozymes (more than 60% homology of human CES1A1). Thus, they could divide into eight subfamilies. Most of all CES1 family, except CES1G, are mainly expressed in liver. The CES 1A subfamily includes the major forms of human, monkey and rabbit CES, and the CES1B includes the major isoforms of rat, mouse and hamster CES, and CES1C includes the major isoforms of dog, cat and pig CES [1,4,5,23,40,46,57,58,59,60]. The CES 1H subfamily includes CES RL1 (CES1H4), mouse ES 4(Ces1H1) and Hydrolase B (CES1H3) and C (CES1H2), which catalyze long-chain acyl-CoA hydrolysis [3,4,10,22,25,61]. Members of the CES 1G family are not retain in ER, which CES isozymes are secrete to blood from liver [24,58]. It is interested that CES1G family isozymes only found in rats and mice, not found in human. Although high level of hydrolase activity can be detected in the blood of the rats and mice, no such activity is detected in the blood of humans. On the other hand, the CES2 family includes human intestinal CES (CES2A1) [28,30,44,62,63], rCES2 (CES2A10) [64], rat intestinal CES, mCES2 (Ces2A6) [2], rabbit form2 [65] and hamster AT51 (CES2A11) [66], which are mainly expressed in small intestines. The CES3 family includes ES-male (CES3A2) and human CES3 (CES3A1) [67,68]. Human CES3 (CES3A1) has about 40% amino acid sequence identity with both CEA1A1 and CES2A1, and is expressed in the liver and gastrointestinal tract at an extremely low level in comparison with CES1A1 and CES2A1 [68]. The CES4 family includes carboxylesterase-like urinary excreted protein (CAUXIN) (CES4A2), which is excrete as a major urinary protein in cat urine [69,70]. The CES 5 family includes 46.5-kDa CES isozymes [71,72], which have a different structure from the structures of isozyme in other CES families. ES 46.5-kDa from mouse liver [38] and amide hydrolase of monkey liver [19] probably belong to this family. These groupings are similar to the results of phylogenic analysis (Figure 2).
Structure and Catalytic Mechanism of CES Isozymes
It has been shown that several proteins of the ER lumen have a common carboxy-terminal sequence, KDEL-COOH, and that the structural motif is essential for retention of the protein in the luminal side of the ER through the KDEL receptor bound to the ER membrane [73,74,75]. Korza and Ozols [76] and Ozols [65] have established the primary structures of two microsomal esterases purified from rabbit liver and designated them 60-kDa esterase forms 1 and form 2, respectively. These two forms of CES have the consensus sequence for the ER retention tetra-peptide (HTEL or HIEL in the one-letter code). The HXEL-COOH motif is also essential for retention of the protein in the luminal side of the ER through the KDEL receptor bound to ER membrane [73,74,75]. Robbi et al. [60] reported cDNA cloning of rat liver CES1B4 (ES-10). That was the first report to show that cDNA of liver CES has the consensus sequence of the ER retention tetrapeptide (HVEL-COOH). Later, Robbi and Beaufay [77] isolated a cDNA clone of another rat liver CES1D2 (ES-3) which encode the consensus sequence of the ER retention tetrapeptide (HTEL-COOH). The other clone encoded egasyn, an accessory protein of β-glucuronidase in the liver microsomes [78]. Egasyn is identical to CES, and it binds β-glucuronidase via its CES active site. In the case of mouse rat and mouse, the carboxyl terminal amino acid sequence of clone rat CES60KDa(CES1G1) and mouse Es-N (Ces1G2) is HTEHK-COOH, which could not bound to KDEL-receptor, and these isozymes are secreted to blood [58].
CES have a signal peptide of 17 to 22 amino acid residues of N-terminal amino acid, including hydrophobic amino acid. In CES1 family, exon1 encode a signal peptide [79,80]. In a case of CES1 family, a bulky aromatic residue (Trp) followed by a small neutral residue (Gly) directly precedes the cleavage site [81].
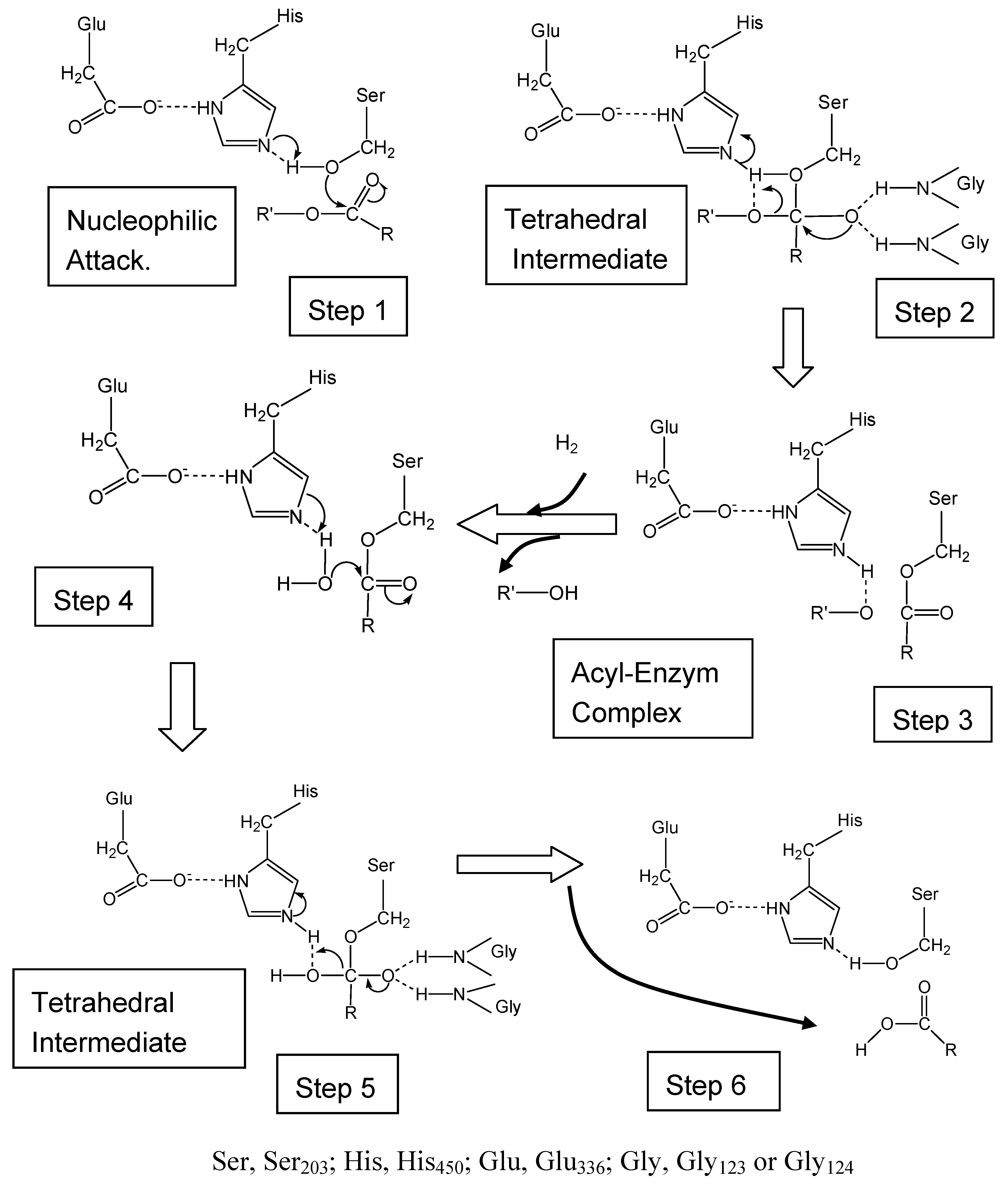
CES have four Cys residues that may be involved in specific disulfide bonds. Among them, Cys98 is the most highly conserved residue in many CES isozymes. Cygler et al. [82] reported an important alignment of a collection of related amino acid sequences of esterase, lipase and related proteins based on X-ray structures of Torpedo californica acetylcholinesterase and Geotrichum candidum lipase. According to these authors, Ser203, Glu336 and His450 form a catalytic triad, and Gly124-Gly125 may be part of an oxyanion hole. These residues are also highly conserved among CES isozymes. As a result we have started mutation analysis [11]. Site-specific mutation of Ser203 to Thr203, Glu336 to Ala336, or His450 to Ala 450 greatly reduced the CES activity towards substrates. Therefore, the mutagenesis confirmed a role of Glu336 and His450 in forming a putative charge relay system with active site Ser203 [11].
Frey et al. [83] reported that the formation of low barrier hydrogen bonds between His and Asp (Glu for CES) facilitates nucleophilic attack by the β-OH group of Ser on the acyl carbonyl group of peptide in chymotrypsin. The catalytic triad in the tetrahedral addition intermediate is stabilized by the low barrier hydrogen bonds. According to their theory, we speculated that the low barrier hydrogen bond between Glu336 and His 450 facilitates nucleophilic attack by the β-OH group of Ser203 on the carbonyl group of the substrate in CES (Figure 3). The mechanism of CES could thus be divided into next steps. 1) The enzyme substrate complex form, positioning the substrate in the correct orientation for reaction. 2) Hydrolysis of the ester bond starts with an attack by the oxygen atom of the hydroxy group of Ser203 on the carbonyl carbon atom of the ester bond. 3) The hydrogen bonds between the negatively charged oxygen of the tetrahedral intermediate and the N-H group of Gly123 and Gly124 stabilize the negatively charged oxygen (O-). This configuration, in which negatively charged carboxyl oxygen is hydrogen bonded to two N-H group, is called oxyanion hole. In the general acid-catalyzed step, the ester bond breaks, and the leaving group picks up a proton from the imidazolium ion of His450. The acyl portion of the original ester bond remains bound to the enzyme as an acyl-enzyme intermediate. The alcohol component (R’-OH) diffused away, completing the acylation stage of the hydrolytic reaction. 4) A water molecule attacks the acyl-enzyme intermediate to give a second tetrahedral intermediate. 5) His450 then donates the proton to the oxygen atom of Ser203, which then releases the acid component of the substrate. The acid component diffused away and the enzyme is ready for catalysis. The tetrahedral transition state is stabilized by the formation of low barrier hydrogen bonds between His450 and Glu336. The low barrier hydrogen bonds facilitated mechanism includes weak hydrogen bonds between the oxyanion (O-) and peptide N-H bonds contributed by Gly123 and Gly124, which stabilize the tetrahedral adduct on the substrate side of the transition state (Figure 3). Formation of the acyl-enzyme complex in the next step requires removal of a proton from His450, so that the tetrahedral intermediate is disrupted in the acyl-enzyme intermediate. When the unbound portion of the alcohol group of the first product of the substrate has diffused away, a second step which the deacylation step is essentially the reverse of the acylation step occurs, with a water molecule substituting for the alcohol group of the original substrate (Figure 3).
Figure 3.
Proposed mechanism for the action of CES. Conformation of the Ser-His-Glu catalytic triad in CES.
Figure 3.
Proposed mechanism for the action of CES. Conformation of the Ser-His-Glu catalytic triad in CES.

Ser, Ser203; His, His450; Glu, Glu336; Gly, Gly123 or Gly124
It is of interest that the sequences required for the hydrolytic capability at the catalytic triad (Glu, His, Ser) of CES, acetylcholine esterase, butyrylcholin esterase, and cholesterol esterase are highly conserved. This is a common structure of α,β-hydrolase-fold families, which are responsible for the hydrolysis of endogenous and exogenous compounds.
Furthermore, these elements are strongly conserved among orthologous CESs of the mouse, rat, rabbit, monkey and human. A three-dimensional model for human CES has been proposed on the basis of crystal structure coordinates of acetylcholine esterase and overlapping active sites with pancreatic lipase and CES [84]. The modeled structure shares the overall folding and topology of the proteins identified in the recently published crystal structures of the rabbit [85] and human CES [86,87]. CES has a three-dimensional α,β-hydrolase-fold structure, which is a structural feature of all lipases [88]. In general, the structure of CES may be viewed as comprising a central catalytic domain surrounded by α−β and regulatory domains [85,86,87]. In essence, the α,β-hydrolase-fold consists of a central β-sheet surrounded by a variable number of α-helices and accommodates a catalytic triad composed of Ser, His and a carboxylic acid. This suggests that the catalytic function of these proteins is conserved across species. The catalytic triad is located at the bottom of about 25 Å deep active site cleft, approximately in the center of the molecule and comprises a large flexible pocket on one side of Ser203 and a small rigid pocket on the opposite side [86]. The orientation and location of the active site provides an ideal hydrophobic environment for the hydrolysis of a wide variety of hydrophobic substrates [86]. The small rigid active site pocket is adjacent to the oxyanion hole formed by Gly123-124 and is lined by several hydrophobic residues [86]. Short acyl chains would be easily accommodated within the small rigid pocket. The larger flexible active site pocket is lined by several non-polar residues and could accommodate larger or polycyclic molecules such as cholesterol. The large pocket is adjacent to a side door secondary pore that would permit small molecules (substrates and reaction products) to enter and exit the active site [86]. Longer acyl chains may be oriented for catalysis in such a way that they extend through the side door. Indeed, the presence of a hydrophobic residue at position 423 in mice Ces1B2 and 425 in humans CES1A1 is necessary for efficient hydrolysis of hydrophobic substrates, as mutation of Met present in position 423 of the related rat lung CES (CES1B4) to Ile increased the CES activity towards a more hydrophobic substrate without affecting activity towards short-chain esters [89].
Most CES isozymes are glycoproteins, and the carbohydrate chain is required for the enzyme activity of CESs [10,18,86,90,91]. Human CES2A1 contains a glycosylation site at two different positions (Asn103 and Asn267), while CES1A1 contains only one glycosylation site at Asn79. This glycosylation site is modified by a carbohydrate chain with first N-acetylglucosamine and terminal sialic acid and appears to be involved in the stabilization of the CES1A1 trimer by packing into the adjacent monomer in its crystal structure [86]. According to the X-ray crystal structure of human CES1, this residue lines the flexible pocket adjacent to the side door [86]. Given the wide range of substrates that CESs are known to hydrolyze, the large flexible pocket confers the ability to hydrolyze many structurally distinct compounds, whereas the rigid pocket is much more selective with regard to the substrates that may be accommodated.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Alcohol Substituent | Acyl Substituent | Substrate Specficity |
|---|---|---|---|
| Cocaine (methyl ester) | CH3OH |  | CES1 |
| Meperidine | CH3CH2OH |  | CES1 |
| Methylphenidate | CH3OH |  | CES1 |
| Temocapril | C2H5OH |  | CES1 |
| Oseltamivir | C2H5OH |  | CES1 |
| Cocaine (benzoyl ester) |  |  | CES2>>CES1 |
| CPT-11 |  |  | CES2>>CES1 |
| Heroin |  | CH3COOH | CES2>>CES1 |
| Methylprednisolone 21-Hemisuccinate |  | HOOC-CH2-CH2-COONa | CES2>>CES1 |
Structure-activity Relationships of Substrates with CES1 and CES2 Families
Recent studies have shown that there are some differences between these families in terms of substrate specificity, tissue distribution, immunological properties, and gene regulation [10]. For example, the preferential substrates for CES1A1, a human CES1 family isozyme, are thought to be compounds esterified by small alcohols, while those for CES2A1, a human CES2 family isozyme, are thought to be compounds esterified by relatively large alcohols (Table 1). CES1A1, but not CES2A1, hydrolyzed the methyl ester of cocaine and the ethyl esters of temocapril, meperidine and delapril [1,40,41,54,92]. More recently, Sun et al. [93] reported that CES1A1 has a high catalytic efficiency for both d- and l-enantiomers of methylphenidate. No catalytic activity was detected with CES2 and CES3 for either enantiomer. The catalytic efficiency of CES1A1 for l-methylphenidate (kcat/Km= 7.7 mM-1min-1) is greater than that for d-methylphenidate (kcat/Km= 1.3-2.1 mM-1min-1). Hence, the catalytic efficiency of CES1A1 for methylphenidate enantiomers agrees with stereoselective clearance of methylphenidate reported in human subjects. Both enantiomers of methylphenidate can fit into the three-dimensional model of CES1A1 to form productive complexes in the active site [93]. Oseltamivir as an anti-influenza viral agent is an ester prodrug, and its antiviral activity is achieved by its hydrolytic metabolite: oseltamivir carboxylate. Shi et al. [94] reported that human liver microsomes rapidly hydrolyzed oseltamivir, but no hydrolysis was detected with intestinal microsomes or plasma. Recombinant CES1A1, but not CES2A1, hydrolyzed oseltamivir and produced similar kinetic parameters as the liver microsomes [94]. We also reported that mouse mCES1(CES MH1), a mouse CES1 family isozyme, hydrolyzed temocapril, which esterified a small alcohol, similar to human CES1 isozyme [1].
In contrast to the specificity of CES1 for the methyl ester of cocaine, only CES2 hydrolyzed the benzoyl ester of cocaine [54,92]. For the remaining substrates that could be hydrolyzed by both enzymes, CES2 exhibited higher catalytic efficiency than did CES1 for heroin; enzymatic conversion of 6-acetylmorphine to morphine was not known before the isolation and characterization of CES2 [50]. CPT-11 and heroin bearing a small acyl moiety and a bulky alcohol group are good substrates for the CES2 isozyme (Table 1). On the other hand, we also reported that rat rCES2 (also called CES RL4), a rat CES2 family isozyme, hydrolyzed methylprednisolone hemisuccinate, which esterified a large alcohol as did the human CES2 family [64]. It has been suggested that although these two CES families exhibit broad substrate specificity for ester, carbamate, or amide hydrolysis, these CES isozymes do exhibit distinct catalytic efficiencies that correlate with the relative size of the substrate substituents versus that of the enzyme active sites.
Tissue specific expression of CES1 and CES2 was examined by northern blots, RT-PCR and real time PCR analysis. As shown in Table 2, human CES1A was highly expressed in liver and lung, human CES2A was highly expressed in small intestine and kidney. Knowledge of these substrate structure-activity relationships and the tissue distribution of CES isozymes is critical for predicting the metabolism and the pharmacokinetics and pharamcodynamics of ester drugs or prodrugs.
| Species | Isozyme | Liver | Small intestine | Kidney | Lung |
|---|---|---|---|---|---|
| Mouse | CES1 CES2 | +++ +++ | - +++ | +++ +++ | +++ - |
| Rat | CES1 CES2 | +++ - | - +++ | +++ - | +++ - |
| Hamster | CES1 CES2 | +++ +++ | - +++ | +++ - | NT NT |
| Guinea Pig | CES1 CES2 | +++ - | +++ + | ++ - | NT NT |
| Beagle Dog | CES1 CES2 | +++ ++ | - - | NT NT | +++ + |
| Monkey | CES1 CES2 | +++ + | ++ +++ | - + | NT NT |
| Human | CES1 CES2 | +++ + | - +++ | + +++ | +++ - |
-, undetectable, +, weakly expressed, ++, moderately expressed, +++, strongly expressed, NT, not tested
Gene Structure and Regulation of CES Isozymes
Both the murine [10] and human [80,95] CES1 genes span about 30 kb and contain 14 small exons. Recently, sequencing of the mouse and human genomes have been completed, enabling detailed sequence comparisons. Previously published sequences of individual exons, splice junctions, size of the introns and restriction sites within the murine and human CES genes are consistent with their respective genes sequenced by the mouse and human genome projects. Therefore, the organization of the CES gene is evolutionarily conserved in mice and humans. In previous studies, the human CES gene have been mapped to chromosome 16 at 16q13–q22.1 [18,57]. This region is syntenic to a region of mouse chromosome 8 at 8C5 [57]. The murine CES Es22 [79] and Es-N [58] have been previously mapped to chromosome 8. The completion of the mouse genome sequencing project unambiguously demonstrated that the murine CES gene was located on the minus strand of chromosome 8 at 8C5 in a cluster of six CES genes that spans 260.6 kb in total. These six CES genes are presumed to have originated from repeated gene duplications of a common ancestral gene that encoded a CES [80], and subsequent evolutionary divergence occurred.
Recently, we have identified a mouse liver microsomal acylcarnitine hydrolase, mCES2, as a member of the CES 2 family [2]. It has been revealed that this enzyme is significantly induced by di(2-ethylhexyl)phthalate and shows medium- and long-chain acylcarnitine hydrolase activity [2]. In addition, we have found that mCES2 is expressed in various tissues with higher levels of expression in the liver, kidney and small intestine. It was shown that three transcription factors, specificity protein (Sp) 1, Sp3 and upstream stimulatory factor 1, could bind to the promoter region of the mCES2 gene, leading to a synergistic transactivation of the promoter [96]. Although this mechanism may explain the ubiquitous tissue expression profiles of mCES2, it is unlikely to contribute to the higher levels of mCES2 expression in the liver, kidney and small intestine. Therefore, it is thought that there exists another mechanism controlling this tissue-specific transcription of the mCES2 gene [96]. More recently, we have shown that hepatocyte nuclear factor-4 alpha (HNF-4α) can strongly enhance mCES2 gene transcription and that the involvement of HNF-4α accounts for the high expression level of mCES2 in the liver [97]. These findings are notable when physiological roles of mCES2 are studied, since HNF-4α is involved in various hepatic functions, such as glucose and cholesterol metabolism and drug metabolism. In addition, we found that bile acid can repress mCES2 gene transcription by repressing HNF-4α-mediated transactivation [97].
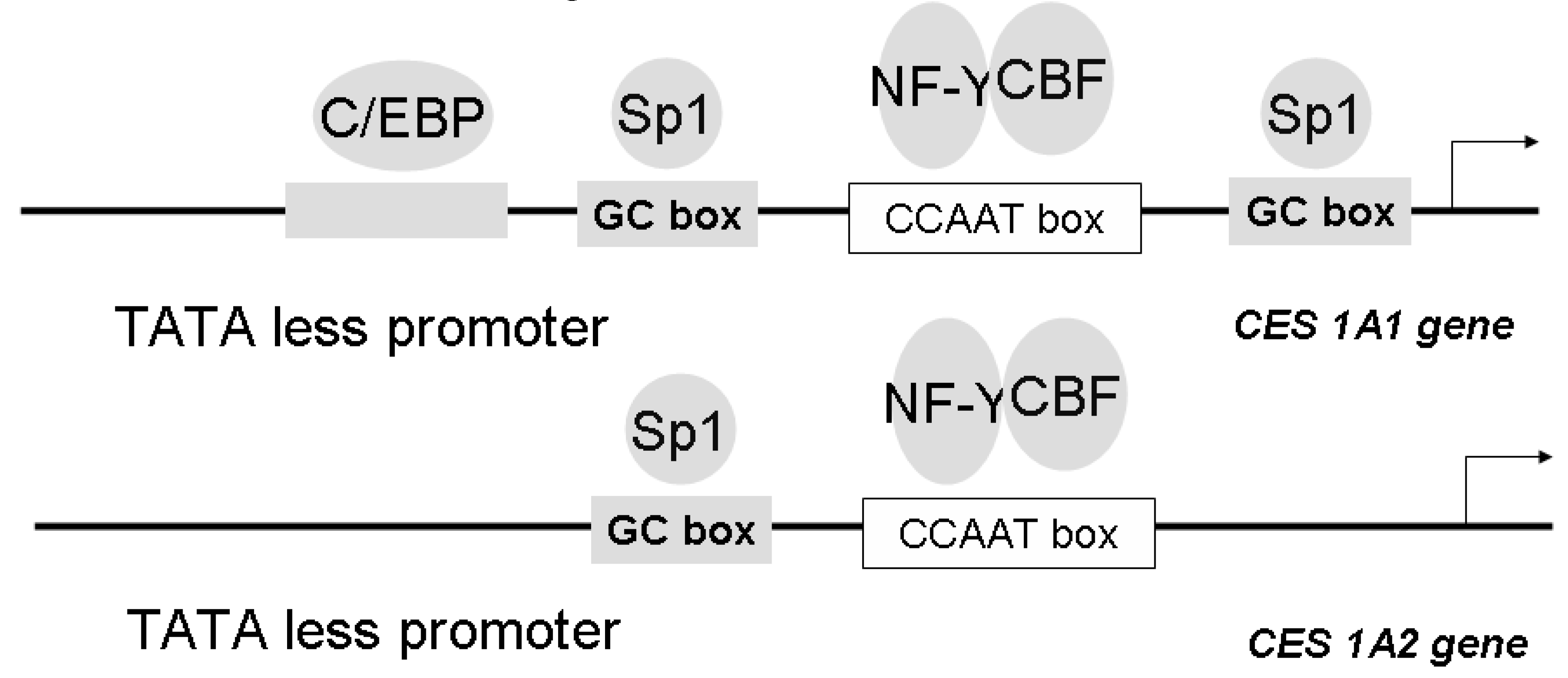
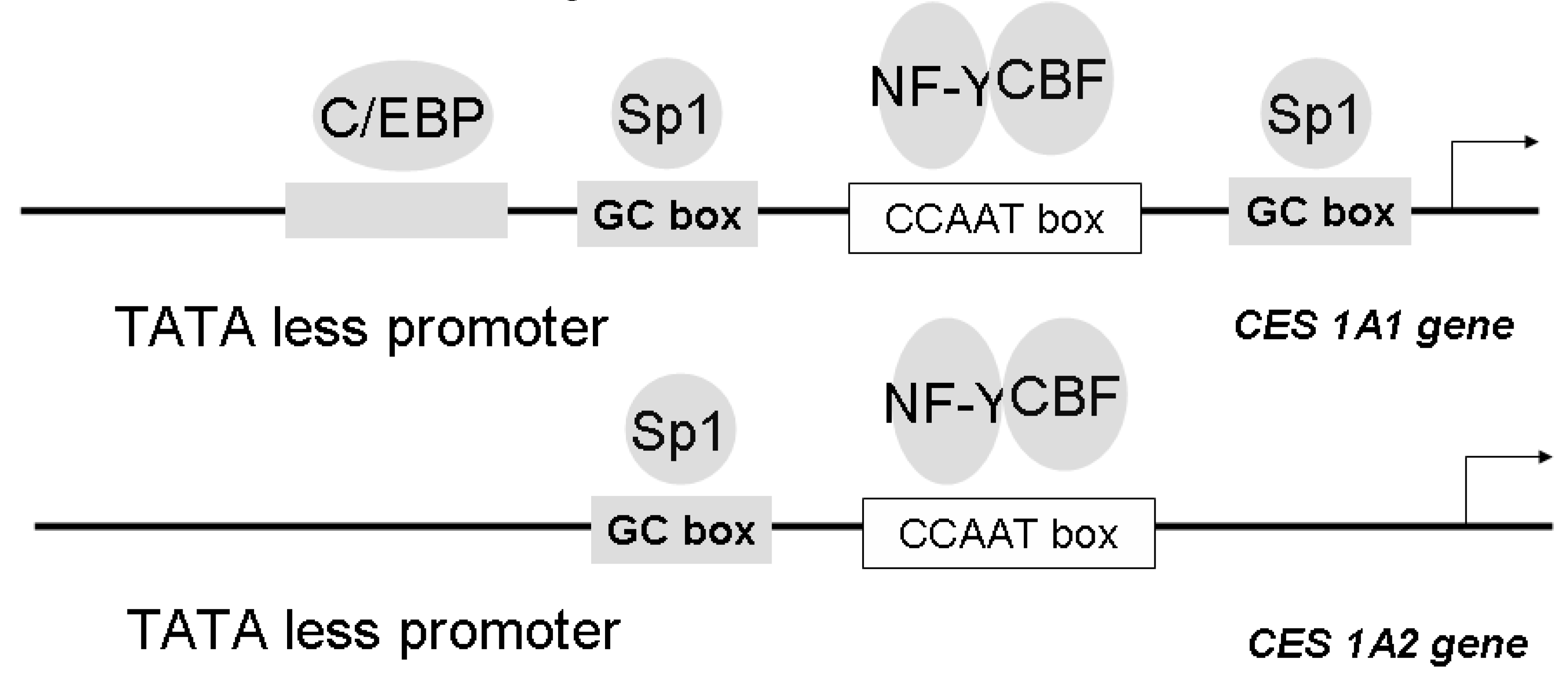
We have also isolated and characterized two genes encoding the human CES1A1 (AB119997) and CES 1A2 (AB119998), and we also cloned and sequenced the 5' flanking region of each gene in order to elucidate the structure of the promoter [98]. It is noteworthy that both the CES1A1 and CES1A2 genes are located on chromosome 16q13-q22 with a tail-to-tail structure. A comparison of the nucleotide sequences of CES1A1 and CES 1A2 genes revealed about 98% homology in 30 Kbp. There are only six nucleotide differences resulting in four amino acid differences in the open reading frame, and all of the differences existed in exon 1. Gene duplication has generally been viewed as a necessary source of material for the origin of evolutionary novelties, and duplicate genes evolve new functions. The majority of gene duplicates are silenced within a few million years, with the small number of survivors subsequently being subjected to strong purifying selection. Although duplicate genes may only rarely evolve new functions, the stochastic silencing of such genes may play a significant role in the passive origin of new species. Since exon 1 of the CES1 gene encodes a signal peptide region, intracellular localization of the CES1 gene product was preliminary investigated using a signal peptide/EYFP-ER chimera protein-expressing system. It was interesting that the CES1A1 signal peptide/EYFP-ER chimera protein was localized to the endoplasmic reticulum, whereas the CES1A2 signal peptide/EYFP-ER chimera protein was distributed in the endoplasmic reticulum and cytosol. On the other hand, CES1A2 mRNA was found to be expressed only in human adult liver, although CES1A1 is expressed in human adult liver and fetal liver (Hosokawa et al. submitted for publication). These results suggested that CES1A1 and CES1A2 have different intracellular localizations and different expression profiles in liver differentiation. We investigated the transcriptional regulation of these two CES genes. Reporter gene assays and electrophoretic mobility shift assays demonstrated that Sp1 and C/EBPα could bind to each responsive element of the CES1A1 promoter but that Sp1 and C/EBP could not bind to the responsive element of the CES1A2 promoter (Figure 4) [98].
The expression level of CES1A1 mRNA is much higher than that of CES1A2 mRNA in the liver [98]. Since CES1A1 is highly variable in the individual liver [33], it was thought that these result provide information on individual variation of human CES1.
Figure 4.
Structure of the 5’ flanking region of CES 1A1 and CES 1A2 genes. Sp1 and C/EBPα could bind to each responsive element of the CES1A1 promoter but that Sp1 and C/EBP could not bind to the 5’ flanking region of the CES1A2 promoter. NF-Y, nuclear factor Y, CBF, CCAAT-binding factor
Figure 4.
Structure of the 5’ flanking region of CES 1A1 and CES 1A2 genes. Sp1 and C/EBPα could bind to each responsive element of the CES1A1 promoter but that Sp1 and C/EBP could not bind to the 5’ flanking region of the CES1A2 promoter. NF-Y, nuclear factor Y, CBF, CCAAT-binding factor
Conclusions
Multiple CES play an important role in the hydrolytic biotransformation of a vast number of structurally diverse drugs. These enzymes are major determinants of the pharmacokinetic behavior of most therapeutic agents containing an ester or amide bond. There are several factors that influence CES activity, either directly or at the level of enzyme regulation. In the clinical field, drug elimination is decreased and the incidence of drug-drug interactions increases when two or more drugs compete for hydrolysis by the same CES isozyme. Exposure to environmental pollutants or to lipophilic drugs can result in induction of CES activity. Several drug-metabolizing enzymes, such as CYP, UGT and SULT, have been extensively studied to clarify the substrate specificity using molecular cloning and cell expression systems. In the present review also described the structure and substrate specificity of CES isozymes, and tissue-specific expression profile of CES isozymes. Therefore, successful design of ester-containing drugs will be greatly improved by further detailed analysis for the mechanism of action and substrate recognition site of human CES isozymes. Further experimentation for an understanding of detailed substrate specificity of prodrugs for CES isozymes and its hydrolysates will help us to design ideal prodrugs.
Acknowledgements
The author would like to thank Professor Teruko Imai, Graduate School of Pharmaceutical Sciences, Kumamoto University, and many colleagues for their tremendous encouragement and helpful discussions.
References
- Furihata, T.; Hosokawa, M.; Koyano, N.; Nakamura, T.; Satoh, T.; Chiba, K. Identification of di-(2-ethylhexyl) phthalate-induced carboxylesterase 1 in C57BL/6 mouse liver microsomes: purification, cDNA cloning, and baculovirus-mediated expression. Drug Metab. Dispos. 2004, 32, 1170–7. [Google Scholar] [CrossRef]
- Furihata, T.; Hosokawa, M.; Nakata, F.; Satoh, T.; Chiba, K. Purification, molecular cloning, and functional expression of inducible liver acylcarnitine hydrolase in C57BL/6 mouse, belonging to the carboxylesterase multigene family. Arch. Biochem. Biophys. 2003, 416, 101–9. [Google Scholar] [CrossRef]
- Hosokawa, M.; Maki, T.; Satoh, T. Characterization of molecular species of liver microsomal carboxylesterases of several animal species and humans. Arch. Biochem. Biophys. 1990, 277, 219–27. [Google Scholar] [CrossRef]
- Hosokawa, M.; Satoh, T. Molecular aspect of the inter-species variation in carboxylesterase, 7th North American ISSX Meeting, San Diego, CA, USA; 1996.
- Hosokawa, M.; Suzuki, K.; Takahashi, D.; Mori, M.; Satoh, T.; Chiba, K. Purification, molecular cloning, and functional expression of dog liver microsomal acyl-CoA hydrolase: a member of the carboxylesterase multigene family. Arch. Biochem. Biophys. 2001, 389, 245–53. [Google Scholar] [CrossRef]
- Maki, T.; Hosokawa, M.; Satoh, T.; Sato, K. Changes in carboxylesterase isoenzymes of rat liver microsomes during hepatocarcinogenesis. Jpn. J. Cancer Res. 1991, 82, 800–6. [Google Scholar] [CrossRef]
- Mentlein, R.; Berge, R. K.; Heymann, E. Identity of purified monoacylglycerol lipase, palmitoyl-CoA hydrolase and aspirin-metabolizing carboxylesterase from rat liver microsomal fractions. A comparative study with enzymes purified in different laboratories. Biochem. J. 1985, 232, 479–83. [Google Scholar]
- Mentlein, R.; Heiland, S.; Heymann, E. Simultaneous purification and comparative characterization of six serine hydrolases from rat liver microsomes. Arch. Biochem. Biophys. 1980, 200, 547–559. [Google Scholar] [CrossRef]
- Mentlein, R.; Heymann, E. Hydrolysis of ester- and amide-type drugs by the purified isoenzymes of nonspecific carboxylesterase from rat liver. Biochem. Pharmacol. 1984, 33, 1243–8. [Google Scholar]
- Hosokawa, M.; Furihata, T.; Yaginuma, Y.; Yamamoto, N.; Koyano, N.; Fujii, A.; Nagahara, Y.; Satoh, T.; Chiba, K. Genomic structure and transcriptional regulation of the rat, mouse, and human carboxylesterase genes. Drug Metab. Rev. 2007, 39, 1–15. [Google Scholar]
- Satoh, T.; Hosokawa, M. The mammalian carboxylesterases: from molecules to functions. Ann. Rev. Pharmacol. Toxicol. 1998, 38, 257–88. [Google Scholar] [CrossRef]
- Satoh, T.; Hosokawa, M. Structure, function and regulation of carboxylesterases. Chem. Biol. Interact. 2006, 162, 195–211. [Google Scholar] [CrossRef]
- Hosokawa, M.; Hirata, K.; Nakata, F.; Suga, T.; Satoh, T. Species differences in the induction of hepatic microsomal carboxylesterases caused by dietary exposure to di(2-ethylhexyl)phthalate, a peroxisome proliferator. Drug Metab. Dispos. 1994, 22, 889–94. [Google Scholar]
- Hosokawa, M.; Maki, T.; Satoh, T. Multiplicity and regulation of hepatic microsomal carboxylesterases in rats. Mol. Pharmacol. 1987, 31, 579–84. [Google Scholar]
- Brzezinski, M. R.; Abraham, T. L.; Stone, C. L.; Dean, R. A.; Bosron, W. F. Purification and characterization of a human liver cocaine carboxylesterase that catalyzes the production of benzoylecgonine and the formation of cocaethylene from alcohol and cocaine. Biochem. Pharmacol. 1994, 48, 1747–55. [Google Scholar] [CrossRef]
- Brzezinski, M. R.; Spink, B. J.; Dean, R. A.; Berkman, C. E.; Cashman, J. R.; Bosron, W. F. Human liver carboxylesterase hCE-1: binding specificity for cocaine, heroin, and their metabolites and analogs. Drug Metab. Dispos. 1997, 25((9)), 1089–96. [Google Scholar]
- Ellinghaus, P.; Seedorf, U.; Assmann, G. Cloning and sequencing of a novel murine liver carboxylesterase cDNA. Biochim. Biophys. Acta 1998, 1397, 175–9. [Google Scholar] [CrossRef]
- Kroetz, D. L.; McBride, O. W.; Gonzalez, F. J. Glycosylation-dependent activity of baculovirus-expressed human liver c arboxylesterases: cDNA cloning and characterization of two highly similar enzyme forms. Biochemistry 1993, 32, 11606–17. [Google Scholar] [CrossRef]
- Kusano, K.; Seko, T.; Tanaka, S.; Shikata, Y.; Ando, T.; Ida, S.; Hosokawa, M.; Satoh, T.; Yuzuriha, T.; Horie, T. Purification and characterization of monkey liver amidohydrolases and it's relationship to a metabolic polymorphism of E6123, a platelet activating factor receptor antagonist. Drug Metab. Dispos. 1996, 24, 1186–91. [Google Scholar]
- Langmann, T.; Aslanidis, C.; Schuierer, M.; Schmitz, G. Differentiation-dependent expression of a human carboxylesterase in monocytic cells and transcription factor binding to the promoter. Biochem. Biophys. Res. Commun. 1997, 230, 215–9. [Google Scholar] [CrossRef]
- Morgan, E. W.; Yan, B.; Greenway, D.; Petersen, D. R.; Parkinson, A. Purification and characterization of two rat liver microsomal carboxylesterases (hydrolase A and B). Arch. Biochem. Biophys. 1994, 315, 495–512. [Google Scholar] [CrossRef]
- Yan, B.; Yang, D.; Brady, M.; Parkinson, A. Rat kidney carboxylesterase. Cloning, sequencing, cellular localization, and relationship to rat liver hydrolase. J. Biol. Chem. 1994, 269, 29688–96. [Google Scholar]
- Yan, B.; Yang, D.; Brady, M.; Parkinson, A. Rat testicular carboxylesterase: cloning, cellular localization, and relationship to liver hydrolase A. Arch. Biochem. Biophys. 1995, 316, 899–908. [Google Scholar] [CrossRef]
- Yan, B.; Yang, D.; Bullock, P.; Parkinson, A. Rat serum carboxylesterase. Cloning, expression, regulation, and evidence of secretion from liver. J. Biol. Chem. 1995, 270, 19128–34. [Google Scholar] [CrossRef]
- Yan, B.; Yang, D.; Parkinson, A. Cloning and expression of hydrolase C, a member of the rat carboxylesterase family. Arch. Biochem. Biophys. 1995, 317, 222–34. [Google Scholar] [CrossRef]
- Prueksaritanont, T.; Gorham, L. M.; Hochman, J. H.; Tran, L. O.; Vyas, K. P. Comparative studies of drug-metabolizing enzymes in dog, monkey, and human small intestines, and in Caco-2 cells. Drug Metab. Dispos. 1996, 24, 634–42. [Google Scholar]
- Inoue, M.; Morikawa, M.; Tsuboi, M.; Sugiura, M. Species difference and characterization of intestinal esterase on the hydrolizing activity of ester-type drugs. Jpn. J. Pharmacol. 1979, 29, 9–16. [Google Scholar] [CrossRef]
- Imai, T.; Taketani, M.; Shii, M.; Hosokawa, M.; Chiba, K. Substrate specificity of carboxylesterase isozymes and their contribution to hydrolase activity in human liver and small intestine. Drug Metab. Dispos. 2006, 34, 1734–41. [Google Scholar] [CrossRef]
- Mansbach, C. M., 2nd; Nevin, P. Intracellular movement of triacylglycerols in the intestine. J. Lipid Res. 1998, 39, 963–8. [Google Scholar]
- Schwer, H.; Langmann, T.; Daig, R.; Becker, A.; Aslanidis, C.; Schmitz, G. Molecular cloning and characterization of a novel putative carboxylesterase, present in human intestine and liver. Biochem. Biophys. Res. Commun. 1997, 233, 117–20. [Google Scholar] [CrossRef]
- Derbel, M.; Hosokawa, M.; Satoh, T. Differences in the induction of carboxylesterase RL4 in rat liver microsomes by various perfluorinated fatty acids, metabolically inert derivatives of fatty acids. Biol. Pharm. Bull. 1996, 19, 765–767. [Google Scholar]
- Hattori, K.; Igarashi, M.; Itoh, M.; Tomisawa, H.; Tateishi, M. Specific induction by glucocorticoids of steroid esterase in rat hepatic microsomes and its release into serum. Biochem. Pharmacol. 1992, 43, 1921–7. [Google Scholar] [CrossRef]
- Hosokawa, M.; Endo, Y.; Fujisawa, M.; Hara, S.; Iwata, N.; Sato, Y.; Satoh, T. Interindividual variation in carboxylesterase levels in human liver microsomes. Drug Metab. Dispos. 1995, 23, 1022–1027. [Google Scholar]
- Hosokawa, M.; Hattori, K.; Igarashi, M.; Satoh, T.; Ohkawara, S.; Igarashi, T.; Ueno, K.; Ohshima, T.; Kitagawa, H. Effects of pituitary hormones on carboxylesterase and drug metabolizing enzymes in rat liver microsomes. Jpn. J. Pharmacol. 1984, 36, 73. [Google Scholar] [CrossRef]
- Hosokawa, M.; Satoh, T. Differences in the induction of carboxylesterase isozymes in rat liver microsomes by perfluorinated fatty acids. Xenobiotica 1993, 23, 1125–33. [Google Scholar] [CrossRef]
- Lehner, R.; Kuksis, A. Purification of an acyl-CoA hydrolase from rat intestinal microsomes. A candidate acyl-enzyme intermediate in glycerolipid acylation. J. Biol. Chem. 1993, 268, 24726–33. [Google Scholar]
- Mentlein, R.; Schumann, M.; Heymann, E. Comparative chemical and immunological characterization of five lipolytic enzymes (carboxylesterases) from rat liver microsomes. Arch. Biochem. Biophys. 1984, 234, 612–21. [Google Scholar] [CrossRef]
- Watanabe, K.; Kayano, Y.; Matsunaga, T.; Yamamoto, I.; Yoshimura, H. Purification and characterization of a novel 46.5-kilodalton esterase from mouse hepatic microsomes. Biochem. Mol. Biol. Int. 1993, 31, 25–30. [Google Scholar]
- Geshi, E.; Kimura, T.; Yoshimura, M.; Suzuki, H.; Koba, S.; Sakai, T.; Saito, T.; Koga, A.; Muramatsu, M.; Katagiri, T. A single nucleotide polymorphism in the carboxylesterase gene is associated with the responsiveness to imidapril medication and the promoter activity. Hypertens. Res. 2005, 28, 719–25. [Google Scholar] [CrossRef]
- Mori, M.; Hosokawa, M.; Ogasawara, Y.; Tsukada, E.; Chiba, K. cDNA cloning, characterization and stable expression of novel human brain carboxylesterase. FEBS Lett. 1999, 458, 17–22. [Google Scholar] [CrossRef]
- Takai, S.; Matsuda, A.; Usami, Y.; Adachi, T.; Sugiyama, T.; Katagiri, Y.; Tatematsu, M.; Hirano, K. Hydrolytic profile for ester- or amide-linkage by carboxylesterases pI 5.3 and 4.5 from human liver. Biol. Pharm. Bull. 1997, 20, 869–73. [Google Scholar]
- Danks, M. K.; Morton, C. L.; Pawlik, C. A.; Potter, P. M. Overexpression of a rabbit liver carboxylesterase sensitizes human tumor cells to CPT-11. Cancer Res. 1998, 58, 20–2. [Google Scholar]
- Guichard, S. M.; Morton, C. L.; Krull, E. J.; Stewart, C. F.; Danks, M. K.; Potter, P. M. Conversion of the CPT-11 metabolite APC to SN-38 by rabbit liver carboxylesterase. Clin. Cancer Res. 1998, 4, 3089–94. [Google Scholar]
- Humerickhouse, R.; Lohrbach, K.; Li, L.; Bosron, W. F.; Dolan, M. E. Characterization of CPT-11 hydrolysis by human liver carboxylesterase isoforms hCE-1 and hCE-2. Cancer Res. 2000, 60, 1189–92. [Google Scholar]
- Kojima, A.; Hackett, N. R.; Ohwada, A.; Crystal, R. G. In vivo human carboxylesterase cDNA gene transfer to activate the prodrug CPT-11 for local treatment of solid tumors. J. Clin. Invest. 1998, 101, 1789–96. [Google Scholar] [CrossRef]
- Potter, P. M.; Pawlik, C. A.; Morton, C. L.; Naeve, C. W.; Danks, M. K. Isolation and partial characterization of a cDNA encoding a rabbit liver carboxylesterase that activates the prodrug irinotecan (CPT-11). Cancer Res. 1998, 58, 2646–51. [Google Scholar]
- Sanghani, S. P.; Quinney, S. K.; Fredenburg, T. B.; Sun, Z.; Davis, W. I.; Murry, D. J.; Cummings, O. W.; Seitz, D. E.; Bosron, W. F. Carboxylesterases expressed in human colon tumor tissue and their role in CPT-11 hydrolysis. Clin. Cancer Res. 2003, 9, 4983–91. [Google Scholar]
- Satoh, T.; Hosokawa, M.; Atsumi, R.; Suzuki, W.; Hakusui, H.; Nagai, E. Metabolic activation of CPT-11, 7-ethyl-10-[4-(1-piperidino)-1- piperidino]carbonyloxycamptothecin, a novel antitumor agent, by c arboxylesterase. Biol. Pharm. Bull. 1994, 17, 662–4. [Google Scholar]
- Tabata, T.; Katoh, M.; Tokudome, S.; Nakajima, M.; Yokoi, T. Identification of the cytosolic carboxylesterase catalyzing the 5'-deoxy-5-fluorocytidine formation from capecitabine in human liver. Drug Metab. Dispos. 2004, 32, 1103–10. [Google Scholar] [CrossRef]
- Kamendulis, L. M.; Brzezinski, M. R.; Pindel, E. V.; Bosron, W. F.; Dean, R. A. Metabolism of cocaine and heroin is catalyzed by the same human liver carboxylesterases. J. Pharmacol. Exp. Ther. 1996, 279, 713–7. [Google Scholar]
- Zhang, J.; Burnell, J. C.; Dumaual, N.; Bosron, W. F. Binding and hydrolysis of meperidine by human liver carboxylesterase hCE-1. J. Pharmacol. Exp. Ther. 1999, 290, 314–8. [Google Scholar]
- Imai, T.; Yoshigae, Y.; Hosokawa, M.; Chiba, K.; Otagiri, M. Evidence for the involvement of a pulmonary first-pass effect via carboxylesterase in the disposition of a propranolol ester derivative after intravenous administration. J. Pharmacol. Exp. Ther. 2003, 307, 1234–42. [Google Scholar] [CrossRef]
- Ohtsuka, K.; Inoue, S.; Kameyama, M.; Kanetoshi, A.; Fujimoto, T.; Takaoka, K.; Araya, Y.; Shida, A. Intracellular conversion of irinotecan to its active form, SN-38, by native carboxylesterase in human non-small cell lung cancer. Lung Cancer 2003, 41, 187–198. [Google Scholar]
- Pindel, E. V.; Kedishvili, N. Y.; Abraham, T. L.; Brzezinski, M. R.; Zhang, J.; Dean, R. A.; Bosron, W. F. Purification and cloning of a broad substrate specificity human liver carboxylesterase that catalyzes the hydrolysis of cocaine and heroin. J. Biol. Chem. 1997, 272, 14769–75. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, G.; McLeod, H. L. Comprehensive evaluation of carboxylesterase-2 expression in normal human tissues using tissue array analysis. Appl. Immunohistochem. Mol. Morphol. 2002, 10, 374–80. [Google Scholar] [CrossRef]
- Aldridge, W. N. The esterases: perspectives and problems. Chem. Biol. Interact. 1993, 87, 5–13. [Google Scholar] [CrossRef]
- Zschunke, F.; Salmassi, A.; Kreipe, H.; Buck, F.; Parwaresch, M. R.; Radzun, H. J. cDNA cloning and characterization of human monocyte/macrophage serine esterase-1. Blood 1991, 78, 506–12. [Google Scholar]
- Ovnic, M.; Tepperman, K.; Medda, S.; Elliott, R. W.; Stephenson, D. A.; Grant, S. G.; Ganschow, R. E. Characterization of a murine cDNA encoding a member of the carboxylesterase multigene family. Genomics 1991, 9, 344–54. [Google Scholar] [CrossRef]
- Robbi, M.; Beaufay, H. Biosynthesis of rat liver pI-6.1 esterase, a carboxylesterase of the cisternal space of the endoplasmic reticulum. Biochem. J. 1987, 248, 545–50. [Google Scholar]
- Robbi, M.; Beaufay, H.; Octave, J. N. Nucleotide sequence of cDNA coding for rat liver pI 6.1 esterase (ES-10), a carboxylesterase located in the lumen of the endoplasmic reticulum. Biochem. J. 1990, 269, 451–8. [Google Scholar]
- Robbi, M.; Van Schaftingen, E.; Beaufay, H. Cloning and sequencing of rat liver carboxylesterase ES-4 (microsomal palmitoyl-CoA hydrolase). Biochem. J. 1996, 313, 821–6. [Google Scholar]
- Taketani, M.; Shii, M.; Ohura, K.; Ninomiya, S.; Imai, T. Carboxylesterase in the liver and small intestine of experimental animals and human. Life Sci. 2007, 81, 924–32. [Google Scholar] [CrossRef]
- Yang, J.; Shi, D.; Yang, D.; Song, X.; Yan, B. Interleukin-6 alters the cellular responsiveness to clopidogrel, irinotecan, and oseltamivir by suppressing the expression of carboxylesterases HCE1 and HCE2. Mol. Pharmacol. 2007, 72, 686–94. [Google Scholar] [CrossRef]
- Furihata, T.; Hosokawa, M.; Fujii, A.; Derbel, M.; Satoh, T.; Chiba, K. Dexamethasone-induced methylprednisolone hemisuccinate hydrolase: Its identification as a member of the rat carboxylesterase 2 family and its unique existence in plasma. Biochem. Pharmacol. 2005, 69, 1287–97. [Google Scholar] [CrossRef]
- Ozols, J. Isolation, properties, and the complete amino acid sequence of a second form of 60-kDa glycoprotein esterase. J. Biol. Chem. 1989, 264, 12533–12545. [Google Scholar]
- Sone, T.; Isobe, M.; Takabatake, E.; Wang, C. Y. Cloning and sequence analysis of a hamster liver cDNA encoding a novel putative carboxylesterase. Biochim. Biophys. Acta 1994, 1207, 138–42. [Google Scholar]
- Aida, K.; Moore, R.; Negishi, M. Cloning and nucleotide sequence of a novel, male-predominant carboxylesterase in mouse liver. Biochim. Biophys. Acta 1993, 1174, 72–4. [Google Scholar] [CrossRef]
- Sanghani, S. P.; Quinney, S. K.; Fredenburg, T. B.; Davis, W. I.; Murry, D. J.; Bosron, W. F. Hydrolysis of irinotecan and its oxidative metabolites, 7-ethyl-10-[4-N-(5-aminopentanoic acid)-1-piperidino]carbonyloxy camptothecin and 7-ethyl-10-[4-(1-piperidino)-1-amino]- carbonyloxy camptothecin, by human carboxylesterases CES1A1, CES2, and a newly expressed carboxylesterase isoenzyme, CES3. Drug Metab. Dispos. 2004, 32, 505–11. [Google Scholar] [CrossRef]
- Miyazaki, M.; Yamashita, T.; Hosokawa, M.; Taira, H.; Suzuki, A. Species-, sex-, and age-dependent urinary excretion of cauxin, a mammalian carboxylesterase. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2006, 145, 270–7. [Google Scholar]
- Miyazaki, M.; Yamashita, T.; Suzuki, Y.; Saito, Y.; Soeta, S.; Taira, H.; Suzuki, A. A major urinary protein of the domestic cat regulates the production of felinine, a putative pheromone precursor. Chem. Biol. 2006, 13, 1071–9. [Google Scholar] [CrossRef]
- Probst, M. R.; Beer, M.; Beer, D.; Jeno, P.; Meyer, U. A.; Gasser, R. Human liver arylacetamide deacetylase. J. Biol. Chem. 1994, 269, 21650–21656. [Google Scholar]
- Probst, M. R.; Jeno, P.; Meyer, U. A. Purification and characterization of a human liver arylacetamide deacetylase. Biochem. Biophys. Res. Commun. 1991, 177, 453–9. [Google Scholar] [CrossRef]
- Pelham, H. R. The retention signal for soluble proteins of the endoplasmic reticulum. Trends Biochem. Sci. 1990, 15, 483–486. [Google Scholar] [CrossRef]
- Robbi, M.; Beaufay, H. The COOH terminus of several liver carboxylesterases targets these enzymes to the lumen of the endoplasmic reticulum. J. Biol. Chem. 1991, 266, 20498–503. [Google Scholar]
- Tang, B. K.; Kalow, W. Variable activation of lovastatin by hydrolytic enzymes in human plasma and liver. 4. Eur. J. Clin. Pharmacol. 1995, 47, 449–51. [Google Scholar]
- Korza, G.; Ozols, J. Complete covalent structure of 60-kDa esterase isolated from 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced rabbit liver. J. Biol. Chem. 1988, 263, 3486–3495. [Google Scholar]
- Robbi, M.; Beaufay, H. Cloning and sequencing of rat liver carboxylesterase ES-3 (egasyn). Biochem. Biophys. Res. Commun. 1994, 203, 1404–11. [Google Scholar] [CrossRef]
- Medda, S.; Takeuchi, K.; Devore-Carter, D.; von Deimling, O.; Heymann, E.; Swank, R. T. An accessory protein identical to mouse egasyn is complexed with rat microsomal beta-glucuronidase and is identical to rat esterase-3. J. Biol. Chem. 1987, 262, 7248–53. [Google Scholar]
- Ovnic, M.; Swank, R. T.; Fletcher, C.; Zhen, L.; Novak, E. K.; Baumann, H.; Heintz, N.; Ganschow, R. E. Characterization and functional expression of a cDNA encoding egasyn (esterase-22): the endoplasmic reticulum-targeting protein of beta- glucuronidase. Genomics 1991, 11, 956–67. [Google Scholar] [CrossRef]
- Shibata, F.; Takagi, Y.; Kitajima, M.; Kuroda, T.; Omura, T. Molecular cloning and characterization of a human carboxylesterase gene. Genomics 1993, 17, 76–82. [Google Scholar] [CrossRef]
- von Heijne, G. Patterns of amino acids near signal-sequence cleavage sites. Eur. J. Biochem. 1983, 133, 17–21. [Google Scholar] [CrossRef]
- Cygler, M.; Schrag, J. D.; Sussman, J. L.; Harel, M.; Silman, I.; Gentry, M. K.; Doctor, B. P. Relationship between sequence conservation and three-dimensional structure in a large family of esterases, lipases, and related proteins. Protein Sci. 1993, 2, 366–82. [Google Scholar]
- Frey, PA; Whitt, SA; Tobin, JB. A low-barrier hydrogen bond in the catalytic triad of serine proteases. Science 1994, 264, 1927–1930. [Google Scholar]
- Alam, M.; Vance, D. E.; Lehner, R. Structure-function analysis of human triacylglycerol hydrolase by site-directed mutagenesis: identification of the catalytic triad and a glycosylation site. Biochemistry 2002, 41, 6679–87. [Google Scholar] [CrossRef]
- Bencharit, S.; Morton, C. L.; Howard-Williams, E. L.; Danks, M. K.; Potter, P. M.; Redinbo, M. R. Structural insights into CPT-11 activation by mammalian carboxylesterases. Nat. Struct. Biol. 2002, 9, 337–42. [Google Scholar] [CrossRef]
- Bencharit, S.; Morton, C. L.; Hyatt, J. L.; Kuhn, P.; Danks, M. K.; Potter, P. M.; Redinbo, M. R. Crystal structure of human carboxylesterase 1 complexed with the Alzheimer's drug tacrine: from binding promiscuity to selective inhibition. Chem. Biol. 2003, 10, 341–9. [Google Scholar] [CrossRef]
- Bencharit, S.; Morton, C. L.; Xue, Y.; Potter, P. M.; Redinbo, M. R. Structural basis of heroin and cocaine metabolism by a promiscuous human drug-processing enzyme. Nat. Struct. Biol. 2003, 10, 349–56. [Google Scholar] [CrossRef]
- Wong, H.; Schotz, M. C. The lipase gene family. J. Lipid Res. 2002, 43, 993–9. [Google Scholar] [CrossRef]
- Wallace, T. J.; Ghosh, S.; McLean Grogan, W. Molecular cloning and expression of rat lung carboxylesterase and its potential role in the detoxification of organophosphorus compounds. Am. J. Respir. Cell. Mol. Biol. 1999, 20, 1201–8. [Google Scholar] [CrossRef]
- Hosokawa, M. Differences in the functional roles of hepatic microsomal carboxylesterase isozymes in various mammals and humans. Xenobiotic Metab. Dispos. 1990, 5, 185–195. [Google Scholar]
- Imai, T. Human carboxylesterase isozymes: catalytic properties and rational drug design. Drug Metab. Pharmacokinet. 2006, 21, 173–85. [Google Scholar] [CrossRef]
- Satoh, T.; Taylor, P.; Bosron, W. F.; Sanghani, S. P.; Hosokawa, M.; La Du, B. N. Current progress on esterases: from molecular structure to function. Drug Metab. Dispos. 2002, 30, 488–93. [Google Scholar] [CrossRef]
- Sun, Z.; Murry, D. J.; Sanghani, S. P.; Davis, W. I.; Kedishvili, N. Y.; Zou, Q.; Hurley, T. D.; Bosron, W. F. Methylphenidate is stereoselectively hydrolyzed by human carboxylesterase CES1A1. J. Pharmacol. Exp. Ther. 2004, 310, 469–76. [Google Scholar] [CrossRef]
- Shi, D.; Yang, J.; Yang, D.; LeCluyse, E. L.; Black, C.; You, L.; Akhlaghi, F.; Yan, B. Anti-influenza prodrug oseltamivir is activated by carboxylesterase human carboxylesterase 1, and the activation is inhibited by antiplatelet agent clopidogrel. J. Pharmacol. Exp. Ther. 2006, 319, 1477–84. [Google Scholar] [CrossRef]
- Langmann, T.; Becker, A.; Aslanidis, C.; Notka, F.; Ullrich, H.; Schwer, H.; Schmitz, G. Structural organization and characterization of the promoter region of a human carboxylesterase gene. Biochim. Biophys. Acta 1997, 1350, 65–74. [Google Scholar]
- Furihata, T.; Hosokawa, M.; Satoh, T.; Chiba, K. Synergistic role of specificity proteins and upstream stimulatory factor 1 in transactivation of the mouse carboxylesterase 2/microsomal acylcarnitine hydrolase gene promoter. Biochem. J. 2004, 384, 101–10. [Google Scholar] [CrossRef]
- Furihata, T.; Hosokawa, M.; Masuda, M.; Satoh, T.; Chiba, K. Hepatocyte nuclear factor-4alpha plays pivotal roles in the regulation of mouse carboxylesterase 2 gene transcription in mouse liver. Arch. Biochem. Biophys. 2006, 447, 107–17. [Google Scholar] [CrossRef]
- Hosokawa, M.; Furihata, T.; Yaginuma, Y.; Yamamoto, N.; Watanabe, N.; Tsukada, E.; Ohhata, Y.; Kobayashi, K.; Satoh, T.; Chiba, K. Structural organization and characterization of the regulatory element of the human carboxylesterase (CES1A1 and CES1A2) genes. Drug Metab. Pharmacokinet. 2008, 23, 73–84. [Google Scholar] [CrossRef]
- Samples Availability: Contact the author.
© 2008 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Hosokawa, M. Structure and Catalytic Properties of Carboxylesterase Isozymes Involved in Metabolic Activation of Prodrugs. Molecules 2008, 13, 412-431. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules13020412
AMA Style
Hosokawa M. Structure and Catalytic Properties of Carboxylesterase Isozymes Involved in Metabolic Activation of Prodrugs. Molecules. 2008; 13(2):412-431. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules13020412
Chicago/Turabian StyleHosokawa, Masakiyo. 2008. "Structure and Catalytic Properties of Carboxylesterase Isozymes Involved in Metabolic Activation of Prodrugs" Molecules 13, no. 2: 412-431. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules13020412