Soft Alkyl Ether Prodrugs of a Model Phenolic Drug: The Effect of Incorporation of Ethyleneoxy Groups on Transdermal Delivery

Abstract
:1. Introduction
2. Results and Discussion
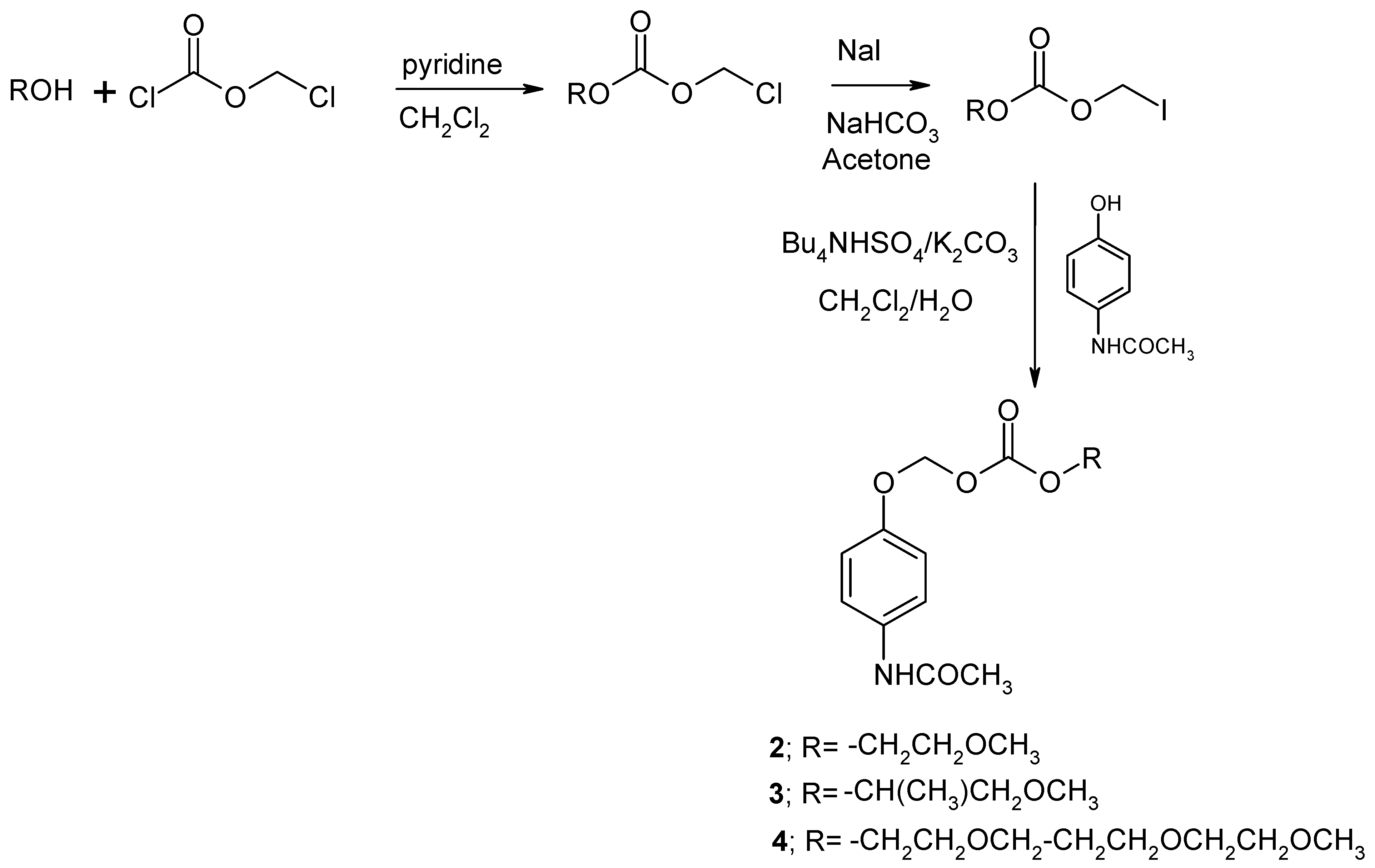
2.1. Synthesis and physicochemical properties of the AOCOM and NANAOCAM prodrugs
2.2. Diffusion cell experiments
3. Experimental
3.1. General
3.2. Syntheses
3.2.1. Procedure for the synthesis of the AOCOM prodrugs 2 to 4 [20]
3.2.2. Procedure for the synthesis of the NANAOCOM prodrug 5 [15]
3.3. Physicochemical properties and analysis
3.4. Diffusion cell experiments
4. Conclusions
References
- Dhareshwar, S.S.; Stella, V.J. Prodrugs of Alcohols and Phenols. 3.2. In Prodrugs: Challenges and Rewards, Part 2; Stella, V.J., Borchardt, R.T., Hageman, M.J., Oliyai, R., Maag, H., Tilley, J.W., Eds.; Springer Science: New York, NY, USA, 2007; pp. 31–100. [Google Scholar]
- Sloan, K.B. Prodrugs for dermal delivery. Adv. Drug Deliv. Rev. 3 1989, 67–101. [Google Scholar] [CrossRef]
- Bonina, F.P.; Montenegro, L.; DeCapraris, P.; Bousquet, E.; Tirendi, S. 1-Alkylazacycloalkan-2-one esters as prodrugs of indomethacin for improved delivery through human skin. Int. J. Pharm. 1991, 77, 21–29. [Google Scholar] [CrossRef]
- Roberts, W.J.; Sloan, K.B. Correlation of aqueous and lipid solubilities with flux for prodrugs of 5-fluorouracil, theophylline, and 6-mercaptopurine: A Potts-Guy approach. J. Pharm. Sci. 1999, 88, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Rautio, J.; Taipale, H.; Gynther, J.; Vepsalainen, J.; Nevalainen, T.; Jarvinen, T. In vitro evaluation of acyloxyalkyl esters as dermal prodrugs of ketoprofen and naproxen. J. Pharm. Sci. 1998, 87, 1622–1628. [Google Scholar] [CrossRef] [PubMed]
- Roberts, W.J.; Sloan, K.B. Application of the transformed Potts-Guy equation to in vivo human skin data. J. Pharm. Sci. 2001, 90, 1318–1323. [Google Scholar] [CrossRef] [PubMed]
- Sloan, K.B.; Wasdo, S.C. Topical Delivery Using Prodrugs 2.1.2. In Prodrugs: Challenges and Rewards, Part 1; Stella, V.J., Borchardt, R.T., Hageman, M.J., Oliyai, R., Maag, H., Tilley, J.W., Eds.; Springer Science: New York, NY, USA, 2006; pp. 83–123. [Google Scholar]
- Juntunen, J.; Majumdar, S.; Sloan, K.B. The effect of water solubility of solutes on their flux through human skin in vitro: A prodrug database integrated into the extended Flynn database. Int. J. Pharm. 2008, 351, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Drustrup, J.; Fullerton, A.; Christrup, L.; Bundgaard, H. Utilization of prodrugs to enhance the transdermal absorption of morphine. Int. J. Pharm. 1991, 71, 104–116. [Google Scholar] [CrossRef]
- Stinchcomb, A.L.; Swaan, P.W.; Ekabo, O.; Harris, K.; Browe, J.; Hammell, D.C.; Cooperman, T.A.; Pearsall, M. Straight-chain naltrexone ester prodrugs: Diffusion and concurrent esterase biotransformation in human skin. J. Pharm. Sci. 2002, 91, 2571–2578. [Google Scholar] [CrossRef] [PubMed]
- Pillai, O.; Hamad, M.O.; Crooks, P.A.; Stinchcomb, A.L. Physicochemical evaluation, in vitro human skin diffusion, and concurrent biotransformation of 3-O-alkyl carbonate prodrugs of naltrexone. Pharm. Res. 2004, 21, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
- Valiveti, S.; Paudel, K.S.; Hammell, D.C.; Hamad, M.O.; Chen, J.; Crooks, P.A.; Stinchcomb, A.L. In vitro/in vivo correlation of transdermal naltrexone prodrugs. Pharm. Res. 2005, 22, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Wasdo, S.C.; Sloan, K.B. Topical delivery of a model phenolic drug: Alkyloxycarbonyl prodrugs of acetaminophen. Pharm. Res. 2004, 21, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Bonina, F.P.; Montenegro, L.; DeCapraris, P.; Palagiano, F.; Trapani, G.; Liso, G. In vitro and in vivo evaluation of polyoxyethene indomethacin esters as dermal prodrugs. J. Controlled Release 1995, 34, 223–232. [Google Scholar] [CrossRef]
- Majumdar, S.; Sloan, K.B. Synthesis and topical delivery of N-alkyl-N-alkyloxycarbonyl-aminomethyl prodrugs of a model phenolic drug: Acetaminophen. Int. J. Pharm. 2007, 337, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.D.; Sloan, K.B. Evaluation of alkyloxycarbonyloxymethyl (AOCOM) ethers as novel prodrugs of phenols for topical delivery: AOCOM prodrugs of acetaminophen. Int. J. Pharm. 2009, 371, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.D.; Sloan, K.B. In vitro evaluation of alkylcarbonyloxymethyl (ACOM) ethers as novel prodrugs of phenols for topical delivery: ACOM prodrugs of acetaminophen. Int. J. Pharm. 2008, 346, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Sloan, K.B.; Hashida, M.; Alexander, J.; Bodor, N.; Higuchi, T. Prodrugs of 6-thiopurines: Enhanced delivery through the skin. J. Pharm. Sci. 1983, 72, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Sloan, K.B. Functional Groups Consideration in the Development of Prodrug Approaches to Solving Topical Delivery Problems. In Prodrugs: Topical and Ocular Drug Delivery; Sloan, K.B., Ed.; Marcel Dekker, Inc.: New York, NY, USA, 1992; pp. 17–116. [Google Scholar]
- Thomas, J.D.; Sloan, K.B. Overcoming steric effects in the coupling reaction of alkyloxycarbonyloxymethyl (AOCOM) halides with phenols: An efficient synthesis of AOCOM phenolic prodrugs. Tetrahedron Lett. 2007, 48, 109–112. [Google Scholar] [CrossRef]
- Majumdar, S.; Sloan, K.B. Practical syntheses of N-alkyl-N-alkyloxycarbonylaminomethyl prodrugs of acetaminophen, theophylline, and 6-mercaptopurine. Synth. Commun. 2006, 36, 3537–3548. [Google Scholar] [CrossRef]
- Kasting, G.B.; Smith, R.L.; Cooper, E.R. Effect of Lipid Solubility and Molecular Size on Percutaneous Absorption. In Pharmacology and the Skin; Shroot, B., Schaefer, H., Eds.; Karger: Basel, Switzerland, 1987; Vol. 1, pp. 138–153. [Google Scholar]
- Bieze, T.W.N.; Barnes, A.C.; Huige, C.J.M.; Enderby, J.E.; Leyte, J.C. Distribution of water around poly(ethylene oxide): A neutron diffraction study. J. Phys. Chem. 1994, 743, 49–52. [Google Scholar] [CrossRef]
- Beall, H.D.; Getz, J.J.; Sloan, K.B. The estimation of relative water solubility for prodrugs that are unstable in water. Int. J. Pharm. 1993, 93, 37–47. [Google Scholar] [CrossRef]
- Sloan, K.B.; Koch, S.A.M.; Siver, K.; Flowers, F.P. The use of solubility parameters of drug and vehicle to predict flux. J. Invest. Derm. 1986, 87, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Sloan, K.B.; Beall, H.D.; Villanueva, R.; Weimar, W.R. The effect of receptor phase composition on the permeability of hairless mouse skin in diffusion cell experiments. Int. J. Pharm. 1991, 73, 97–104. [Google Scholar] [CrossRef]
- Sloan, K.B.; Wasdo, S.; Ezike-Mkparu, U.; Murray, T.J.; Nichels, D.; Singh, S.; Shanks, T.; Tovar, J.; Ulmer, K.; Warranis, R. Topical delivery of 5-fluorouracil (5-FU) and 6-mercaptopurine (6-MP) by their alkylcarbonyloxymethyl (ACOM) prodrugs from water: Vehicle effects on design of prodrugs. Pharm. Res. 2003, 20, 639–645. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |



{kind=link}
{kind=link}

| Compound | X | n | R | R’ | MW | mp oC | SIPMd | SAQd |
| APAP, 1 | 151 | 167-170 | 1.9 | 100 | ||||
| 2 | O | 1 | H | CH3 | 283 | 69-70 | 10.43 | 31.1 |
| 3 | O | 1 | CH3 | CH3 | 297 | 80-82 | 14.01 | 7.68 |
| 4 | O | 3 | H | CH3 | 371 | oil | 12.14 | 184.0 |
| 5 | N(CH3) | 1 | H | CH3 | 296 | oil | 4.16e | 66.1 |
| 6 | O | 0 | - | C2H5 | 253 | 83-85 | 20.7 | 7.76 |
| 7 | O | 0 | - | C3H7 | 267 | 68-69 | 45.8 | 2.00 |
| 8 | O | 0 | - | C10H21 | 365 | 54-56 | 130 | 0.0056 |
| 9 | N(CH3) | 0 | - | C2H5 | 266 | 75-77 | 19.9 | 15.85 |
| 10 | N(CH3) | 0 | - | C3H7 | 280 | 59-60 | 40.7 | 8.91 |
| 11 | N(CH3) | 0 | - | C4H9 | 294 | oil | 67.6 | 3.63 |
| 12 | - | 1 | H | CH3 | 253 | 78-81 | 10.23 | 34.67 |
| 13 | - | 1 | CH3 | CH3 | 267 | 120-123 | 3.38 | 3.31 |
| Compound a,b,c | KIPM:4.0 | CLogP | EXP JMMIPM | % Parent Drug | log EXP JMMIPMd | log EXP-log PRE JMMIPMe |
| 1 | - | 0.49 | 0.51 | - | -0.29 | -0.49 |
| 2 | 0.364 | 0.93 | 0.72 | 38 | -0.14 | -0.13 |
| 3 | 1.41 | 1.24 | 0.433 | 65 | -0.36 | -0.08 |
| 4 | 0.027 | 0.66 | 0.379 | 52 | -0.42 | -0.61 |
| 5 | 0.063 | 1.51 | 0.696 | 10 | -0.16 | -0.08 |
| 6 | 2.76 | 1.51 | 0.66 | 54 | -0.18 | -0.12 |
| 7 | 9.21 | 1.85 | 0.283 | 75 | -0.55 | -0.31 |
| 8 | 26500 | 5.56 | 0.00739 | 100 | -2.13 | -0.1 |
| 9 | 1.20 | 1.80 | 0.51 | 23 | -0.29 | -0.32 |
| 10 | 4.89 | 2.33 | 0.43 | 22 | -0.37 | -0.27 |
| 11 | 18.6 | 2.86 | 0.62 | 24 | -0.21 | -0.24 |
| 12 | 0.50 | 0.54 | 0.776 | 49 | -0.11 | -0.19 |
| 13 | 1.44 | 0.85 | 0.087 | 100 | -1.06 | -0.36 |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Thomas, J.D.; Majumdar, S.; Sloan, K.B. Soft Alkyl Ether Prodrugs of a Model Phenolic Drug: The Effect of Incorporation of Ethyleneoxy Groups on Transdermal Delivery. Molecules 2009, 14, 4231-4245. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules14104231
Thomas JD, Majumdar S, Sloan KB. Soft Alkyl Ether Prodrugs of a Model Phenolic Drug: The Effect of Incorporation of Ethyleneoxy Groups on Transdermal Delivery. Molecules. 2009; 14(10):4231-4245. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules14104231
Chicago/Turabian StyleThomas, Joshua Denver, Susruta Majumdar, and Kenneth Berry Sloan. 2009. "Soft Alkyl Ether Prodrugs of a Model Phenolic Drug: The Effect of Incorporation of Ethyleneoxy Groups on Transdermal Delivery" Molecules 14, no. 10: 4231-4245. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules14104231