Bystander or No Bystander for Gene Directed Enzyme Prodrug Therapy

Abstract
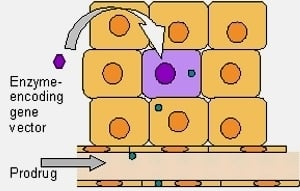
:
1. Chemotherapy
1.1. History of Modern Chemotherapy
1.2. Characteristics of Chemotherapy
2. Principals of Gene Directed Enzyme Prodrug Therapy

{kind=link}
{kind=link}
| Enzyme (origin) | Prodrug | Cytotoxin | Bystander | References |
|---|---|---|---|---|
| Carboxylesterase (human, rabbit, rat) | Irinotecan | SN-38 | high | [43] |
| Capecitabine | 5-FU | |||
| Paclitaxel-2-ethylcarbonate | Paclitaxel | |||
| dpVP-16 | VP16 | |||
| Carboxypeptidase A (human, rat) | Methotrexate-α-peptides | Methotrexate | high | [44] |
| Carboxypeptidase G2 (Pseudomonas R16) | CMDA | CMBA | high | [45,46,47] |
| ZD-2767P | Phenol-bis-iodo nitrogen mustard | |||
| Self-immolative prodrugs | Alkylating agents, anthracycline antibiotics | |||
| Cytochrome P450 (Human: CYP2B1, 2B6, 2C8, 2C9, 2C18, 3A),(Rat: CYP2B10), (Rabbit: CYP4B1 ±CYPOR), (Dog: CYP2B11) | Oxaza phosphorines: CPA and IFO | Alkylating agents (4-hydroxy forms) | high | [48,49] |
| Acetaminophen | NABQI (N-acetyl benzoquinone imine) | low | ||
| Cytosine deaminase (±UPRT) (E. coli, S. cerevisiae) | 5-FC | 5-FU | high | [50,51,52,53] |
| Horseradish peroxidase (plant: horseradish) | Indole-3-acetic acid and derivatives | 3-methylene-2-oxindole | high | [54] |
| Acetominophen | NABQI | low | ||
| NADPH-cytochrome P450 reductase (human) | Tirapazamine, EO9, RSU1069/ misonidazole | Reduced metabolites | medium | [55] |
| Nitroreductase (E. coli NfsB, NfsA and other reductases) | CB1954 and analogues | Alkylating agents (N-acetoxy derivatives) | high to very high | [56] |
| Self-immolative prodrugs | Alkylating agents, pyrazolidines, enediynes | |||
| 2-fluoroadenine | ||||
| Metronidazole | Alkylating agent | very low | [57] | |
| Pyrimidine nucleoside phosphorylase (human) | 5’-deoxy-5-fluorouridine | 5-FU | high | [58] |
| Thymidine kinase (Herpes simplex virus, Varicella zoster virus, Equine herpes virus) | Modified purine and pyrimidine nucleosides: GCV, E-GCV, ACV, valacyclovir, araM, araT, BVDU | Mono phosphorylated nucleotide analogues | high, dependent on gap junctions | [59,60] |
| FIAU, purine and pyrimidine nucleosides, araM | Monophosphorylated nucleotide analogues | |||
| Thymidine phosphorylase (human) | Pyrimidine analogues e.g. 5’-DFUR | 5-fluoro deoxyuridine monophosphate | high | [61] |
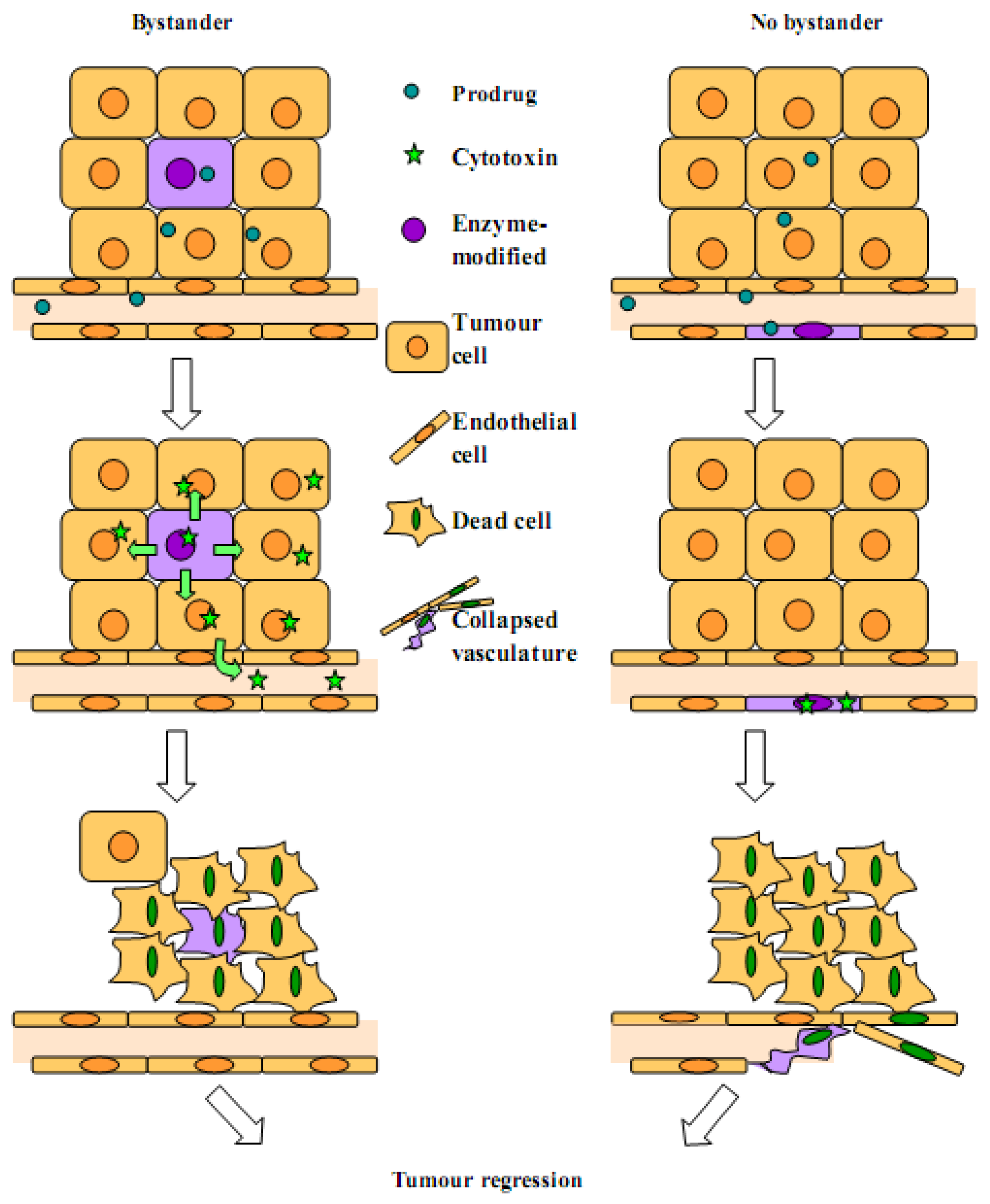
2.1. Bystander Effect
2.2. Arguments for a Strong Local Bystander Effect

2.3. Arguments for a Reduced Local Bystander Effect
3. Combinations of Enzyme Prodrug Therapy
3.1. Thymidine Kinase and Ganciclovir
3.1.1. Mode of Action
3.1.2. Clinical Trials
3.1.3. Enzyme Modifications and Drug Development
3.2. Cytosine Deaminase and 5-FC
3.2.1. Mode of Action
3.2.2. Clinical Trials
3.2.3. Enzyme Modifications and Drug Development
3.3. Nitroreductase and CB1954
3.3.1. Mode of Action
3.3.2. Clinical Trials
3.3.3. Enzyme Modifications and Drug Development
4. Conclusions
Acknowledgements
- Sample Availability: Not applicable.
References and Notes
- Lissauer, H. Zwei Fälle von Leukämie. Berliner Klinische Wochenschrift 1865, 2, 403–404. [Google Scholar]
- Breasted, J. Tumourous ulcers in the breast, perhaps resulting from injury. In The Edwin Smith Surgical Papyrus; University of Chicago Press: Chicago, IL, USA, 1930; pp. 363–369. [Google Scholar]
- Chabner, B.; Roberts, T., Jr. Timeline: Chemotherapy and the war on cancer. Nat. Rev. Cancer 2005, 5, 65–72. [Google Scholar] [CrossRef]
- Berenblum, I.; Riley, S. The modifying influence of dichloroethyl sulphide on the induction of tumours in mice by tar. J. Pathol. Bacteriol. 1929, 32, 424–434. [Google Scholar]
- Krumbhaar, E. Role of the blood and the bone marrow in certain forms of gas poisoning. I. Peripheral blood changes and their significance. JAMA 1919, 72, 39–41. [Google Scholar] [CrossRef]
- Berenblum, I.; Riley, S. Experimental inhibition of tumour induction by mustard gas and other compounds. J. Pathol. Bacteriol. 1935, 40, 549–558. [Google Scholar] [CrossRef]
- Gilman, A. The initial clinical trial of nitrogen mustard. Am. J. Surg. 1963, 105, 574–578. [Google Scholar] [CrossRef]
- Scheithauer, W.; Rosen, H.; Kornek, G.; Sebesta, C.; Depisch, D. Randomised comparison of combination chemotherapy plus supportive care with supportive care alone in patients with metastatic colorectal cancer. BMJ 1993, 306, 752–755. [Google Scholar] [CrossRef]
- Thirion, P.; Michiels, S.; Pignon, J.; Buyse, M.; Braud, A.; Carlson, R.; O'Connell, M.; Sargent, P.; Piedbois, P. Modulation of fluorouracil by leucovorin in patients with advanced colorectal cancer: An updated meta-analysis. J. Clin. Oncol. 2004, 22, 3766–3775. [Google Scholar] [CrossRef]
- Douillard, J.; Cunningham, D.; Roth, A.; Navarro, M.; James, R.; Karasek, P.; Jandik, P.; Iveson, T.; Carmichael, J.; Alakl, M.; Gruia, G.; Awad, L.; Rougier, P. Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: A multicentre randomised trial. Lancet 2000, 355, 1041–1047. [Google Scholar]
- Saltz, L.; Cox, J.; Blanke, C.; Rosen, L.; Fehrenbacher, L.; Moore, M.; Maroun, J.; Ackland, S.; Locker, P.; Pirotta, N.; Elfring, G.; Miller, L. Irinotecan plus fluorouracil and leucovorin for metastatic colorectal cancer. Irinotecan Study Group. N. Engl. J. Med. 2000, 343, 905–914. [Google Scholar] [CrossRef]
- Goldberg, R.; Sargent, D.; Morton, R.; Fuchs, C.; Ramanathan, R.; Williamson, S.; Findlay, B.; Pitot, H.; Alberts, S. A randomized controlled trial of fluorouracil plus leucovorin, irinotecan, and oxaliplatin combinations in patients with previously untreated metastatic colorectal cancer. J. Clin. Oncol. 2004, 22, 23–30. [Google Scholar]
- Saltz, L.B.; Clarke, S.; Diaz-Rubio, E.; Scheithauer, W.; Figer, A.; Wong, R.; Koski, S.; Lichinitser, M.; Yang, T.S.; Rivera, F.; Couture, F.; Sirzen, F.; Cassidy, J. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: A randomized phase III study. J. Clin. Oncol. 2008, 26, 2013–2019. [Google Scholar] [CrossRef]
- Boven, E.; Venema-Gaberscek, E.; Erkelens, C.; Bissery, M.; Pinedo, H. Antitumor activity of taxotere (RP 56976, NSC 628503), a new taxol analog, in experimental ovarian cancer. Ann. Oncol. 1993, 4, 321–324. [Google Scholar]
- Colvin, M. The alkylating agents. In Pharmacologic Principles of Cancer Treatment; Chabner, B., Ed.; W.B. Saunders Company: Philadelphia, PA, USA, 1982; pp. 276–308. [Google Scholar]
- Denny, W.; Baguley, B. Dual topoisomerase I/II inhibitors in cancer therapy. Curr. Top. Med. Chem. 2003, 3, 1349–1364. [Google Scholar]
- Fuertes, M.; Castilla, J.; Alonso, C.; Perez, J. Cisplatin biochemical mechanism of action: From cytotoxicity to induction of cell death through interconnections between apoptotic and necrotic pathways. Curr. Med. Chem. 2003, 10, 257–266. [Google Scholar] [CrossRef]
- Kaye, S. New antimetabolites in cancer chemotherapy and their clinical impact. Br. J. Cancer 1998, 78, 1–7. [Google Scholar] [CrossRef]
- Sullivan, D.; Chow, K.; Glisson, B.; Ross, W. Role of proliferation in determining sensitivity to topoisomerase II-active chemotherapeutic agents. NCI Monogr. 1987, 4, 73–78. [Google Scholar]
- Yoshii, Y.; Maki, Y.; Tsuboi, K.; Tomono, Y.; Nakagawa, K.; Hoshino, T. Estimation of growth fraction with bromodeoxyuridine in human central nervous system tumors. J. Neurosurg. 1986, 65, 659–663. [Google Scholar] [CrossRef]
- Chatelut, E.; Delord, J.; Canal, P. Toxicity patterns of cytotoxic drugs. Invest. New Drugs 2003, 21, 141–148. [Google Scholar] [CrossRef]
- Peterson, D.; Cariello, A. Mucosal damage: A major risk factor for severe complications after cytotoxic therapy. Semin. Oncol. 2004, 31, 35–44. [Google Scholar] [CrossRef]
- Feeney, K.; Cain, M.; Nowak, A. Chemotherapy induced nausea and vomiting - prevention and treatment. Aust. Fam. Physician 2007, 36, 702–706. [Google Scholar]
- Klumpp, T.; Goldberg, S.; Magdalinski, A.; Mangan, K. Phase II study of high-dose cyclophosphamide, etoposide, and carboplatin (CEC) followed by autologous hematopoietic stem cell rescue in women with metastatic or high-risk non-metastatic breast cancer: Multivariate analysis of factors affecting survival and engraftment. Bone Marrow Transplant. 1997, 20, 273–281. [Google Scholar]
- Curtis, R.; Boice, J.; Stovall, M.; Bernstein, L.; Greenberg, R.; Flannery, J.; Schwartz, A.; Weyer, P.; Moloney, W.; Hoover, R. Risk of leukemia after chemotherapy and radiation treatment for breast cancer. N. Engl. J. Med. 1992, 326, 1745–1751. [Google Scholar] [CrossRef]
- Yahalom, J.; Portlock, C. Long-term cardiac and pulmonary complications of cancer therapy. Hematol. Oncol. Clin. North Am. 2008, 22, 305–318. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Thomlinson, R.; Gray, L. The histological structure of some human lung cancers and possible implications for radiotherapy. Br. J. Cancer 1955, 9, 539–549. [Google Scholar] [CrossRef]
- Brown, J.; Giaccia, A. The unique physiology of solid tumors: Opportunities (and problems) for cancer therapy. Cancer Res. 1998, 58, 1408–1416. [Google Scholar]
- Chaplin, D.; Olive, P.; Durand, R. Intermittent blood flow in a murine tumor: Radiobiological effects. Cancer Res. 1987, 47, 597–601. [Google Scholar]
- Padera, T.; Stoll, B.; Tooredman, J.; Capen, D.; di Tomaso, E.; Jain, R. Pathology: Cancer cells compress intratumour vessels. Nature 2004, 427, 695. [Google Scholar] [CrossRef]
- Hirst, D.; Denekamp, J. Tumour cell proliferation in relation to the vasculature. Cell Tiss. Kinet. 1979, 12, 31–42. [Google Scholar]
- Tannock, I. The relation between cell proliferation and the vascular system in a transplanted mouse mammary tumour. Br. J. Cancer 1968, 22, 258–273. [Google Scholar] [CrossRef]
- Gillies, R. Causes and consequences of hypoxia and acidity in tumors - Novartis Foundation symposium. Trends Mol. Med. 2001, 7, 47–49. [Google Scholar] [CrossRef]
- Jain, R. Barriers to drug delivery in solid tumors. Sci. Am. 1994, 271, 58–65. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Mekapati, S.; Kurup, A. QSAR and ADME. Bioorg. Med. Chem. 2004, 12, 3391–3400. [Google Scholar] [CrossRef]
- Dachs, G.U.; Tupper, J.; Tozer, G.M. From bench to bedside for gene-directed enzyme prodrug therapy of cancer. Anticancer Drugs 2005, 16, 349–359. [Google Scholar] [CrossRef]
- Hamstra, D.; Lee, K.; Tychewicz, J.; Schepkin, V.; Moffat, B.; Chen, M.; Dornfeld, K.; Lawrence, T.; Chenevert, T.; Ross, B.; Gelovani, J.; Rehemtulla, A. The use of 19F spectroscopy and diffusion-weighted MRI to evaluate differences in gene-dependent enzyme prodrug therapies. Mol. Ther. 2004, 10, 916–928. [Google Scholar] [CrossRef]
- Aghi, M.; Hochberg, F.; Breakefield, X.O. Prodrug activation enzymes in cancer gene therapy. J. Gene. Med. 2000, 2, 148–164. [Google Scholar] [CrossRef]
- Hunt, M.A.; Dachs, G.U.; Currie, M.J. Vascular targeted gene therapy in cancer treatmen. In New gene therapy and cancer research; Gustafsson, W.B., Ed.; Nova Science Publishers, Inc.: New York,NY,USA, 2008; pp. 145–182. [Google Scholar]
- Niculescu-Duvaz, I.; Springer, C.J. Introduction to the background, principles, and state of the art in suicide gene therapy. Meth. Mol. Med. 2004, 90, 1–27. [Google Scholar]
- Portsmouth, D.; Hlavaty, J.; Renner, M. Suicide genes for cancer therapy. Mol. Aspects Med. 2007, 28, 4–41. [Google Scholar] [CrossRef]
- Oosterhoff, D.; Witlox, M.A.; van Beusechem, V.W.; Haisma, H.J.; Schaap, G.R.; Bras, J.; Kruyt, F.A.; Molenaar, B.; Boven, E.; Wuisman, P.I.; Pinedo, H.M.; Gerritsen, W.R. Gene-directed enzyme prodrug therapy for osteosarcoma: Sensitization to CPT-11 in vitro and in vivo by adenoviral delivery of a gene encoding secreted carboxylesterase-2. Mol. Cancer Ther. 2003, 2, 765–771. [Google Scholar]
- Hao, X.K.; Liu, J.Y.; Yue, Q.H.; Wu, G.J.; Bai, Y.J.; Yin, Y. In vitro and in vivo prodrug therapy of prostate cancer using anti-gamma-Sm-scFv/hCPA fusion protein. Prostate 2006, 66, 858–866. [Google Scholar] [CrossRef]
- Davies, L.C.; Friedlos, F.; Hedley, D.; Martin, J.; Ogilvie, L.M.; Scanlon, I.J.; Springer, C.J. Novel fluorinated prodrugs for activation by carboxypeptidase G2 showing good in vivo antitumor activity in gene-directed enzyme prodrug therapy. J. Med. Chem. 2005, 48, 5321–5328. [Google Scholar] [CrossRef]
- Friedlos, F.; Davies, L.; Scanlon, I.; Ogilvie, L.M.; Martin, J.; Stribbling, S.M.; Spooner, R.A.; Niculescu-Duvaz, I.; Marais, R.; Springer, C.J. Three new prodrugs for suicide gene therapy using carboxypeptidase G2 elicit bystander efficacy in two xenograft models. Cancer Res. 2002, 62, 1724–1729. [Google Scholar]
- Schepelmann, S.; Ogilvie, L.M.; Hedley, D.; Friedlos, F.; Martin, J.; Scanlon, I.; Chen, P.; Marais, R.; Springer, C.J. Suicide gene therapy of human colon carcinoma xenografts using an armed oncolytic adenovirus expressing carboxypeptidase G2. Cancer Res. 2007, 67, 4949–4955. [Google Scholar] [CrossRef]
- Braybrooke, J.P.; Slade, A.; Deplanque, G.; Harrop, R.; Madhusudan, S.; Forster, M.D.; Gibson, R.; Makris, A.; Talbot, D.C.; Steiner, J.; White, L.; Kan, O.; Naylor, S.; Carroll, M.W.; Kingsman, S.M.; Harris, A.L. Phase I study of MetXia-P450 gene therapy and oral cyclophosphamide for patients with advanced breast cancer or melanoma. Clin. Cancer Res. 2005, 11, 1512–1520. [Google Scholar] [CrossRef]
- Salmons, B.; Lohr, M.; Gunzburg, W.H. Treatment of inoperable pancreatic carcinoma using a cell-based local chemotherapy: Results of a phase I/II clinical trial. J. Gastroenterol. 2003, 38 Suppl. 15, 78–84. [Google Scholar]
- Freytag, S.O.; Stricker, H.; Pegg, J.; Paielli, D.; Pradhan, D.G.; Peabody, J.; DePeralta-Venturina, M.; Xia, X.; Brown, S.; Lu, M.; Kim, J.H. Phase I study of replication-competent adenovirus-mediated double-suicide gene therapy in combination with conventional-dose three-dimensional conformal radiation therapy for the treatment of newly diagnosed, intermediate- to high-risk prostate cancer. Cancer Res. 2003, 63, 7497–7506. [Google Scholar]
- Nemunaitis, J.; Cunningham, C.; Senzer, N.; Kuhn, J.; Cramm, J.; Litz, C.; Cavagnolo, R.; Cahill, A.; Clairmont, C.; Sznol, M. Pilot trial of genetically modified, attenuated Salmonella expressing the E. coli cytosine deaminase gene in refractory cancer patients. Cancer Gene Ther. 2003, 10, 737–744. [Google Scholar] [CrossRef]
- Nemunaitis, J.; Cunningham, C.; Tong, A.W.; Post, L.; Netto, G.; Paulson, A.S.; Rich, D.; Blackburn, A.; Sands, B.; Gibson, B.; Randlev, B.; Freeman, S. Pilot trial of intravenous infusion of a replication-selective adenovirus (ONYX-015) in combination with chemotherapy or IL-2 treatment in refractory cancer patients. Cancer Gene Ther. 2003, 10, 341–352. [Google Scholar] [CrossRef]
- Pandha, H.S.; Martin, L.A.; Rigg, A.; Hurst, H.C.; Stamp, G.W.; Sikora, K.; Lemoine, N.R. Genetic prodrug activation therapy for breast cancer: A phase I clinical trial of erbB-2-directed suicide gene expression. J. Clin. Oncol. 1999, 17, 2180–2189. [Google Scholar]
- Tupper, J.; Greco, O.; Tozer, G.M.; Dachs, G.U. Analysis of the horseradish peroxidase/indole-3-acetic acid combination in a three-dimensional tumor model. Cancer Gene Ther. 2004, 11, 508–513. [Google Scholar] [CrossRef]
- Cowen, R.L.; Williams, K.J.; Chinje, E.C.; Jaffar, M.; Sheppard, F.C.D.; Telfer, B.A.; Wind, N.S.; Stratford, I.J. Hypoxia targeted gene therapy to increase the efficacy of tirapazamine as an adjuvant to radiotherapy: Reversing tumor radioresistance and effecting cure. Cancer Res. 2004, 64, 1396–1402. [Google Scholar]
- Palmer, D.H.; Mautner, V.; Mirza, D.; Oliff, S.; Gerritsen, W.; van der Sijp, J.R.; Hubscher, S.; Reynolds, G.; Bonney, S.; Rajaratnam, R.; Hull, D.; Horne, M.; Ellis, J.; Mountain, A.; Hill, S.; Harris, P.A.; Searle, P.F.; Young, L.S.; James, N.D.; Kerr, D.J. Virus-directed enzyme prodrug therapy: Intratumoral administration of a replication-deficient adenovirus encoding nitroreductase to patients with resectable liver cancer. J. Clin. Oncol. 2004, 22, 1546–1552. [Google Scholar]
- Bridgewater, J.A.; Knox, R.J.; Pitts, J.D.; Collins, M.K.; Springer, C.J. The bystander effect of the nitroreductase/CB1954 enzyme/prodrug system is due to a cell-permeable metabolite. Hum. Gene Ther. 1997, 8, 709–717. [Google Scholar] [CrossRef]
- Nagata, T.; Nakamori, M.; Iwahashi, M.; Yamaue, H. Overexpression of pyrimidine nucleoside phosphorylase enhances the sensitivity to 5'-deoxy-5-fluorouridine in tumour cells in vitro and in vivo. Eur. J. Cancer 2002, 38, 712–717. [Google Scholar] [CrossRef]
- Immonen, A.; Vapalahti, M.; Tyynela, K.; Hurskainen, H.; Sandmair, A.; Vanninen, R.; Langford, G.; Murray, N.; Yla-Herttuala, S. AdvHSV-tk gene therapy with intravenous ganciclovir improves survival in human malignant glioma: A randomised, controlled study. Mol. Ther. 2004, 10, 967–972. [Google Scholar] [CrossRef]
- Rainov, N.G. A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum. Gene Ther. 2000, 11, 2389–2401. [Google Scholar] [CrossRef]
- Evrard, A.; Cuq, P.; Robert, B.; Vian, L.; Pelegrin, A.; Cano, J.P. Enhancement of 5-fluorouracil cytotoxicity by human thymidine-phosphorylase expression in cancer cells: In vitro and in vivo study. Int. J. Cancer 1999, 80, 465–470. [Google Scholar] [CrossRef]
- Rainov, N.G. A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum. Gene Ther. 2000, 11, 2389–2401. [Google Scholar] [CrossRef]
- Freeman, S.M.; Abboud, C.N.; Whartenby, K.A.; Packman, C.H.; Koeplin, D.S.; Moolten, F.L.; Abraham, G.N. The "bystander effect": Tumor regression when a fraction of the tumor mass is genetically modified. Cancer Res. 1993, 53, 5274–5283. [Google Scholar]
- Wilson, W.R.; Pullen, S.M.; Hogg, A.; Helsby, N.A.; Hicks, K.O.; Denny, W.A. Quantitation of bystander effects in nitroreductase suicide gene therapy using three-dimensional cell cultures. Cancer Res. 2002, 62, 1425–1432. [Google Scholar]
- Kuriyama, S.; Tsujinoue, H.; Yoshiji, H. Immune response to suicide gene therapy. Methods Mol. Med. 2004, 90, 353–369. [Google Scholar]
- Kianmanesh, A.R.; Perrin, H.; Panis, Y.; Fabre, M.; Nagy, H.J.; Houssin, D.; Klatzmann, D. A "distant" bystander effect of suicide gene therapy: Regression of nontransduced tumors together with a distant transduced tumor. Hum. Gene Ther. 1997, 8, 1807–1814. [Google Scholar] [CrossRef]
- Pierrefite-Carle, V.; Baque, P.; Gavelli, A.; Mala, M.; Chazal, M.; Gugenheim, J.; Bourgeon, A.; Milano, G.; Staccini, P.; Rossi, B. Cytosine deaminase/5-fluorocytosine-based vaccination against liver tumors: Evidence of distant bystander effect. J. Natl. Cancer Inst. 1999, 91, 2014–2019. [Google Scholar] [CrossRef]
- Ram, Z.; Culver, K.W.; Walbridge, S.; Blaese, R.M.; Oldfield, E.H. In situ retroviral-mediated gene transfer for the treatment of brain tumors in rats. Cancer Res. 1993, 53, 83–88. [Google Scholar]
- Moolten, F.L. Tumor chemosensitivity conferred by inserted herpes thymidine kinase genes: Paradigm for a prospective cancer control strategy. Cancer Res. 1986, 46, 5276–5281. [Google Scholar]
- Grignet-Debrus, C.; Cool, V.; Baudson, N.; Velu, T.; Calberg-Bacq, C.M. The role of cellular- and prodrug-associated factors in the bystander effect induced by the Varicella zoster and Herpes simplex viral thymidine kinases in suicide gene therapy. Cancer Gene Ther. 2000, 7, 1456–1468. [Google Scholar]
- Mesnil, M.; Yamasaki, H. Bystander effect in herpes simplex virus-thymidine kinase/ganciclovir cancer gene therapy: Role of gap-junctional intercellular communication. Cancer Res. 2000, 60, 3989–3999. [Google Scholar]
- Mesnil, M.; Piccoli, C.; Tiraby, G.; Willecke, K.; Yamasaki, H. Bystander killing of cancer cells by herpes simplex virus thymidine kinase gene is mediated by connexins. Proc. Natl. Acad. Sci. USA 1996, 93, 1831–1835. [Google Scholar] [CrossRef]
- Nishida, M.; Futami, S.; Morita, I.; Maekawa, K.; Murota, S.I. Hypoxia-reoxygenation inhibits gap junctional communication in cultured human umbilical vein endothelial cells. Endothelium 2000, 7, 279–286. [Google Scholar]
- Touraine, R.L.; Ishii-Morita, H.; Ramsey, W.J.; Blaese, R.M. The bystander effect in the HSVtk/ganciclovir system and its relationship to gap junctional communication. Gene Ther. 1998, 5, 1705–1711. [Google Scholar]
- Haustermans, K.; Hofland, I.; Van de Pavert, L.; Geboes, K.; Varia, M.; Raleigh, J.; Begg, A.C. Diffusion limited hypoxia estimated by vascular image analysis: Comparison with pimonidazole staining in human tumors. Radiother. Oncol. 2000, 55, 325–333. [Google Scholar] [CrossRef]
- Trotter, M.J.; Acker, B.D.; Chaplin, D.J. Histological evidence for nonperfused vasculature in a murine tumor following hydralazine administration. Int. J. Radiat. Oncol. Biol. Phys. 1989, 17, 785–789. [Google Scholar] [CrossRef]
- Morita, I.; Zhang, Y.W.; Murota, S.I. Eicosapentaenoic acid protects endothelial cell function injured by hypoxia/reoxygenation. Ann. N. Y. Acad. Sci. 2001, 947, 394–397. [Google Scholar]
- Jimenez, T.; Fox, W.P.; Naus, C.C.; Galipeau, J.; Belliveau, D.J. Connexin over-expression differentially suppresses glioma growth and contributes to the bystander effect following HSV-thymidine kinase gene therapy. Cell Commun. Adhes. 2006, 13, 79–92. [Google Scholar] [CrossRef]
- Park, J.Y.; Elshami, A.A.; Amin, K.; Rizk, N.; Kaiser, L.R.; Albelda, S.M. Retinoids augment the bystander effect in vitro and in vivo in herpes simplex virus thymidine kinase/ganciclovir-mediated gene therapy. Gene Ther. 1997, 4, 909–917. [Google Scholar]
- Elliott, G.; O'Hare, P. Intercellular trafficking and protein delivery by a herpesvirus structural protein. Cell 1997, 88, 223–233. [Google Scholar] [CrossRef]
- Dilber, M.S.; Phelan, A.; Aints, A.; Mohamed, A.J.; Elliott, G.; Smith, C.I.; O'Hare, P. Intercellular delivery of thymidine kinase prodrug activating enzyme by the herpes simplex virus protein, VP22. Gene Ther. 1999, 6, 12–21. [Google Scholar] [CrossRef]
- Qiu, Z.; Harms, J.S.; Zhu, J.; Splitter, G.A. Bovine herpesvirus tegument protein VP22 enhances thymidine kinase/ganciclovir suicide gene therapy for neuroblastomas compared to herpes simplex virus VP22. J. Virol. 2004, 78, 4224–4233. [Google Scholar] [CrossRef]
- Denekamp, J. Vascular attack as a therapeutic strategy for cancer. Cancer Metastasis Rev. 1990, 9, 267–282. [Google Scholar] [CrossRef]
- Tozer, G.M.; Kanthou, C.; Baguley, B.C. Disrupting tumour blood vessels. Nat. Rev. Cancer 2005, 5, 423–435. [Google Scholar] [CrossRef]
- Tozer, G.M.; Prise, V.E.; Wilson, J.; Cemazar, M.; Shan, S.; Dewhirst, M.W.; Barber, P.R.; Vojnovic, B.; Chaplin, D.J. Mechanisms associated with tumor vascular shut-down induced by combretastatin A-4 phosphate: Intravital microscopy and measurement of vascular permeability. Cancer Res. 2001, 61, 6413–6422. [Google Scholar]
- Tozer, G.M.; Prise, V.E.; Wilson, J.; Locke, R.J.; Vojnovic, B.; Stratford, M.R.; Dennis, M.F.; Chaplin, D.J. Combretastatin A-4 phosphate as a tumor vascular-targeting agent: Early effects in tumors and normal tissues. Cancer Res. 1999, 59, 1626–1634. [Google Scholar]
- Galbraith, S.M.; Chaplin, D.J.; Lee, F.; Stratford, M.R.; Locke, R.J.; Vojnovic, B.; Tozer, G.M. Effects of combretastatin A4 phosphate on endothelial cell morphology in vitro and relationship to tumour vascular targeting activity in vivo. Anticancer Res. 2001, 21, 93–102. [Google Scholar]
- Hay, M.P.; Pruijn, F.B.; Gamage, S.A.; Liyanage, H.D.; Kovacs, M.S.; Patterson, A.V.; Wilson, W.R.; Brown, J.M.; Denny, W.A. DNA-targeted 1,2,4-benzotriazine 1,4-dioxides: Potent analogues of the hypoxia-selective cytotoxin tirapazamine. J. Med. Chem. 2004, 47, 475–488. [Google Scholar] [CrossRef]
- Helsby, N.A.; Atwell, G.J.; Yang, S.; Palmer, B.D.; Anderson, R.F.; Pullen, S.M.; Ferry, D.M.; Hogg, A.; Wilson, W.R.; Denny, W.A. Aziridinyldinitrobenzamides: Synthesis and structure-activity relationships for activation by E. coli nitroreductase. J. Med. Chem. 2004, 47, 3295–3307. [Google Scholar] [CrossRef]
- Tupper, J.; Tozer, G.M.; Dachs, G.U. Use of horseradish peroxidase for gene-directed enzyme prodrug therapy with paracetamol. Br. J. Cancer 2004, 90, 1858–1862. [Google Scholar]
- Dachs, G.; Hunt, M.; Patterson, A.; Ackerley, D.; Currie, M.; Robinson, B. Vascular targeted gene therapy of cancer. J. Vasc. Res. 2009, 46 Suppl. 2, 1–43. [Google Scholar]
- Degreve, B.; De Clercq, E.; Balzarini, J. Bystander effect of purine nucleoside analogues in HSV-1 tk suicide gene therapy is superior to that of pyrimidine nucleoside analogues. Gene Ther. 1999, 6, 162–170. [Google Scholar] [CrossRef]
- Noble, S.; Faulds, D. Ganciclovir. An update of its use in the prevention of cytomegalovirus infection and disease in transplant recipients. Drugs 1998, 56, 115–146. [Google Scholar] [CrossRef]
- Elion, G.B. The biochemistry and mechanism of action of acyclovir. J. Antimicrob. Chemother. 1983, 12 Suppl. B, 9–17. [Google Scholar] [CrossRef]
- Karkas, J.D.; Germershausen, J.; Tolman, R.L.; MacCoss, M.; Wagner, A.F.; Liou, R.; Bostedor, R. Stereochemical considerations in the enzymatic phosphorylation and antiviral activity of acyclonucleosides. I. Phosphorylation of 2'-nor-2'-deoxyguanosine. Biochim. Biophys. Acta 1987, 911, 127–135. [Google Scholar] [CrossRef]
- Balzarini, J.; Bohman, C.; De Clercq, E. Differential mechanism of cytostatic effect of (E)-5-(2-bromovinyl)-2'-deoxyuridine, 9-(1,3-dihydroxy-2-propoxymethyl)guanine, and other antiherpetic drugs on tumor cells transfected by the thymidine kinase gene of herpes simplex virus type 1 or type 2. J. Biol. Chem. 1993, 268, 6332–6337. [Google Scholar]
- Herman, J.R.; Adler, H.L.; Aguilar-Cordova, E.; Rojas-Martinez, A.; Woo, S.; Timme, T.L.; Wheeler, T.M.; Thompson, T.C.; Scardino, P.T. In situ gene therapy for adenocarcinoma of the prostate: A phase I clinical trial. Hum. Gene Ther. 1999, 10, 1239–1249. [Google Scholar] [CrossRef]
- Klatzmann, D.; Valery, C.A.; Bensimon, G.; Marro, B.; Boyer, O.; Mokhtari, K.; Diquet, B.; Salzmann, J.L.; Philippon, J. A phase I/II study of herpes simplex virus type 1 thymidine kinase "suicide" gene therapy for recurrent glioblastoma. Study Group on Gene Therapy for Glioblastoma. Hum. Gene Ther. 1998, 9, 2595–2604. [Google Scholar] [CrossRef]
- Schellingerhout, D.; Rainov, N.G.; Breakefield, X.O.; Weissleder, R. Quantitation of HSV mass distribution in a rodent brain tumor model. Gene Ther. 2000, 7, 1648–1655. [Google Scholar] [CrossRef]
- Ayala, G.; Satoh, T.; Li, R.; Shalev, M.; Gdor, Y.; Aguilar-Cordova, E.; Frolov, A.; Wheeler, T.M.; Miles, B.J.; Rauen, K.; Teh, B.S.; Butler, E.B.; Thompson, T.C.; Kadmon, D. Biological response determinants in HSV-tk + ganciclovir gene therapy for prostate cancer. Mol. Ther. 2006, 13, 716–728. [Google Scholar]
- Nasu, Y.; Saika, T.; Ebara, S.; Kusaka, N.; Kaku, H.; Abarzua, F.; Manabe, D.; Thompson, T.C.; Kumon, H. Suicide gene therapy with adenoviral delivery of HSV-tK gene for patients with local recurrence of prostate cancer after hormonal therapy. Mol. Ther. 2007, 15, 834–840. [Google Scholar]
- Traversari, C.; Marktel, S.; Magnani, Z.; Mangia, P.; Russo, V.; Ciceri, F.; Bonini, C.; Bordignon, C. The potential immunogenicity of the TK suicide gene does not prevent full clinical benefit associated with the use of TK-transduced donor lymphocytes in HSCT for hematologic malignancies. Blood 2007, 109, 4708–4715. [Google Scholar] [CrossRef]
- Field, A.K.; Davies, M.E.; DeWitt, C.; Perry, H.C.; Liou, R.; Germershausen, J.; Karkas, J.D.; Ashton, W.T.; Johnston, D.B.; Tolman, R.L. 9-([2-hydroxy-1-(hydroxymethyl)-ethoxy]methyl)guanine: A selective inhibitor of herpes group virus replication. Proc. Natl. Acad. Sci. USA 1983, 80, 4139–4143. [Google Scholar]
- Munir, K.M.; French, D.C.; Dube, D.K.; Loeb, L.A. Herpes thymidine kinase mutants with altered catalytic efficiencies obtained by random sequence selection. Protein Eng. 1994, 7, 83–89. [Google Scholar] [CrossRef]
- Black, M.E.; Newcomb, T.G.; Wilson, H.M.; Loeb, L.A. Creation of drug-specific herpes simplex virus type 1 thymidine kinase mutants for gene therapy. Proc. Natl. Acad. Sci. USA 1996, 93, 3525–3529. [Google Scholar] [CrossRef]
- Black, M.E.; Kokoris, M.S.; Sabo, P. Herpes simplex virus-1 thymidine kinase mutants created by semi-random sequence mutagenesis improve prodrug-mediated tumor cell killing. Cancer Res. 2001, 61, 3022–3026. [Google Scholar]
- Wiewrodt, R.; Amin, K.; Kiefer, M.; Jovanovic, V.P.; Kapoor, V.; Force, S.; Chang, M.; Lanuti, M.; Black, M.E.; Kaiser, L.R.; Albelda, S.M. Adenovirus-mediated gene transfer of enhanced Herpes simplex virus thymidine kinase mutants improves prodrug-mediated tumor cell killing. Cancer Gene Ther. 2003, 10, 353–364. [Google Scholar] [CrossRef]
- Kokoris, M.S.; Sabo, P.; Black, M.E. In vitro evaluation of mutant HSV-1 thymidine kinases for suicide gene therapy. Anticancer Res. 2000, 20, 959–963. [Google Scholar]
- Ardiani, A.; Sanchez-Bonilla, M.; Black, M.E. Fusion enzymes containing HSV-1 thymidine kinase mutants and guanylate kinase enhance prodrug sensitivity in vitro and in vivo. Cancer Gene Ther. 2009.
- Solaroli, N.; Johansson, M.; Persoons, L.; Balzarini, J.; Karlsson, A. Substrate specificity of feline and canine herpesvirus thymidine kinase. Antiviral Res. 2008, 79, 128–132. [Google Scholar] [CrossRef]
- Beerens, A.M.; Rots, M.G.; Bermudez, B.; de Vries, E.F.; Haisma, H.J. Secretion of thymidine kinase to increase the effectivity of suicide gene therapy results in the loss of enzymatic activity. J. Drug Target 2008, 16, 26–35. [Google Scholar] [CrossRef]
- Kajiwara, E.; Kawano, K.; Hattori, Y.; Fukushima, M.; Hayashi, K.; Maitani, Y. Long-circulating liposome-encapsulated ganciclovir enhances the efficacy of HSV-TK suicide gene therapy. J. Control. Release 2007, 120, 104–110. [Google Scholar] [CrossRef]
- Balzarini, J.; Degreve, B.; Andrei, G.; Neyts, J.; Sandvold, M.; Myhren, F.; de Clercq, E. Superior cytostatic activity of the ganciclovir elaidic acid ester due to the prolonged intracellular retention of ganciclovir anabolites in herpes simplex virus type 1 thymidine kinase gene-transfected tumor cells. Gene Ther. 1998, 5, 419–426. [Google Scholar]
- Hayashi, K.; Lee, J.B.; Maitani, Y.; Toyooka, N.; Nemoto, H.; Hayashi, T. The role of a HSV thymidine kinase stimulating substance, scopadulciol, in improving the efficacy of cancer gene therapy. J. Gene Med. 2006, 8, 1056–1067. [Google Scholar] [CrossRef]
- Zhang, Q.; Ni, Q.; Gan, J.; Shen, Z.; Luo, J.; Jin, C.; Zhang, N.; Zhang, Y. p14ARF upregulation of p53 and enhanced effects of 5-fluorouracil in pancreatic cancer. Chin. Med. J. (Engl.) 2003, 116, 1150–1155. [Google Scholar]
- Scholer, H.J. [Chemotherapy of mycoses of the inner organs]. Schweiz. Med. Wochenschr. 1968, 98, 602–611. [Google Scholar]
- Springer, C.; Niculescu-Duvaz, I. Gene-directed enzyme prodrug therapy (GDEPT): Choice of prodrugs. Adv. Drug Deliv. Rev. 1996, 22, 351–364. [Google Scholar] [CrossRef]
- Kurozumi, K.; Tamiya, T.; Ono, Y.; Otsuka, S.; Kambara, H.; Adachi, Y.; Ichikawa, T.; Hamada, H.; Ohmoto, T. Apoptosis induction with 5-fluorocytosine/cytosine deaminase gene therapy for human malignant glioma cells mediated by adenovirus. J. Neurooncol. 2004, 66, 117–127. [Google Scholar] [CrossRef]
- Huber, B.E.; Austin, E.A.; Richards, C.A.; Davis, S.T.; Good, S.S. Metabolism of 5-fluorocytosine to 5-fluorouracil in human colorectal tumor cells transduced with the cytosine deaminase gene: Significant antitumor effects when only a small percentage of tumor cells express cytosine deaminase. Proc. Natl. Acad. Sci. USA 1994, 91, 8302–8306. [Google Scholar] [CrossRef]
- Haack, K.; Linnebacher, M.; Eisold, S.; Zoller, M.; von Knebel Doeberitz, M.; Gebert, J. Induction of protective immunity against syngeneic rat cancer cells by expression of the cytosine deaminase suicide gene. Cancer Gene Ther. 2000, 7, 1357–1364. [Google Scholar] [CrossRef]
- Crystal, R.G.; Hirschowitz, E.; Lieberman, M.; Daly, J.; Kazam, E.; Henschke, C.; Yankelevitz, D.; Kemeny, N.; Silverstein, R.; Ohwada, A.; Russi, T.; Mastrangeli, A.; Sanders, A.; Cooke, J.; Harvey, B.G. Phase I study of direct administration of a replication deficient adenovirus vector containing the E. coli cytosine deaminase gene to metastatic colon carcinoma of the liver in association with the oral administration of the pro-drug 5-fluorocytosine. Hum. Gene Ther. 1997, 8, 985–1001. [Google Scholar] [CrossRef]
- Haberkorn, U.; Oberdorfer, F.; Gebert, J.; Morr, I.; Haack, K.; Weber, K.; Lindauer, M.; van Kaick, G.; Schackert, H.K. Monitoring gene therapy with cytosine deaminase: In vitro studies using tritiated-5-fluorocytosine. J. Nucl. Med. 1996, 37, 87–94. [Google Scholar]
- Nemunaitis, J.; Cunningham, C.; Senzer, N.; Kuhn, J.; Cramm, J.; Litz, C.; Cavagnolo, R.; Cahill, A.; Clairmont, C.; Sznol, M. Pilot trial of genetically modified, attenuated Salmonella expressing the E. coli cytosine deaminase gene in refractory cancer patients. Cancer Gene Ther. 2003, 10, 737–744. [Google Scholar] [CrossRef]
- Austin, E.A.; Huber, B.E. A first step in the development of gene therapy for colorectal carcinoma: Cloning, sequencing, and expression of Escherichia coli cytosine deaminase. Mol. Pharmacol. 1993, 43, 380–387. [Google Scholar]
- Ge, K.; Xu, L.; Zheng, Z.; Xu, D.; Sun, L.; Liu, X. Transduction of cytosine deaminase gene makes rat glioma cells highly sensitive to 5-fluorocytosine. Int. J. Cancer 1997, 71, 675–679. [Google Scholar] [CrossRef]
- Kievit, E.; Bershad, E.; Ng, E.; Sethna, P.; Dev, I.; Lawrence, T.S.; Rehemtulla, A. Superiority of yeast over bacterial cytosine deaminase for enzyme/prodrug gene therapy in colon cancer xenografts. Cancer Res. 1999, 59, 1417–1421. [Google Scholar]
- Kaliberov, S.A.; Market, J.M.; Gillespie, G.Y.; Krendelchtchikova, V.; Della Manna, D.; Sellers, J.C.; Kaliberova, L.N.; Black, M.E.; Buchsbaum, D.J. Mutation of Escherichia coli cytosine deaminase significantly enhances molecular chemotherapy of human glioma. Gene Ther. 2007, 14, 1111–1119. [Google Scholar] [CrossRef]
- Mahan, S.D.; Ireton, G.C.; Knoeber, C.; Stoddard, B.L.; Black, M.E. Random mutagenesis and selection of Escherichia coli cytosine deaminase for cancer gene therapy. Protein Eng. Des. Sel. 2004, 17, 625–633. [Google Scholar] [CrossRef]
- Mahan, S.D.; Ireton, G.C.; Stoddard, B.L.; Black, M.E. Alanine-scanning mutagenesis reveals a cytosine deaminase mutant with altered substrate preference. Biochemistry 2004, 43, 8957–8964. [Google Scholar] [CrossRef]
- Fuchita, M.; Ardiani, A.; Zhao, L.; Serve, K.; Stoddard, B.L.; Black, M.E. Bacterial cytosine deaminase mutants created by molecular engineering show improved 5-fluorocytosine-mediated cell killing in vitro and in vivo. Cancer Res. 2009, 69, 4791–4799. [Google Scholar] [CrossRef]
- Adachi, Y.; Tamiya, T.; Ichikawa, T.; Terada, K.; Ono, Y.; Matsumoto, K.; Furuta, T.; Hamada, H.; Ohmoto, T. Experimental gene therapy for brain tumors using adenovirus-mediated transfer of cytosine deaminase gene and uracil phosphoribosyltransferase gene with 5-fluorocytosine. Hum. Gene Ther. 2000, 11, 77–89. [Google Scholar] [CrossRef]
- Koyama, F.; Sawada, H.; Hirao, T.; Fujii, H.; Hamada, H.; Nakano, H. Combined suicide gene therapy for human colon cancer cells using adenovirus-mediated transfer of escherichia coli cytosine deaminase gene and Escherichia coli uracil phosphoribosyltransferase gene with 5-fluorocytosine. Cancer Gene Ther. 2000, 7, 1015–1022. [Google Scholar]
- Chung-Faye, G.A.; Chen, M.J.; Green, N.K.; Burton, A.; Anderson, D.; Mautner, V.; Searle, P.F.; Kerr, D.J. In vivo gene therapy for colon cancer using adenovirus-mediated, transfer of the fusion gene cytosine deaminase and uracil phosphoribosyltransferase. Gene Ther. 2001, 8, 1547–1554. [Google Scholar] [CrossRef]
- Erbs, P.; Regulier, E.; Kintz, J.; Leroy, P.; Poitevin, Y.; Exinger, F.; Jund, R.; Mehtali, M. In vivo cancer gene therapy by adenovirus-mediated transfer of a bifunctional yeast cytosine deaminase/uracil phosphoribosyltransferase fusion gene. Cancer Res. 2000, 60, 3813–3822. [Google Scholar]
- Graepler, F.; Lemken, M.L.; Wybranietz, W.A.; Schmidt, U.; Smirnow, I.; Gross, C.D.; Spiegel, M.; Schenk, A.; Graf, H.; Lauer, U.A.; Vonthein, R.; Gregor, M.; Armeanu, S.; Bitzer, M.; Lauer, U.M. Bifunctional chimeric SuperCD suicide gene -YCD: YUPRT fusion is highly effective in a rat hepatoma model. World J. Gastroenterol. 2005, 11, 6910–6919. [Google Scholar]
- Unger, M.M.; Wahl, J.; Ushmorov, A.; Buechele, B.; Simmet, T.; Debatin, K.M.; Beltinger, C. Enriching suicide gene bearing tumor cells for an increased bystander effect. Cancer Gene Ther. 2007, 14, 30–38. [Google Scholar]
- Lemken, M.L.; Graepler, F.; Wolf, C.; Wybranietz, W.A.; Smirnow, I.; Schmidt, U.; Gregor, M.; Bitzer, M.; Lauer, U.M. Fusion of HSV-1 VP22 to a bifunctional chimeric SuperCD suicide gene compensates for low suicide gene transduction efficiencies. Int. J. Oncol. 2007, 30, 1153–1161. [Google Scholar]
- Simpson, G.R.; Han, Z.; Liu, B.; Wang, Y.; Campbell, G.; Coffin, R.S. Combination of a fusogenic glycoprotein, prodrug activation, and oncolytic herpes simplex virus for enhanced local tumor control. Cancer Res. 2006, 66, 4835–4842. [Google Scholar] [CrossRef]
- Lee, K.C.; Hamstra, D.A.; Bullarayasamudram, S.; Bhojani, M.S.; Moffat, B.A.; Dornfeld, K.J.; Ross, B.D.; Rehemtulla, A. Fusion of the HSV-1 tegument protein vp22 to cytosine deaminase confers enhanced bystander effect and increased therapeutic benefit. Gene Ther. 2006, 13, 127–137. [Google Scholar] [CrossRef]
- Wybranietz, W.A.; Gross, C.D.; Phelan, A.; O'Hare, P.; Spiegel, M.; Graepler, F.; Bitzer, M.; Stahler, P.; Gregor, M.; Lauer, U.M. Enhanced suicide gene effect by adenoviral transduction of a VP22-cytosine deaminase (CD) fusion gene. Gene Ther. 2001, 8, 1654–1664. [Google Scholar] [CrossRef]
- Cobb, L.M.; Connors, T.A.; Elson, L.A.; Khan, A.H.; Mitchley, B.C.; Ross, W.C.; Whisson, M.E. 2,4-dinitro-5-ethyleneiminobenzamide (CB 1954): A potent and selective inhibitor of the growth of the Walker carcinoma 256. Biochem. Pharmacol. 1969, 18, 1519–1527. [Google Scholar] [CrossRef]
- Knox, R.J.; Boland, M.P.; Friedlos, F.; Coles, B.; Southan, C.; Roberts, J.J. The nitroreductase enzyme in Walker cells that activates 5-(aziridin-1-yl)-2,4-dinitrobenzamide (CB 1954) to 5-(aziridin-1-yl)-4-hydroxylamino-2-nitrobenzamide is a form of NAD(P)H dehydrogenase (quinone) (EC 1.6.99.2). Biochem. Pharmacol. 1988, 37, 4671–4677. [Google Scholar] [CrossRef]
- Anlezark, G.M.; Melton, R.G.; Sherwood, R.F.; Coles, B.; Friedlos, F.; Knox, R.J. The bioactivation of 5-(aziridin-1-yl)-2,4-dinitrobenzamide (CB1954)--I. Purification and properties of a nitroreductase enzyme from Escherichia coli--a potential enzyme for antibody-directed enzyme prodrug therapy (ADEPT). Biochem. Pharmacol. 1992, 44, 2289–2295. [Google Scholar] [CrossRef]
- Zenno, S.; Koike, H.; Tanokura, M.; Saigo, K. Gene cloning, purification, and characterization of NfsB, a minor oxygen-insensitive nitroreductase from Escherichia coli, similar in biochemical properties to FRase I, the major flavin reductase in Vibrio fischeri. J. Biochem. 1996, 120, 736–744. [Google Scholar] [CrossRef]
- Djeha, A.H.; Hulme, A.; Dexter, M.T.; Mountain, A.; Young, L.S.; Searle, P.F.; Kerr, D.J.; Wrighton, C.J. Expression of Escherichia coli B nitroreductase in established human tumor xenografts in mice results in potent antitumoral and bystander effects upon systemic administration of the prodrug CB1954. Cancer Gene Ther. 2000, 7, 721–731. [Google Scholar]
- Helsby, N.A.; Ferry, D.M.; Patterson, A.V.; Pullen, S.M.; Wilson, W.R. 2-Amino metabolites are key mediators of CB 1954 and SN 23862 bystander effects in nitroreductase GDEPT. Br. J. Cancer 2004, 90, 1084–1092. [Google Scholar] [CrossRef]
- Helsby, N.A.; Wheeler, S.J.; Pruijn, F.B.; Palmer, B.D.; Yang, S.; Denny, W.A.; Wilson, W.R. Effect of nitroreduction on the alkylating reactivity and cytotoxicity of the 2,4-dinitrobenzamide-5-aziridine CB 1954 and the corresponding nitrogen mustard SN 23862: Distinct mechanisms of bioreductive activation. Chem. Res. Toxicol. 2003, 16, 469–478. [Google Scholar] [CrossRef]
- Clark, A.J.; Iwobi, M.; Cui, W.; Crompton, M.; Harold, G.; Hobbs, S.; Kamalati, T.; Knox, R.; Neil, C.; Yull, F.; Gusterson, B. Selective cell ablation in transgenic mice expression E. coli nitroreductase. Gene Ther. 1997, 4, 101–110. [Google Scholar]
- Cui, W.; Gusterson, B.; Clark, A.J. Nitroreductase-mediated cell ablation is very rapid and mediated by a p53-independent apoptotic pathway. Gene Ther. 1999, 6, 764–770. [Google Scholar] [CrossRef]
- Drabek, D.; Guy, J.; Craig, R.; Grosveld, F. The expression of bacterial nitroreductase in transgenic mice results in specific cell killing by the prodrug CB1954. Gene Ther. 1997, 4, 93–100. [Google Scholar]
- Chung-Faye, G.; Palmer, D.; Anderson, D.; Clark, J.; Downes, M.; Baddeley, J.; Hussain, S.; Murray, P.I.; Searle, P.; Seymour, L.; Harris, P.A.; Ferry, D.; Kerr, D.J. Virus-directed, enzyme prodrug therapy with nitroimidazole reductase: A phase I and pharmacokinetic study of its prodrug, CB1954. Clin. Cancer Res. 2001, 7, 2662–2668. [Google Scholar]
- Palmer, D.H.; Mautner, V.; Mirza, D.; Oliff, S.; Gerritsen, W.; van der Sijp, J.R.; Hubscher, S.; Reynolds, G.; Bonney, S.; Rajaratnam, R.; Hull, D.; Horne, M.; Ellis, J.; Mountain, A.; Hill, S.; Harris, P.A.; Searle, P.F.; Young, L.S.; James, N.D.; Kerr, D.J. Virus-directed enzyme prodrug therapy: Intratumoral administration of a replication-deficient adenovirus encoding nitroreductase to patients with resectable liver cancer. J. Clin. Oncol. 2004, 22, 1546–1552. [Google Scholar]
- Workman, P.; White, R.A.; Talbot, K. CB 1954 revisited. I. Disposition kinetics and metabolism. Cancer Chemother. Pharmacol. 1986, 16, 1–8. [Google Scholar] [CrossRef]
- de Poorter, J.; Hoeben, R.; Obermann, W.; Huizinga, T.; Nelissen, R. Gene therapy for the treatment of hip prosthesis loosening: Adverse events in a phase 1 clinical study. Hum. Gene Ther. 2008, 19, 1029–1038. [Google Scholar] [CrossRef]
- Jarrom, D.; Jaberipour, M.; Guise, C.P.; Daff, S.; White, S.A.; Searle, P.F.; Hyde, E.I. Steady-state and stopped-flow kinetic studies of three Escherichia coli NfsB mutants with enhanced activity for the prodrug CB1954. Biochemistry 2009, 48, 7665–7672. [Google Scholar] [CrossRef]
- Grohmann, M.; Paulmann, N.; Fleischhauer, S.; Vowinckel, J.; Priller, J.; Walther, D.J. A mammalianized synthetic nitroreductase gene for high-level expression. BMC Cancer 2009, 9, 301. [Google Scholar] [CrossRef]
- Vass, S.O.; Jarrom, D.; Wilson, W.R.; Hyde, E.I.; Searle, P.F. E. coli NfsA: An alternative nitroreductase for prodrug activation gene therapy in combination with CB1954. Br. J. Cancer 2009, 100, 1903–1911. [Google Scholar] [CrossRef]
- Mitchell, D.J.; Minchin, R.F.E. coli nitroreductase/CB1954 gene-directed enzyme prodrug therapy: Role of arylamine N-acetlytransferase 2. Cancer Gene Ther. 2008, 15, 758–764. [Google Scholar] [CrossRef]
- Palmer, B.D.; Wilson, W.R.; Cliffe, S.; Denny, W.A. Hypoxia-selective antitumor agents. 5. Synthesis of water-soluble nitroaniline mustards with selective cytotoxicity for hypoxic mammalian cells. J. Med. Chem. 1992, 35, 3214–3222. [Google Scholar] [CrossRef]
- Anlezark, G.M.; Melton, R.G.; Sherwood, R.F.; Wilson, W.R.; Denny, W.A.; Palmer, B.D.; Knox, R.J.; Friedlos, F.; Williams, A. Bioactivation of dinitrobenzamide mustards by an E. coli B nitroreductase. Biochem. Pharmacol. 1995, 50, 609–618. [Google Scholar] [CrossRef]
- Denny, W.A.; Wilson, W.R. Bioreducible mustards: A paradigm for hypoxia-selective prodrugs of diffusible cytotoxins (HPDCs). Cancer Metastasis Rev. 1993, 12, 135–151. [Google Scholar] [CrossRef]
- Hicks, K.O.; Myint, H.; Patterson, A.V.; Pruijn, F.B.; Siim, B.G.; Patel, K.; Wilson, W.R. Oxygen dependence and extravascular transport of hypoxia-activated prodrugs: Comparison of the dinitrobenzamide mustard PR-104A and tirapazamine. Int. J. Radiat. Oncol. Biol. Phys. 2007, 69, 560–571. [Google Scholar] [CrossRef]
- Searle, P.F.; Chen, M.J.; Hu, L.; Race, P.R.; Lovering, A.L.; Grove, J.I.; Guise, C.; Jaberipour, M.; James, N.D.; Mautner, V.; Young, L.S.; Kerr, D.J.; Mountain, A.; White, S.A.; Hyde, E.I. Nitroreductase: A prodrug-activating enzyme for cancer gene therapy. Clin. Exp. Pharmacol. Physiol. 2004, 31, 811–816. [Google Scholar] [CrossRef]
- Palmer, B.D.; Wilson, W.R.; Atwell, G.J.; Schultz, D.; Xu, X.Z.; Denny, W.A. Hypoxia-selective antitumor agents. 9. Structure-activity relationships for hypoxia-selective cytotoxicity among analogues of 5-[N,N-bis(2-chloroethyl)amino]-2,4-dinitrobenzamide. J. Med. Chem. 1994, 37, 2175–2184. [Google Scholar] [CrossRef]
- Wilson, W.; Palmer, B.; Pullen, S.; Cliffe, S.; Denny, W. SN 23862, an analogue of CB 1954 with decreased DT diaphorase sensitivity and improved hypoxia-selective cytotoxicity. In 40th Annual Meeting of the Radiation Research Society, Salt Lake City, UT, USA, 1992.
- Patterson, A.V.; Ferry, D.M.; Edmunds, S.J.; Gu, Y.; Singleton, R.S.; Patel, K.; Pullen, S.M.; Hicks, K.O.; Syddall, S.P.; Atwell, G.J.; Yang, S.; Denny, W.A.; Wilson, W.R. Mechanism of action and preclinical antitumor activity of the novel hypoxia-activated DNA cross-linking agent PR-104. Clin. Cancer Res. 2007, 13, 3922–3932. [Google Scholar] [CrossRef]
- Singleton, D.C.; Li, D.; Bai, S.Y.; Syddall, S.P.; Smaill, J.B.; Shen, Y.; Denny, W.A.; Wilson, W.R.; Patterson, A.V. The nitroreductase prodrug SN 28343 enhances the potency of systemically administered armed oncolytic adenovirus ONYX-411(NTR). Cancer Gene Ther. 2007, 14, 953–967. [Google Scholar] [CrossRef]
- Hu, L.; Yu, C.; Jiang, Y.; Han, J.; Li, Z.; Browne, P.; Race, P.R.; Knox, R.J.; Searle, P.F.; Hyde, E.I. Nitroaryl phosphoramides as novel prodrugs for E. coli nitroreductase activation in enzyme prodrug therapy. J. Med. Chem. 2003, 46, 4818–4821. [Google Scholar] [CrossRef]
- Jiang, Y.; Han, J.; Yu, C.; Vass, S.O.; Searle, P.F.; Browne, P.; Knox, R.J.; Hu, L. Design, synthesis, and biological evaluation of cyclic and acyclic nitrobenzylphosphoramide mustards for E. coli nitroreductase activation. J. Med. Chem. 2006, 49, 4333–4343. [Google Scholar] [CrossRef]
- Hay, M.P.; Atwell, G.J.; Wilson, W.R.; Pullen, S.M.; Denny, W.A. Structure-activity relationships for 4-nitrobenzyl carbamates of 5-aminobenz[e]indoline minor groove alkylating agents as prodrugs for GDEPT in conjunction with E. coli nitroreductase. J. Med. Chem. 2003, 46, 2456–2466. [Google Scholar] [CrossRef]
- Hay, M.P.; Wilson, W.R.; Denny, W.A. Nitroarylmethylcarbamate prodrugs of doxorubicin for use with nitroreductase gene-directed enzyme prodrug therapy. Bioorg. Med. Chem. 2005, 13, 4043–4055. [Google Scholar] [CrossRef]
- McFadzean, J. Flagyl, the Story of a Pharmaceutical Discovery; The Parthenon Publishing Group Ltd: London, UK, 1986; pp. 1–117. [Google Scholar]
- Despois, R.; Pinnet-Sindico, S.; Ninet, L.; Preud’homme, J. Three antibiotics of different groups produced by the same strain of Streptomyces. J. Microbiol. 1956, 21, 76. [Google Scholar]
- Muller, M. Mode of action of metronidazole on anaerobic bacteria and protozoa. Surgery 1983, 93, 165–171. [Google Scholar]
- Verdijk, R.M.; Wilke, M.; Beslier, V.; Kloosterman, A.; Brand, A.; Goulmy, E.; Mutis, T. Escherichia coli-nitroreductase suicide gene control of human telomerase reverse transcriptase-transduced minor histocompatibility antigen-specific cytotoxic T cells. Bone Marrow Transplant. 2004, 33, 963–967. [Google Scholar] [CrossRef]
- Curado, S.; Anderson, R.M.; Jungblut, B.; Mumm, J.; Schroeter, E.; Stainier, D.Y. Conditional targeted cell ablation in zebrafish: A new tool for regeneration studies. Dev. Dyn. 2007, 236, 1025–1035. [Google Scholar] [CrossRef]
- Curado, S.; Stainier, D.Y.; Anderson, R.M. Nitroreductase-mediated cell/tissue ablation in zebrafish: A spatially and temporally controlled ablation method with applications in developmental and regeneration studies. Nat. Protoc. 2008, 3, 948–954. [Google Scholar] [CrossRef]
- Pisharath, H. Validation of nitroreductase, a prodrug-activating enzyme, mediated cell death in embryonic zebrafish (Danio rerio). Comp. Med. 2007, 57, 241–246. [Google Scholar]
- Pisharath, H.; Rhee, J.M.; Swanson, M.A.; Leach, S.D.; Parsons, M.J. Targeted ablation of beta cells in the embryonic zebrafish pancreas using E. coli nitroreductase. Mech. Dev. 2007, 124, 218–229. [Google Scholar] [CrossRef]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dachs, G.U.; Hunt, M.A.; Syddall, S.; Singleton, D.C.; Patterson, A.V. Bystander or No Bystander for Gene Directed Enzyme Prodrug Therapy. Molecules 2009, 14, 4517-4545. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules14114517
Dachs GU, Hunt MA, Syddall S, Singleton DC, Patterson AV. Bystander or No Bystander for Gene Directed Enzyme Prodrug Therapy. Molecules. 2009; 14(11):4517-4545. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules14114517
Chicago/Turabian StyleDachs, Gabi U., Michelle A. Hunt, Sophie Syddall, Dean C. Singleton, and Adam V. Patterson. 2009. "Bystander or No Bystander for Gene Directed Enzyme Prodrug Therapy" Molecules 14, no. 11: 4517-4545. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules14114517