1. Introduction
Peptides are increasingly gaining recognition as potential bioactive ingredients in the pharmaceutical industry [
1,
2,
3]. Peptide synthesis depends on the strategies used for protecting the α-amino group and for activating the carboxylic acid group prior to peptide coupling. The two main classes [
4,
5,
6] of carboxylic acid group activation methods are: (i) those that require
in situ activation of the carboxylic acid and (ii) those that require an activated species that has previously been prepared (usually from an
in situ activation step), isolated, purified, and characterized.
The amino group is most commonly protected by preparing the corresponding carbamate derivative. Despite the vast number of reagents reported to date for introducing the protecting group into the N-terminal amino group, there is still no universally active species capable of providing optimal protecting group introduction.
The traditional chloroformate strategy is an extremely powerful approach, providing fast amino group protection [
7,
8,
9]. Nevertheless, in some cases, presence of the free carboxylic acid group can interfere with the reaction and lead to formation of byproducts such as dipeptides and even tripeptides (
Scheme 1) [
10].
Scheme 1.
Mechanism for the formation side products (dipeptides and tripeptides) during the protection of amino acids with haloformates.
Scheme 1.
Mechanism for the formation side products (dipeptides and tripeptides) during the protection of amino acids with haloformates.
Since these side-reactions are associated with the quality of the leaving group, the less reactive species such as the dicarbonates
2 (
Figure 1) [
11,
12,
13,
14] and the succinimidocarbonates
3 (
Figure 1) [
15,
16] have previously been proposed as alternatives to the chloride
1 (
Figure 1). Use of the azide derivatives
4 (
Figure 1), [
17,
18] has also been proposed as an alternative for the chloride to prepare the
N-protection of amino acids, but the explosive nature of azides precludes their use in large-scale synthesis. Moreover, several other approaches, based on the use of other less reactive species such as the 1,2,2,2-tetrachloroethyl [
19,
20], the 5-norbornene-2,3-dicarboximido [
21], the pentafluorophenyl [
22,
23,
24], and the 1-hydroxybenzotriazole [
14,
25,
26] mixed carbonates
5–
8 (
Figure 1), have been proposed.
Recently, ethyl 2-cyano-2-(hydroxyimino)acetate (OxymaPure
®,
12a) has been tested as an additive for use in the carbodiimide approach for the formation of peptide bonds [
27]. OxymaPure
® and its uronium-based phosphium coupling reagents displayed a remarkable capacity to inhibit racemization, together with impressive coupling efficiency, in both automated and manual synthesis, superior to those of
12d and which has recently been reported to exhibit explosive properties [
26] at least comparable to those of HOAt uronium-based phosphonium coupling reagents [
28,
29,
30,
31,
32,
33,
34].
Later, we reported a series of Fmoc/Alloc-oxime carbonate reagents which are easy to prepare, stable, and highly reactive crystalline materials that afford nearly pure Fmoc/Alloc-amino acids in high yields. Among the Fmoc-oxime carbonates that we evaluated for the preparation of Fmoc/Alloc-Gly-OH, the
N-hydroxypicolinimidoyl cyanide derivative
9 (
Figure 1) gave the best results [
35]. More recently, our research group reported the cyanoacetamide-based oximes
10 (
Figure 1), which show unusual ability to afford Fmoc-protected amino acids in high yield, high purity and at lower cost relative to compound
9 [
36].
Figure 1.
Structure of carbonates derivatives.
Figure 1.
Structure of carbonates derivatives.
Herein, we extended our studies for the synthesis of a new family of carbonate derivatives based on OxymaPure®, which are easy to prepare, stable, and have shown high efficiency in N-protection as well as peptide coupling.
3. Experimental
3.1. Materials
The solvents used were of HPLC reagent grade. Melting points were determined with a Mel-Temp apparatus and are uncorrected. Infrared (IR) spectra were recorded on a Perkin-Elmer 1600 series Fourier transform instrument as KBr pellets. Nuclear Magnetic resonance spectra (1H-NMR and 13C-NMR spectra) were recorded on a JOEL 500 MHz and on a Mercury 400 MHz spectrometer with chemical shift values reported in δ units (ppm) relative to an internal standard. Elemental analyses were performed on Perkin-Elmer 2400 elemental analyzer, and the values found were within ±0.3% of the theoretical values. Follow-up of the reactions and checks of the purity of the compounds was done by TLC on silica gel-protected aluminum sheets (Type 60 GF254, Merck, Barcelona, Spain) and the spots were detected by exposure to UV-lamp at λ 254 nm for a few seconds. The compounds were named using ChemDraw Ultra version 11, CambridgeSoft Corporation (Cambridge, MA, USA).
3.2. General Method for Preparation of Ethyloxycarbonyl Derivatives 13(a–d, f)
A solution of ethyloxycarbonyl chloride (11, 0.95 mL, 10 mmol) in CH2Cl2 (30 mL) was added slowly to a solution of sodium carbonate (2.12 g, 20 mmol) and 10 mmol of oximes (12a, 12b), 1-hydroxypyridin-2(1H)-one (12c), or benzotriazole derivatives (12d or 12f) in H2O (20 mL) with stirring at 0 °C. The resulting clear mixture was stirred at 0°C for 30 min and then at room temperature for 2 h. After dilution with CH2Cl2 (50 mL), the organic phase was collected and washed with water and saturated aqueous NaCl (30 mL), dried over anhydrous Na2SO4 and then filtered, and the solvent was then removed on a rotary evaporator. The residue was recrystallized from CH2Cl2/hexane to give the ethyloxycarbonyl derivatives 13(a–d, f).
Ethyl 2-cyano-2-(ethoxycarbonyloxyimino)acetate (13a). The product was obtained as white crystals (1.67 g; 78.17% yield) (m.p. 44–45 °C). IR (KBr): 2241 (w, CN), 1812 (s, CO), 1741 (s, CO, ester) cm−1. 1H-NMR (CDCl3): δ 1.39–1.41 (m, 6H, 2 CH3), 4.43–4.47 (m, 4H, 2 CH2). 13C-NMR (CDCl3): δ 14.14, 14.26, 64.76, 67.33, 106.67, 131.03, 150.87, 156.81. Elemental analysis Calcd for C8H10N2O5: C, 44.86; H, 4.71; N, 13.08. Found: C, 45.08; H, 4.63; N, 13.17.
(Ethoxycarbonyloxy)carbonimidoyl dicyanide (13b). The product was obtained as an oil at room temperature (1.15 g; 68.89% yield). IR (KBr): 2248 (w, CN), 1811 (s, CO, ester) cm−1. 1H-NMR (CDCl3): δ 1.43 (t, 3H, 3J = 7.2 Hz, CH3), 4.49 (q, 2H, 3J = 7.2 Hz, CH2). 13C-NMR (CDCl3): δ 14.18, 68.32, 104.81, 108.03, 114.44, 149.70. Elemental analysis: Calcd for C6H5N3O3: C, 43.12; H, 3.02; N, 25.14. Found: C, 43.25; H, 2.89; N, 25.33.
Ethyl 2-oxopyridin-1(2H)-yl carbonate (13c). The product was obtained as white crystals (1.38 g; 75.47% yield) (m.p. 64–67 °C). IR (KBr): 1792 (s, CO), 1668 (s, CO, amidic) cm−1. 1H-NMR (CDCl3): δ 1.42 (t, 3H, 3J = 7.2 Hz, CH3), 4.42 (q, 2H, 3J = 7.2 Hz, CH2), 6.20 (td, 1H, 3J = 6.8 Hz, 4J = 1.6 Hz, Py-H), 6.72–6.74 (m, 1H, Py-H), 7.36 (td, 1H, 3J = 6.8 Hz, 4J = 2 Hz, Py-H), 7.46 (dd, 1H, 3J = 6.8 Hz, 4J = 2 Hz, Py-H). 13C-NMR (CDCl3): δ 14.25, 67.53, 105.29, 123.18, 135.14, 139.69, 152.45, 157.31.
1H-Benzo[d][1,2,3]triazol-1-yl ethyl carbonate (13d). The product was obtained as white crystals (1.07 g; 51.84% yield) (m.p. 138–139 °C). IR (KBr): 1751 (s, CO) cm−1. 1H-NMR (CDCl3): δ 1.53 (t, 3H, 3J = 7.2 Hz, CH3), 4.63 (q, 2H, 3J = 7.2 Hz, CH2), 7.56 (td, 1H, 3J = 8.4 Hz, 4J = 0.8 Hz, Ar-H), 7.78 (td, 1H, 3J = 8.4 Hz, 4J = 1.2 Hz, Ar-H), 8.00 (d, 1H, 3J = 8.4 Hz, Ar-H), 8.21 (d, 1H, 3J = 8.4 Hz, Ar-H). 13C-NMR (CDCl3): δ = 14.42, 65.67, 115.27, 115.88, 126.46, 132.91, 133.54, 147.52. Elemental analysis: Calcd for C9H9N3O3: C, 52.17; H, 4.38; N, 20.28. Found: C, 51.96; H, 4.54; N, 20.49.
6-Chloro-1H-benzo[d][1,2,3]triazol-1-yl ethyl carbonate (13f). The product was obtained as white crystals (1.81 g; 75.67% yield) (m.p. 144–145 °C). IR (KBr): 1743 (s, CO) cm−1. 1H-NMR (CDCl3): δ 1.53 (t, 3H, 3J = 7.2 Hz, CH3), 4.62 (q, 2H, 3J = 7.2 Hz, CH2), 7.72 (dd, 1H, 3J = 8.8 Hz, 4J = 2 Hz, Ar-H), 8.00 (d, 1H, 4J = 2 Hz, Ar-H), 8.16 (d, 1H, 3J = 8.8 Hz, Ar-H). 13C-NMR (CDCl3): δ 14.42, 65.99, 115.70, 116.32, 132.19, 132.81, 133.78, 147.32. Elemental analysis: Calcd for C9H8ClN3O3: C, 44.74; H, 3.34; N, 17.39. Found: C, 44.53; H, 3.61; N, 17.18.
3.3. 3H-[1,2,3]Triazolo[4,5-b]pyridin-3-yl Ethyl Carbonate (13e)
A solution of HOAt (12e, 0.68 g, 5 mmol) and anhydrous potassium hydroxide (0.3 g, 5.5 mmol) in acetonitrile (5 mL) was cooled to 0 °C. A solution of ethyloxycarbonyl chloride (11, 0.47 mL, 5 mmol) in acetonitrile (5 mL) was slowly added dropwise for 30 min to the solution as it was stirred magnetically. The resulting clear mixture was stirred at room temperature overnight. It was then filtered, and the solvent was removed with a rotary evaporator. The residue was recrystallized from CH2Cl2/hexane to give 3H-[1,2,3]triazolo[4,5-b]pyridin-3-yl ethyl carbonate (13e). The product was obtained as 0.8 g (77.14% yield) of white crystals (m.p. 133–135 °C). IR (KBr): 1744 (s, CO) cm−1. 1H-NMR (DMSO): δ 1.05 (t, 3H, 3J = 7.2 Hz, CH3), 3.43 (q, 2H, 3J = 7.2 Hz, CH2), 7.49–7.53 (m, 1H, Ar-H), 8.52–8.55 (m, 1H, Ar-H), 8.75–8.77 (m, 1H, Ar-H), 13C-NMR (DMSO): δ 18.47, 55.94, 120.62, 124.09, 134.52, 139.50, 151.01. Elemental analysis: Calcd for C8H8N4O3: C, 46.16; H, 3.87; N, 26.91. Found: C, 45.88; H, 4.14; N, 27.19.
3.4. General Method for Preparation of Oxime Carbonate Derivatives 17–19
A solution of chloroformate (10 mmol) [isobutyloxycarbonyl chloride (14), allyloxycarbonyl chloride (15) or benzyloxycarbonyl chloride (16)] in CH2Cl2 (30 mL) was added slowly to a solution (10 mmol) of oxima 12a and sodium carbonate (2.12 g, 20 mmol) in H2O (20 mL) with stirring at 0 °C. The resulting clear mixture was stirred at 0 °C for 30 min and then at room temperature for 2 h. After dilution with CH2Cl2 (50 mL), the organic phase was collected, washed with water and saturated aqueous NaCl (30 mL), and then dried over anhydrous MgSO4. It was then filtered, and the solvent was removed with a rotary evaporator. The residue was recrystallized from CH2Cl2/hexane to give oxime carbonate derivatives 17–19.
Ethyl 2-cyano-2-(isobutoxycarbonyloxyimino)acetate (17). The product was obtained as a white solid (2.42 g; 93% yield) (m.p. 59–60 °C). IR (KBr): 1814 (s, CO), 1758 (s, CO, ester) cm−1. 1H-NMR (CDCl3): δ 1.00 (d, J = 6.8 Hz, 6H, 2 CH3), 1.42 (t, J = 7.2 Hz, 3H, CH3), 2.06–2.13 (m, 1H, CH), 4.17 (d, J = 6.4 Hz, 2H, CH2), 4.50 (q, J = 7.2 Hz, 2H, CH2). 13C-NMR (CDCl3): δ 14.16, 18.89, 27.95, 64.75, 106.70, 130.97, 151.10, 156.84. Elemental analysis: Calcd for C10H14N2O5: C, 49.58; H, 5.83; N, 11.56. Found: C, 49.81; H, 5.57; N, 11.74. The purity of 17 was determined after injection onto reverse-phase HPLC. Conditions: detection at 254 nm Waters 996 PDA detector, Sunfire C18 column 3.5 µm 4.6 ° 100 mm, linear gradient over 14 min of 10 to 100% CH3CN in H2O/0.1% TFA, flow rate 1.0 mL/min. tR [ethyl 2-cyano-2-(isobutoxycarbonyloxyimino)acetate] = 7.38 min; purity 100%.
Ethyl 2-(allyloxycarbonyloxyimino)-2-cyanoacetate (18). The product was obtained as an oily substance that solidified in the refrigerator (2.26 g; 94% yield). IR (KBr): 2211 (w, CN), 1809 (s, CO), 1758 (s, CO, ester) cm−1. 1H-NMR (CDCl3): δ 1.42 (t, 3H, 3J = 7.2 Hz, CH3), 4.48 (q, 2H, 3J = 7.2 Hz, CH2), 4.85–4.87 (m, 2H, CH2), 5.39–5.51 (m, 2H, CH2), 5.96–6.03 (m, 1H, CH). 13C-NMR (CDCl3): δ 14.11, 64.78, 71.23, 106.62, 121.27, 130.07, 131.24, 150.74, 156.73. Elemental analysis. Calcd for C9H10N2O5: C, 47.79; H, 4.46; N, 12.39. Found: C, 47.93; H, 4.61; N, 12.58. The purity of 18 was determined after injection onto reverse-phase HPLC. Conditions: detection at 254 nm Waters 996 PDA detector, Sunfire C18 column 3.5 µm 4.6 ° 100 mm, linear gradient over 14 min of 10 to 100% CH3CN in H2O/0.1% TFA, flow rate 1.0 mL/min. tR [ethyl 2-(allyloxycarbonyloxyimino)-2-cyanoacetate] = 6.69 min; purity 100%.
Ethyl 2-(benzyloxycarbonyloxyimino)-2-cyanoacetate (19). The product was obtained as white crystals (2.76 g; 87% yield) (m.p. 99–100 °C). IR (KBr): 1802 (s, CO), 1743 (s, CO, ester) cm−1. 1H-NMR (CDCl3): δ 1.41 (t, 3H, 3J = 7.2 Hz, CH3), 4.47 (q, 2H, 3J = 7.2 Hz, CH2), 5.38 (s, 2H, CH2), 7.40–7.44 (m, 5H, Ar-H). 13C-NMR (CDCl3): δ 14.17, 64.80, 72.57, 106.63, 129.06, 129.23, 129.63, 131.23, 133.59, 151.00, 156.76. Elemental analysis: Calcd for C13H12N2O5: C, 56.52; H, 4.38; N, 10.14. Found: C, 56.23; H, 4.62; N, 10.41. The purity of 19 was determined after injection onto reverse-phase HPLC. Conditions: detection at 254 nm Waters 996 PDA detector, Sunfire C18 column 3.5 µm 4.6 ° 100 mm, linear gradient over 14 min of 10 to 100% CH3CN in H2O/0.1% TFA, flow rate 1.0 mL/min. tR [ethyl 2-(benzyloxycarbonyloxyimino)-2-cyanoacetate] = 7.31 min; purity 100%.
3.5. Synthesis of 4-(Ethoxycarbonylamino)benzoic Acid (21)
A solution of ethyloxycarbonyl derivative 13(a–f) (1 mmol) in acetone (10 mL) was added dropwise to a stirring solution of 4-aminobenzoic acid 20 (0.14 g, 1 mmol) and sodium carbonate (0.32 g, 3 mmol) in acetone (20 mL) and H2O (10 mL). After stirring overnight, the reaction mixture was concentrated under reduced pressure, and then extracted with CH2Cl2 (20 mL) to remove the unreacted ethyloxycarbonyl derivatives. The reaction mixture was acidified with 1 N HCl (detected with Congo red litmus paper) to give a white solid, which was filtered, washed with water several times, dried and then recrystallized (ethyl acetate/n-hexane) to give a white solid. The purity of 21 was determined by reverse-phase HPLC. Conditions: detection at 220 nm (Waters 996 PDA detector); Sunfire C18 column (3.5 µm 4.6 × 100 mm); linear gradient over 14 min (10 to 100% CH3CN in H2O/0.1% TFA); flow rate 1.0 mL/min. tR [4-(ethoxycarbonylamino)benzoic acid] = 4.18 min. IR (KBr): 3334 (w, NH), 3400–2500 (br, OH, acid), 1704 (s, CO, acidic), 1686 (s, CON) cm−1. 1H-NMR (DMSO): δ 1.24 (t, 3H, 3J = 7.2 Hz, CH3), 4.13 (q, 2H, 3J = 7.2 Hz, CH2), 7.54 (d, 2H, 3J = 8.4 Hz, Ar-H), 7.82 (d, 2H, 3J = 8.4 Hz, Ar-H), 9.93 (s, 1H, NH). 13C-NMR (DMSO): δ 15.11, 61.11, 117.91, 124.91, 131.05, 144.12, 153.00, 167.63.
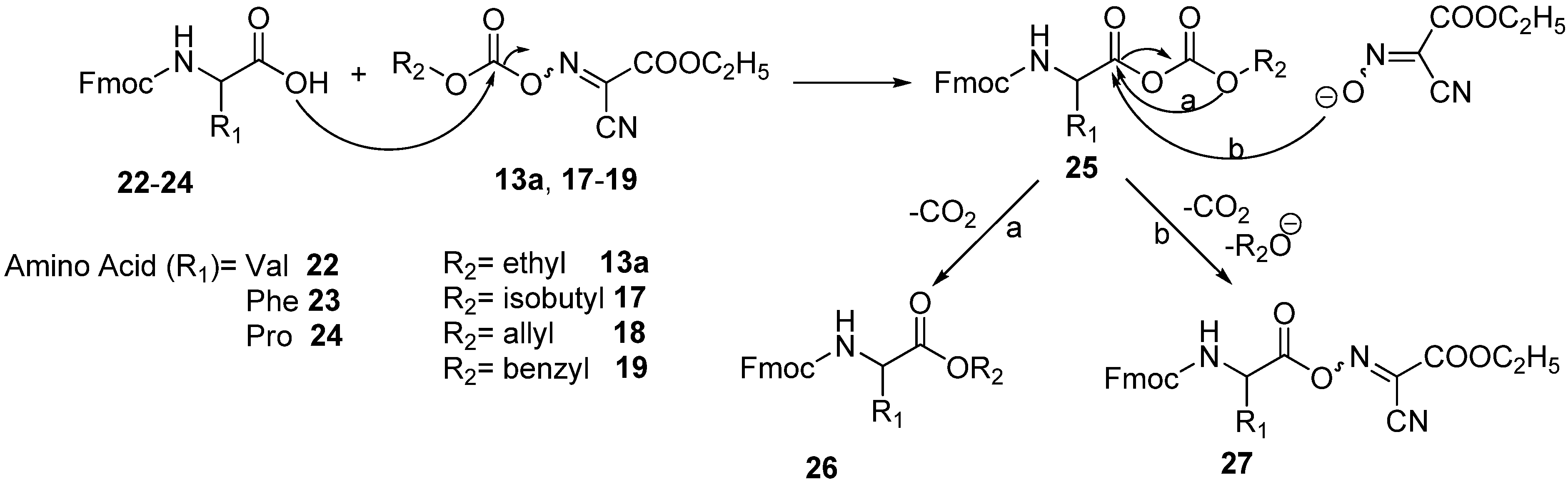
3.7. General Method for the Synthesis of Dipeptide Fmoc-Val-Ala-OMe 28
A solution of Fmoc-Val-OH
22 (0.339 g, 1 mmol) and the appropriate coupling reagent (1 mmol) in DMF (2 mL) was cooled to 0 °C and treated dropwise with pyridine (0.088 mL, 1.1 mmol). The reaction mixture was stirred for pre-activation at different times, depending on the conditions of the entry studied, and then treated with a solution of H-Ala-OMe.HCl (0.139 g, 1 mmol) and pyridine (0.088 mL, 1.1 mmol) in DMF (1 mL). The reaction mixture was stirred overnight. After dilution with 25 mL of ethyl acetate, the organic phase was washed with 5% citric acid (3 × 15 mL), saturated aq. NaHCO
3 (3 × 15 mL) and saturated aq. NaCl (3 × 15 mL), and then dried over anhydrous Na
2SO
4 and filtered. The solvent was removed with a rotary evaporator, and the residue was recrystallized from CH
2Cl
2/hexane to give the dipeptide Fmoc-Val-Ala-OMe
28. The purity of
28 was by reverse-phase HPLC. Conditions: detection at 220 nm (Agilent 1200 PDA detector); Eclipse plus C
18 column (3.5 µm 4.6 × 100 mm); linear gradient over 14 min (10 to 100% CH
3CN in H
2O/0.1% TFA); flow rate 1.0 mL/min.
tR LL [Fmoc-Val-Ala-OMe] = 8.32 min. The results of coupling of Fmoc-Val-OH with H-Ala-OMe using different oximinocarbonate derivatives are shown in
Table 12.
1H-NMR (CDCl
3): δ 0.94–0.98 (m, 6H, 2CH
3), 1.40 (d, 3H,
3J = 6.9 Hz, CH
3), 2.10–2.11 (m, 1H, CH), 3.73 (s, 3H, CH
3), 3.90–4.00 (m, 1H, CH), 4.17–4.21 (m, 1H, CH), 4.34–4.43 (m, 2H, CH
2), 4.55–4.58 (m, 1H, CH), 5.47–5.51 (m, 1H, NH), 6.42–6.45 (m, 1H, NH), 7.25–776 (m, 8H, Ar-H).





{kind=link}
{kind=link}
{kind=link}
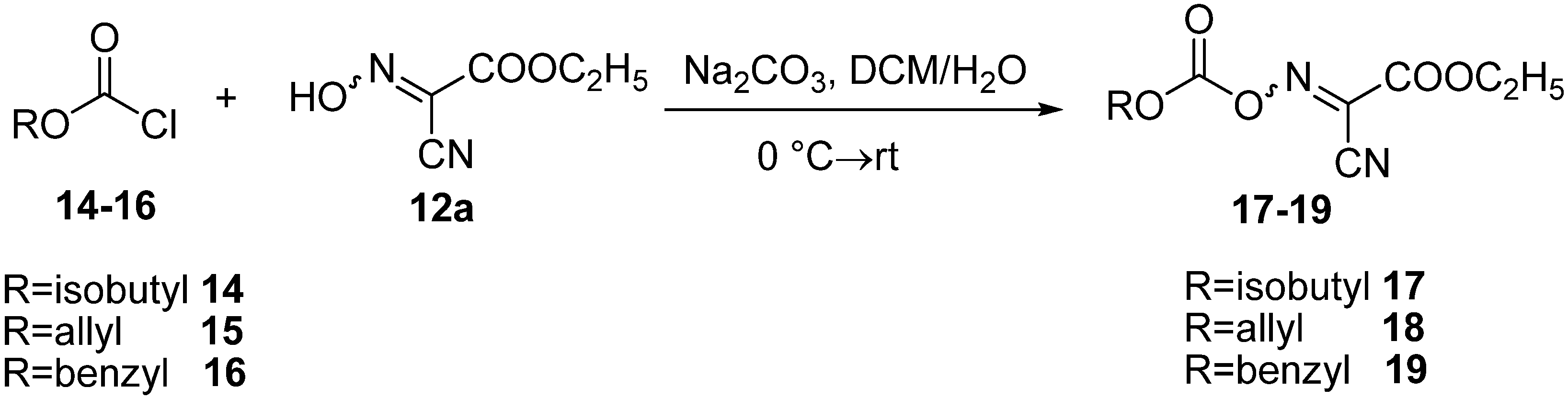{kind=link}
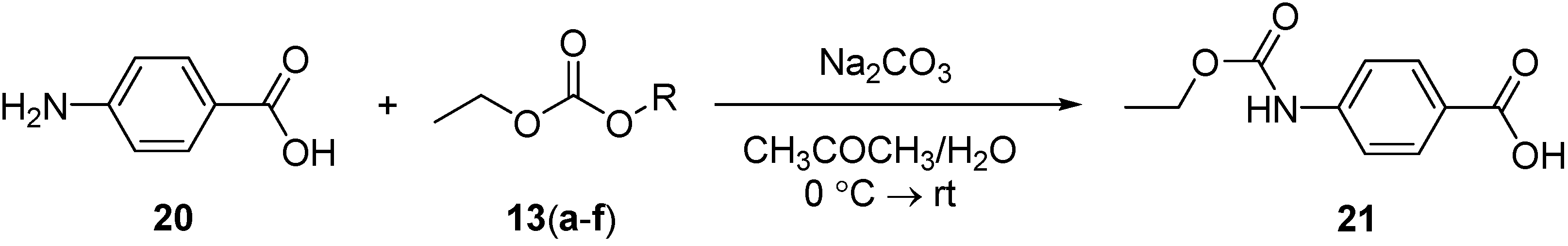{kind=link}
{kind=link}


