Solid Phase versus Solution Phase Synthesis of Heterocyclic Macrocycles


School of Chemistry, University of New South Wales, Sydney, NSW 2052, Australia
*
Author to whom correspondence should be addressed.
Molecules 2013, 18(1), 1111-1121; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules18011111
Submission received: 11 November 2012
/
Revised: 10 January 2013
/
Accepted: 10 January 2013
/
Published: 16 January 2013
(This article belongs to the Special Issue Chemical Protein and Peptide Synthesis)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Comparing a solution phase route to a solid phase route in the synthesis of the cytotoxic natural product urukthapelstatin A (Ustat A) confirmed that a solid phase method is superior. The solution phase approach was tedious and involved cyclization of a ridged heterocyclic precursor, while solid phase allowed the rapid generation of a flexible linear peptide. Cyclization of the linear peptide was facile and subsequent generation of three oxazoles located within the structure of Ustat A proved relatively straightforward. Given the ease with which the oxazole Ustat A precursor is formed via our solid phase approach, this route is amenable to rapid analog synthesis.
1. Introduction
Natural product-based macrocyclic peptides have tremendous potential as drug leads [1,2,3,4,5]. They have numerous pharmacokinetic benefits as drug candidates, including metabolic stability, structural rigidity, high affinity to biological targets and reasonable solubility properties [6,7]. As of 2009, there were 617 peptide drugs or drug candidates, 24% of these are in clinical trials, 65% are in advanced preclinical phases, and 11% are on the market [3]. These peptide drugs are used to treat numerous diseased states including: prostate and breast cancer, HIV infections, osteoporosis, acute coronary syndrome, and they serve as immunosuppressants [8]. Several large peptide-based drugs include: cyclosporin A (MW = 1,185), caspofungin (MW = 1,093), vancomycin (MW = 1,431), and fuzeon (MW = 4,492). Cyclosporin A is an 11 amino acid macrocyclic peptide that is used to suppress the immune system after organ transplants [9]. Caspofungin, vancomycin, and fuzeon are peptide-based antifungal, antibacterial, and anti-HIV drugs, respectively. Aplidine (MW = 1,067) is an eight amino acid peptide-based cancer agent that is currently in clinical trials [10,11,12].
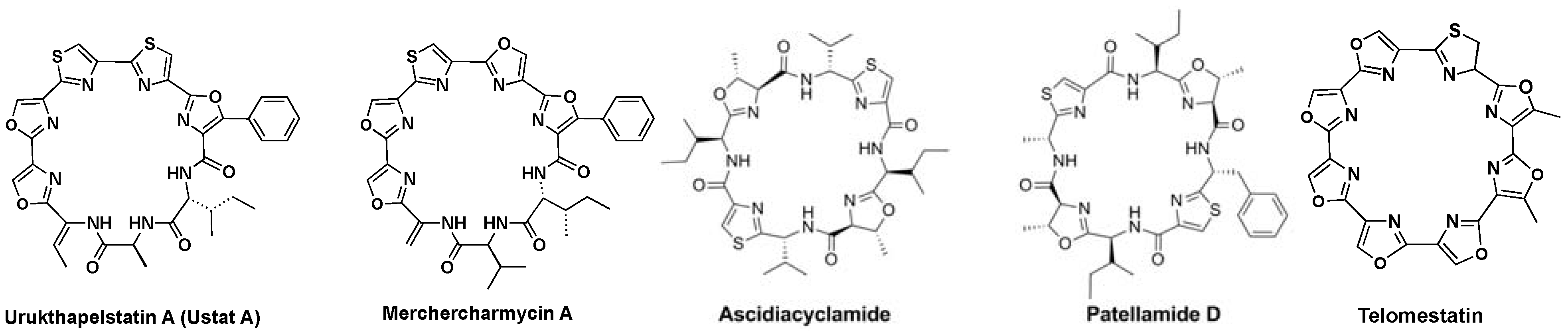
Urukthapelstatin A (Ustat A, Figure 1) falls in a unique class of natural products as it contains directly linked 4,2-azoles [13,14]. Over the last two decades there has been a surge in discoveries of natural products with directly linked azoles, many are now drug candidates [15,16,17]. Mixed 4,2-bisheterocycle tandem pairs are relatively rare in macrocycles, and thus, urukthapelstatin A represents a unique macrocyclic structure. Ustat A has an average GI50 of 5.6 nM on the Japanese 39 cell line panel [13,14], yet it’s mechanism of action remains unknown. As a modified macrocyclic octapeptide, Ustat A resembles several biologically active natural products including: merchercharmycin A (IB-01211), ascidiacyclamide, patellamide D, and telomestatin (Figure 1). IB-01211 was isolated from the marine microorganism strain ES7-008, and it is an antitumor macrocyclic peptide that has an IC50 = 14 nM in Hela S3 cells [18]. The same structure was proposed for merchercharmycin A, which was isolated from a marine-derived Thermoactinomices sp. and has an IC50 = 40 nM and 46 nM in A549 cells and Jurkat cells [19] respectively. Ascidiacyclamide is a cytotoxic cyclic peptide with thiazole and methyl-oxazoline rings [20] and it exhibits cytotoxic activity (IC50 = 14 µM) against leukemia cells [21]. Patellamide D that is a resistance-modifying agent, where it reverses resistance to the anti-cancer agent vinblastine [22]. Finally, the most investigated of these macrocycles is telomestatin. Telomestatin stabilizes the G-quadruplex found in telomeric DNA and inhibits telomerase activity (IC50 = 5 nM) in numerous cancerous cells, inducing apoptosis [23]. Telomestatin is currently in clinical trials [24]. The interesting biological activity displayed by these heterocycles appears related to their planar aromatic structures, which lend rigidity to the ring and provide sites for binding interactions via pi-stacking.
Depending on the natural product structure, using a solution phase approach can be advantageous over a solid-phase approach [29,30,31,32,33,34,35,36,37,38]. Synthesizing Ustat A in solution appeared desirable for several reasons. The first was that the syntheses of merchercharmycin A [25,26] were completed in solution, providing precedent. The second was the ability to explore whether the fragments of the heterocycles were cytotoxic [39]. Our experience and the complexity of Ustat A lead to our decision to initially explore a solution phase synthesis approach [40,41,42,43,44,45,46,47,48].
However, there are also numerous advantages to utilizing solid phase synthesis in building complex peptide-based macrocycles. Our expertise in solid phase synthesis of cyclic peptides has also been extensive [49,50,51]. General advantages of solid phase synthesis are easy purification, rapid generation of linear peptide intermediates, and precedent in the synthesis of large peptides. Given the ease with which peptide linear precursors are produced on solid phase, we opted to employ this approach in addition to a solution phase method. Should the solid-phase approach work, it would simplify analog synthesis. Thus, exploring the synthesis of the macrocycle Ustat A using both a solution phase and solid phase method provided opportunities to evaluate the benefits of both methods to synthesize this natural product.
2. Results and Discussion
2.1. Synthetic Approach
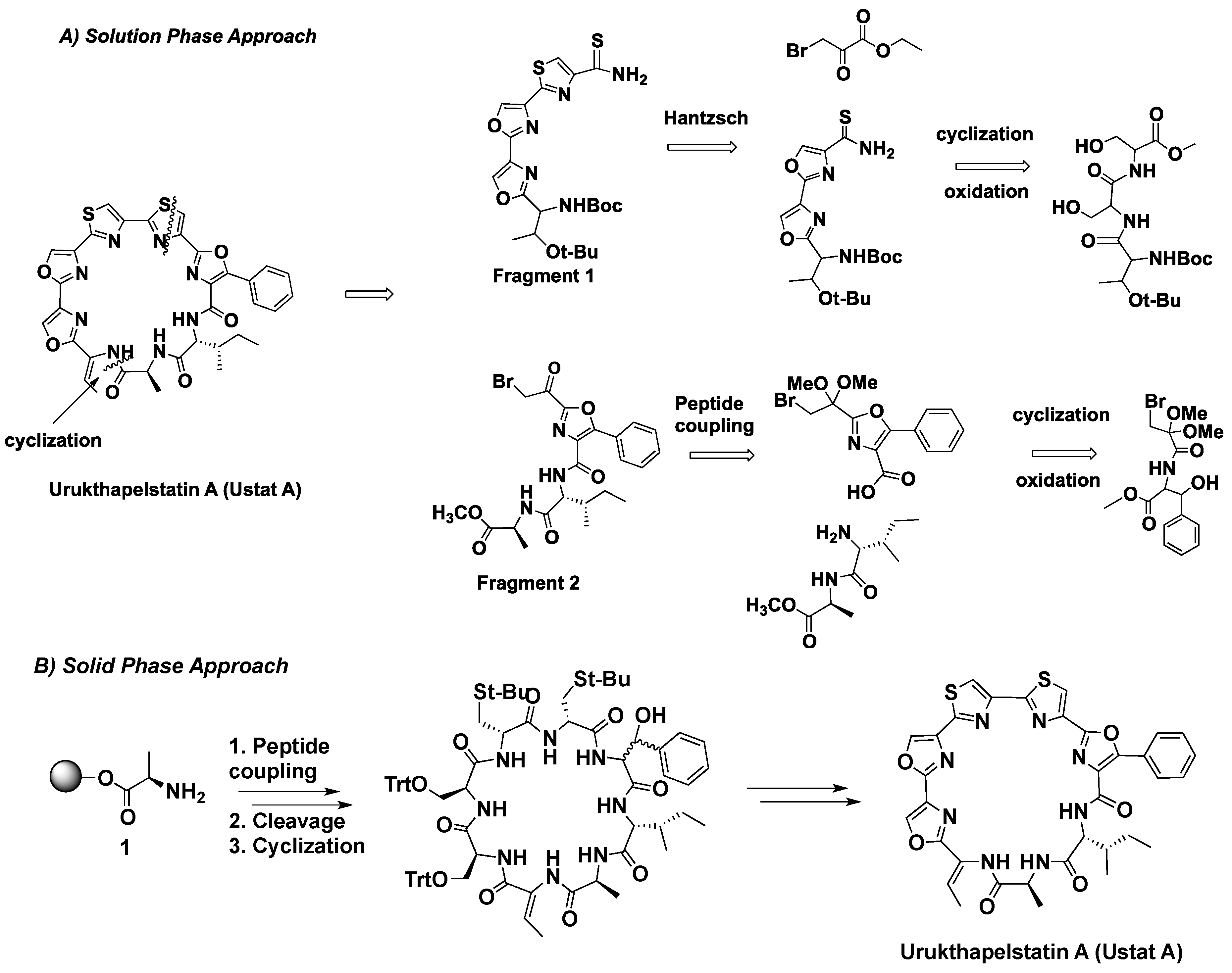
Solution synthesis of Ustat A involved cyclization at the peptide bond after forming the linear heterocyclic peptide (Scheme 1A). Formation of the linear precursor involved a modified Hantzsch thiazole reaction between the thioamide and bromoketone (fragments 1 and 2 respectively). Synthesis of fragment 1 entailed cyclization and oxidation of serine residues into oxazoles and the subsequent modified Hantzsch thiazole reaction between ethyl bromopyruvate and the thioamide located on the dioxazole moiety (Scheme 1A). Solid phase synthesis started with an alanine residue attached to a chlorotrityl resin (Scheme 1B). Sequential coupling of residues, cleavage, and cyclization generates a peptide precursor. Subsequent cyclization and oxidation of the serines and cysteines to heterocycles would generate the desired natural product.
2.2. Solution Phase Synthesis
We have published the formation of the Ustat A linear precursor that was generated in solution [52]. Several macrocyclization trials were performed from C- and N-deprotected linear precursor using these conditions with activating agents. Initially, cyclization of the macrocycle involved standard coupling conditions developed in our lab (Scheme 2, condition a) [42,50]. However, when these conditions were unsuccessful, we followed the conditions described by Hernandez et al. in the synthesis of merchercharmycin A [25,26,28] (Scheme 2, condition b). These conditions were also unsuccessful, and a closer analysis of Hernandez data on the cyclized product suggests that these conditions were also unsuccessful for synthesizing merchercharmycin A [28]. Additional cyclization conditions were attempted including esters activated by pentafluorophenyl diphenylphosphinate (FDPP) as reported by Joullié et al. (Scheme 2, condition c) [53]. All methods showed no trace of cyclized compound as determined by both LCMS and 1H-NMR. The unsuccessful cyclization of the linear heterocyclic Ustat A precursor using numerous conditions indicated that the sequential heterocycles created a molecule that was too rigid to cyclize with the heterocycles in place.
2.3. Solid Phase Synthesis
Synthesis using solid phase where construction of the heterocycles could be done after formation of the large macrocycle was appealing for three reasons. First, it would offer a simple option for substituting serines for cysteines allowing production of thiazoles in place of oxazoles. Given that thiazole fragments show promising biological activity, exchange of oxazoles for thiazoles is attractive [39]. Second, modification of the amino acids within the peptide region would be straightforward using solid phase, and analogs would be rapidly generated. Rapid production of these compounds provides the opportunity to explore structure activity relationships in biological systems. Third, forming the heterocycles after macrocycle formation allows the choice of sequentially closing the heterocycles, providing synthetic options for forming the final heterocyclic analogs.
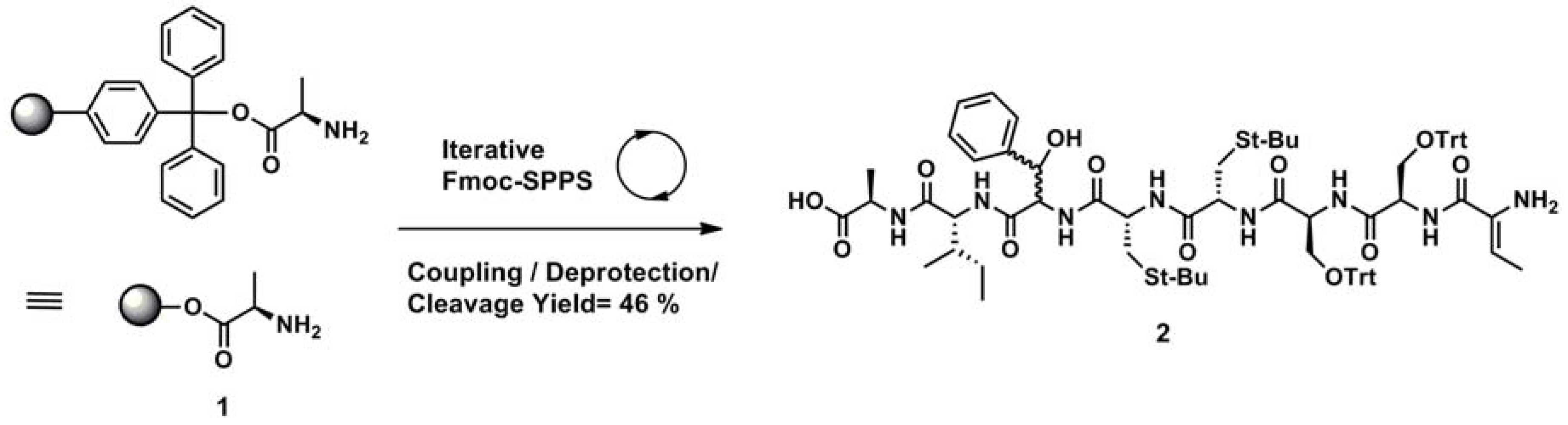
This is the first report of the solid phase synthesis of a Ustat A precursor. Starting with alanine bound to a chlorotrityl resin (1, Scheme 3), we sequentially coupled amino acids using N-hydroxy-benzotriazole (HOBt) and diisopropylcarbodiimide (DIC) in DMF and Fmoc removal using 20% piperidine and 80% DMF (Scheme 3) [50]. Upon generating the linear precursor on solid phase, cleavage to produce the desired compound 2 occurred using trifluoroethanol (TFE):CH2Cl2 (1:1, v/v) in a 46% yield. It was characterized by 1H-NMR, indicating we had successfully generated the linear peptide (Scheme 3).
Cyclizing the linear precursor 2 utilizing our standard conditions [50] generated the cyclized compound 3 (Scheme 4, yield = 36%). Employing triethylsilane (TES):CH2Cl2 with 1.1% trifluoroacetic acid (TFA) in CH2Cl2 selectively removed the trityl protecting group (Trt) from serine in the presence of the tBu protecting groups on cysteines. Oxazoline formation was carried out by intramolecular cyclodehydration of serines using the fluorinating agent diethylamino sulfurtrifluoride (DAST) and pyridine/K2CO3 to afford all three intermediate oxazolines (yield = 47%). The oxazolines were oxidized using bromochloroform (BrCCl3) and 1,8-diazabicyclo[5,4,0]undec-7-ene (DBU) generating compound 4.
3. Experimental
3.1. Materials
All reagents and solvents were commercially available and used without further purification. All reactions were carried out with dry solvents under argon. Silica gel 60 (230–400 mesh) was used for thin-layer chromatography (TLC), and flash chromatography. Reactions were monitored by LCMS (Shimadzu Liquid Chromatograph-Mass Spectrometry) or on TLC using UV light as visualizing method and KMnO4, Bromocresol Green, and Ninhydrin as developing agents. All amino acids are commercially available and solid phase reactions were monitored by the ninhydrin test. 1H- and 13C-NMR spectra were recorded with Bruker Avance III instruments operating at 300 MHz and 600 MHz, respectively. Accurate mass spectra for the novel molecules were recorded using a Thermo LTQ Orbitrap XL high resolution liquid chromatograph-mass spectrometer/mass spectrometer, equipped with a conventional ESI source.
3.2. Synthesis
3.2.1. General Procedure for Solid Couplings
Solid-phase peptide synthesis was produced manually on 0.08 mmol scale in polypropylene peptide synthesis reaction vessels according to the following general procedures: pre-loaded N-deprotected resin was swelled by DMF for 30 min in the synthesis reaction vessel, drained, then subjected to the following coupling cycle for all standard Fmoc AA’s: Fmoc-protected amino acid (3 equiv. relative to resin loading) and HOBt (3 equiv.) dissolved in minimal DMF. DIC (6 equiv.) was added in one portion to resin slurry. The resulting solution was shaken for a minimum of 2 h. Some sample was removed from the resin and washed with IPA. It was checked by the ninhydrin test. Once the N-terminal amino acid residue has been coupled, the peptide-resin is washed with DMF (3 × 1 min). The resin was agitated in 20% piperidine/DMF (2 × 10 min), and then washed with DMF (3 × 1 min), IPA (3 × 1 min), and DMF (5 × 1 min). It was also checked by the ninhydrin test.
3.2.2. Solid Phase Synthesis of Linear Precursor 2
Resin-bound linear precursor compound was generated following from alanine to (2Z)-2-amino-2-butenoic acid via the general procedure for solid coupling method (Section 3.2.1). Ordered Fmoc-AA’s: Resin based alanine, D-allo-isoleucine, racemic-phenyl serine, tBu-cysteine, tBu-cysteine, trityl-serine, trityl-serine, and (2Z)-2-amino-2-butenoic acid [55]. After the last Fmoc deprotection, the resin was washed with DMF (3 × 1 min), IPA (3 × 1 min) and MeOH (3 × 1 min). The linear peptide was cleaved from the resin by swelling and stirring the resin in a solution of TFE/CH2Cl2 (1:1, v/v, 10 mL/g of dried resin) for 24 h. Cleaved peptide solution was then filtered through a Buchner filter, drained resin was then washed with additional CH2Cl2 to fully extract cleaved peptide from resin. Solvents were then collected and removed by rotary evaporation and peptide was dried under high vacuum overnight. Dried peptide was then reconstituted with CH2Cl2 and evaporated multiple times by rotary evaporator before being dried in vacuo overnight to remove residual TFE to give a linear precursor as clear glassy solid to afford compound 2. Yield: 46%; 1H-NMR (300 MHZ, Bruker, CDCl3), δ 7.51–7.08 (m, 35H), 5.46 (m, 1H), 4.73 (m, 1H), 4.58 (m, 1H), 4.58–4.32 (m, 4H), 4.0 (q, 4H, J = 8.31 Hz), 3.62–3.34 (m, 3H), 3.15 (q, 4H, J =7.20 Hz), 3.01–2.83 (m, 3H), 2.14 (m, 1H), 2.01–1.78 (bs, 2H), 1.68 (s, 3H), 1.40–1.25 (m, 18H), 1.05–1.02 (m, 3H), 0.87–0.85 (m, 3H); 13C-NMR (300 MHz, Bruker, CDCl3), δ 197.9, 172.1–169.5 (peptide amide C=O), 143.1, 128.4–126.3 (aromatic carbons, trityl), 126.1, 87,4, 72.4, 62.4, 60.4, 55.2, 54.7, 43.5 (peptide aliphatic carbons), 30.97 (tBu-CH3), 25.9, 22.1, 17.6, 13.8, 11.3, 6.7 (peptide aliphatic carbons); LCMS (ESI): calcd for C80H96N8O12S2Na+ [M+Na+] 1448.78, found 1448.70; HRMS(ESI): calcd for C80H97N8O12S2 [M+H+] 1425.6667, found 1425.6443.
3.2.3. Synthesis of the Cyclized Compound 3
The cyclized compound 3 was synthesized using linear compound 2 (0.8 g) in CH2Cl2 (280 mL, 2 mM) with HATU (0.10 g, 0.5 equiv.), TBTU (0.09 g, 0.5 equiv.), DMTMM (0.07 g, 0.5 equiv.), COMU (0.11 g, 0.5 equiv.) as the coupling reagents with anhydrous DIPEA (0.78 mL, 8 equiv.). The reaction mixture was stirred at room temperature for 6 h. Upon completion, the reaction was washed with aqueous HCl solution (pH = 1, 50 mL × 2), and saturated aqueous NaHCO3 solution (50 mL × 2). The collected organic layer was dried over Na2SO4 and concentrated in vacuo. The obtained crude residue was purified by flash column chromatography (silica gel, EtOAc/hexanes) to yield the desired cyclized compound 3 as a yellow oil (28 mg). The cyclization reaction to afford compound 3 was found to be rapid and efficient by LCMS analysis. Yield: 36%; 1H-NMR (300 MHz, CDCl3,) δ 7.51–7.10 (m, 35H), 5.40 (m, 1H), 4.81 (m, 1H), 4.70 (m, 1H), 4.36–4.21 (m, 4H), 4.20–3.91 (m, 4H), 3.56–3.34 (m, 3H), 3.17–2.86 (m, 7H), 2.38–2.33 (m, 1H), 2.01 (s, 3H), 1.62 (m, 2H), 1.40–1.25 (m, 18H), 0.93–0.88 (m, 6H); 13C-NMR (600 MHz, Bruker, CDCl3), δ 176.8–171.2 (peptide amide C=O), 146.8, 131.6–125.7 (aromatic carbons, trityl), 120.5, 115.1, 66.6, 60.4, 56.9, 54.4, 48.8, 43.9, 36.7, 32.6 (peptide aliphatic carbons), 29.4 (tBu-CH3), 27.1, 25.2, 22.7, 20.6, 14.2 (peptide aliphatic carbons); LCMS (ESI): calcd for C80H95N8O11S2 [M+H+] 1408.78, found 1408.95; HRMS(ESI): calcd for C80H94N8O11S2Na [M+Na] 1429.6381, found 1429.6691.
3.2.4. Synthesis of Oxazole-based Macrocycle 4
Selective removal of Trts: compound 3 (0.28 g) was diluted to a 0.1 M solution in TES/CH2Cl2 (1:1, v/v), followed by the addition of 1.1% TFA in CH2Cl2 until colorless. The reaction was performed at room temperature for 3 h. Upon completion, the reaction mixture was dried, and concentrated to afford the alcohol (quantitative yield).
Oxazoline formation: 3.5 equiv. of DAST (0.14 mL) were added dropwise to the serine macrocyclic compound in CH2Cl2 (3.0 mL, 0.1 M) at −70 °C under Ar. The reaction mixture was stirred for 1 h, followed by the addition of pyridine (3 equiv., 0.07 mL), and K2CO3 (1.5 equiv., 0.06 g) and stirred for an additional 30 min. The solution was then allowed to room temperature for 12 h. Upon completion, the organic solution was partitioned between saturated aqueous NaHCO3 and CH2Cl2. The solvent was removed and the residue was purified by flash column chromatography.
Oxazole formation: Oxidation of the oxazoline fragment was performed with DBU (6 equiv., 0.29 mL) in CH2Cl2 at −40 °C. The solution was stirred for 15 min and BrCCl3 (6 equiv., 0.21 mL) were then added to the reaction mixture, which was allowed to proceed over 12 h. The reaction was worked up using aqueous HCl solution (pH = 1, 50 mL × 2), and saturated aqueous NaHCO3 solution (50 mL × 2). The solvent was removed and the residue was purified by flash column chromatography to afford compound 4. Yield: 56% (2 step); 1H-NMR (600 MHz, CDCl3), δ 7.38–7.24 (m, 5H), 7.19–7.17 (d, 2H, J = 7.48 Hz), 5.38 (m, 1H), 4.39–4.18 (m, 2H), 4.17–4.06 (m, 2H), 4.05–3.91 (m, 4H), 2.36 (s, 3H), 2.06 (m, 1H), 1.80–1.61 (m, 3H), 1.50 (m, 2H), 1.40–1.25 (m, 18H), 0.93–0.88 (m, 6H); 13C-NMR (600 MHz, Bruker, CDCl3), δ 173.5 (C=O), 165.9 (C=O), 146.8, 143.9 (oxazoles-CH), 134.2, 129.5, 129.4–126.3 (aromatic carbons, trityl), 68.1, 67.7, 56.8, 38.9, 34.5, 31.4 (peptide aliphatic carbons), 29.7 (tBu-CH3), 28.9, 24.5, 23.8, 14.1, 11.0 (peptide aliphatic carbons); LCMS (ESI): calcd for C42H54N8O8S2Na+ [M+Na+] 886.05, found 886.10; HRMS(ESI): calcd for C42H55N8O8S2 [M+H+] 863.3584, found 863.3579.
4. Conclusions
We have developed a solid phase method for generating the potential anticancer lead Ustat A. Solid phase is superior to solution phase as it rapidly generates a flexible linear precursor. The linear peptide is easily cyclized (yield = 36%) and conversion of the serines and phenylserine to oxazoles is straightforward. Formation of the thiazoles remains, and synthetic studies to this end are ongoing. In addition, biological evaluation of intermediates 2–4 are in progress and will be reported in due course.
Supplementary Materials
Supplementary materials can be accessed at: https://0-www-mdpi-com.brum.beds.ac.uk/1420-3049/18/1/1111/s1.
Acknowledgments
We thank the University of New South Wales for a scholarship to S.J.K. and support of this research. We thank NIH 1R01CA137873 for providing reagents for this project.
References and Notes
- Jarvis, L.M. Breakthroughs in manufacturing are making large-scale synthesis of peptides a viable proposition. C E News 2006, 84, 23–25. [Google Scholar]
- Chatterjee, J.; Gilon, C.; Hoffman, A.; Kessler, H. N-Methylation of peptides: A new perspective in medicinal chemistry. Acc. Chem. Res. 2008, 41, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Danho, W.; Swistok, J.; Khan, W.; Chu, X.; Cheung, A.; Fry, D.; Sun, H.; Kurylko, G.; Rumennik, L.; Cefalu, J.; et al. Opportunities and challenges of developing peptide drugs in the pharmaceutical industry. In Peptides for Youth, Proceedings of the 20th American Peptide Symposium, Montreal, Canada, 23–28 June 2007; Valle, S.D., Escher, E., Lubell, W.D., Eds.; Springer: New York, NY, USA, 2009; Volume 611, pp. 467–469. [Google Scholar]
- Fletcher, J.M.; Hughes, R.A. Modified low molecular weight cyclic peptides as mimetics of BDNF with improved potency, proteolytic stability and transmembrane. Bioorg. Med. Chem. 2009, 17, 2695–2702. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, J.; Vilcinskas, A. Antimicrobial peptides, the ancient arm of the human immune system. Virulence 2010, 1, 440–464. [Google Scholar] [CrossRef] [PubMed]
- Fairlie, D.P.; Abbenante, G.; March, D.R. Macrocyclic peptidomimetics—Forcing peptides into bioactive conformations. Curr. Med. Chem. 1995, 2, 654–686. [Google Scholar]
- Koehn, F.E.; Carter, G.T. The evolving role of natural products in drug discovery. Nat. Rev. Drug Discov. 2005, 4, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Loffet, A. Peptides as Drugs: Is there a Market? Eur. Pept. Soc. 2002, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Starzl, T.E.; Klintmalm, G.B.; Porter, K.A.; Iwatsuki, S.; Schroter, G.P. Clinical trials of cyclosporin A. N. Engl. J. Med. 1981, 305, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Faivre, S.; Chieze, S.; Delbaldo, C.; Ady-Vago, N.; Guzman, C.; Lopez-Lazaro, L.; Lozahic, S.; Jimeno, J.; Pico, F.; Armand, J.P.; et al. Phase I and pharmacokinetic study of aplidine, a new marine cyclodepsipeptide in patients with advanced malignancies. J. Clin. Oncol. 2005, 23, 7871–7880. [Google Scholar] [CrossRef] [PubMed]
- Maroun, J.A.; Belanger, K.; Seymour, L.; Matthews, S.; Roach, J.; Dionne, J.; Soulieres, D.; Stewart, D.; Goel, R.; Charpentier, D.; et al. Phase I study of Aplidine in a daily35 one-hour infusion every 3 weeks in patients with solid tumors refractory to standard therapy. A National Cancer Institute of Canada Clinical Trials Group study: NCIC CTG IND 115. Ann. Oncol. 2006, 17, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Le Tourneau, C.; Raymond, E.; Faivre, S. Aplidine: A paradigm of how to handle the activity and toxicity of a novel marine anticancer poison. Curr. Pharm. Des. 2007, 13, 3427–3429. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Kanoh, K.; Imanaka, H.; Adachi, K.; Nishizawa, M.; Shizuri, Y. Urukthapelstatin A, a novel cytotoxic substance from marine-derived Mechercharimyces asporophorigenes YM11-542 II physcico-chemical properties and structural elucidation. J. Antibiot. 2007, 60, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Kanoh, K.; Yamori, T.; Kasai, H.; Katsuta, A.; Adachi, K.; Shin-ya, K.; Shizuri, Y. Urukthapelstatin A, a novel cytotoxic substance from marine-derived Mechercharimyces asporophorigenes YM11-542 I fermentation, isolation, and biological properties. J. Antibiot. 2007, 60, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Irschik, H.; Reichenbach, H.; Hofle, G.; Jansen, R. The thuggacins, novel antibacterial macrolides from sorangium cellulosum acting against selected gram-positive bacteria. J. Antibiot. 2007, 60, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Riego, E.; Hernández, D.; Albericio, F.; Álvarez, M. Directly linked polyazoles: Important moieties in natural products. Synthesis 2005, 2005, 1907–1922. [Google Scholar] [CrossRef]
- Ying, Y.; Taori, K.; Kim, H.; Hong, J.; Luesch, H. Total synthesis and molecular target of Largazole, a histone deacetylase inhibitor. J. Am. Chem. Soc. 2008, 130, 8455–8459. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Malet, L.; Canedo, L.; Maria, C.C.; Fernando, R.J. New cytotoxic depsipeptides. WO 2005/000880 A2, 2005. [Google Scholar]
- Kanoh, K.; Matsuo, Y.; Adachi, K.; Imagawa, H.; Nishizawa, M.; Shizuri, Y. Mechercharmycins A and B, cytotoxic substances from marine-derived thermoactinomyces sp. YM3-251. J. Antibiot. 2005, 58, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Inoue, M.; Hamada, Y.; Kato, S.; Shioiri, T. X-ray crystal structure of ascidiacyclamide, a cytotoxic cyclic peptide from ascidian. J. Chem. Soc. Chem. Commun. 1987, 370–371. [Google Scholar] [CrossRef]
- Asano, A.; Minoura, K.; Yamada, T.; Numata, A.; Ishida, T.; Doi, M.; Katsuya, Y.; Mezaki, Y.; Sasaki, M.; Taniguchi, T.; et al. Effect of asymmetric modification on the conformation of ascidiacyclamide analogs. J. Pept. Res. 2002, 60, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.B.; Jacobs, R.S. A marine natural product, patellamide D, reverses multidrug resistance in a human leukemic cell line. Cancer Lett. 1993, 71, 97–102. [Google Scholar] [CrossRef]
- Miyazaki, T.; Pan, Y.; Joshi, K.; Purohit, D.; Hu, B.; Demir, H.; Mazumder, S.; Okabe, S.; Yamori, T.; Viapiano, M.S.; et al. Telomestatin impairs glioma stem cell survival and growth through the disruption of telomeric G-quadruplex and inhibition of the proto-oncogene, c-Myb. Clin. Cancer Res. 2012, 18, 1268–1280. [Google Scholar] [CrossRef] [PubMed]
- Telomestatin. Available online: http://www.clinicaltrials.gov (accessed on 1 October 2012).
- Hernandez, D.; Riego, E.; Albericio, F.; Alvarez, M. Synthesis of natural product derivatives containing 2,4-concatenated oxazoles. Eur. J. Org. Chem. 2008, 3389–3396. [Google Scholar] [CrossRef]
- Hernandez, D.; Vilar, G.; Riego, E.; Canedo, L.M.; Cuevas, C.; Albericio, F.; Alvarez, M. Synthesis of IB-01211, a cyclic peptide containing 2,4-concentrated thia- and oxazoles, vix Hantzsch macrocyclization. Org. Lett. 2007, 9, 809–811. [Google Scholar] [CrossRef] [PubMed]
- Shin-ya, K.; Wierzba, K.; Matsuo, K.; Ohtani, T.; Yamada, Y.; Furihata, K.; Hayakawa, Y.; Seto, H. Synthesis of Telomerastatin. J. Am. Chem. Soc. 2001, 123, 1262–1263. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, D.; Riego, E.; Francesch, A.; Cuevas, C.; Albericio, F.; Alvarez, M. Preparation of penta-azole containing cyclopeptides: challenges in macrocyclization. Tetrahedron 2007, 63, 9862–9870. [Google Scholar] [CrossRef]
- Ward, D.E.; Gai, Y.; Lazny, R.; Pedras, M.S.C. Probing Host-selective Phytoxicity: Synthesis of Destruxin B and several natural analogues. J. Org. Chem. 2001, 66, 7832–7840. [Google Scholar] [CrossRef] [PubMed]
- Tarver, J.; Pfizenmayer, A.J.; Joullié, M.M. Total synthesis of comformationally constrained Didemnin B analogues. Replacement of N,O-dimethyltyrosine with L-1,2,3,4-tetrahydroisoquinoline and L-1,2,3,4-tetrahydro-7-methoxyisoquinoline. J. Org. Chem. 2001, 66, 7575–7587. [Google Scholar] [CrossRef] [PubMed]
- Li, W.R.; Ewing, W.R.; Harris, B.D.; Joullié, M.M. Total synthesis and structural investigations of didemnins A, B, and C. J. Am. Chem. Soc. 1990, 112, 7659–7672. [Google Scholar] [CrossRef]
- Mayer, S.C.; Ramanjulu, J.; Vera, M.D.; Pfizenmayer, A.; Joullié, M.M. Synthesis of new didemnin B analogs for investigations of structure/biological activity relationships. J. Org. Chem. 1994, 59, 5192–5205. [Google Scholar] [CrossRef]
- Vera, M.D.; Pfizenmayer, A.J.; Ding, X.; Ahuja, D.; Toogood, P.L.; Joullié, M.M. Synthesis and biological evaluation of didemnin photoaffinity analogues. Bioorg. Med. Chem. Lett. 2001, 11, 1871–1874. [Google Scholar] [CrossRef]
- Vera, M.D.; Pfizenmayer, A.J.; Ding, X.; Xiao, D.; Joullié, M.M. [Lys3]didemnins as potential affinity ligands. Bioorg. Med. Chem. Lett. 2001, 11, 13–16. [Google Scholar] [CrossRef]
- Xiao, D.; Vera, M.D.; Liang, B.; Joullié, M.M. Total synthesis of a conformationally constrained didemnin B analog. J. Org. Chem. 2001, 66, 2734–2742. [Google Scholar] [CrossRef] [PubMed]
- Ramanjulu, J.; Ding, X.; Li, W.R.; Joullié, M.M. Synthesis of a reduced ring analog of Didemnin B. J. Org. Chem. 1997, 62, 4961–4969. [Google Scholar] [CrossRef]
- Kopp, F.; Stratton, C.F.; Akella, L.B.; Tan, D.S. A diversity-oriented synthesis approach to macrocycles via oxidative ring expansion. Nat. Chem. Biol. 2012, 8, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Campbell, F.; Kilner, C.A.; Wilson, A.J. Expedient synthesis of benzene tricarboxamide macrocycles derived from p-aminobenzoic acid. Tetrahedron Lett. 2010, 51, 1361–1363. [Google Scholar] [CrossRef]
- Kim, S.J.; Lin, C.-C.; Pan, C.-M.; Rananaware, D.P.; Ramsey, D.M.; McAlpine, S.R. A structure-activity relationship study of compounds containing sequential oxazoles and thiazoles. Med. Chem. Commun. 2012, in press. [Google Scholar]
- Carroll, C.L.; Johnston, J.V.C.; Kekec, A.; Brown, J.D.; Parry, E.; Cajica, J.; Medina, I.; Cook, K.M.; Corral, R.; Pan, P.-S.; et al. Synthesis and cytotoxicity of novel Sansalvamide A derivatives. Org. Lett. 2005, 7, 3481–3484. [Google Scholar] [CrossRef] [PubMed]
- Styers, T.J.; Kekec, A.; Rodriguez, R.A.; Brown, J.D.; Cajica, J.; Pan, P.-S.; Parry, E.; Carroll, C.L.; Medina, I.; Corral, R.; et al. Synthesis of Sansalvamide A derivatives and their cytotoxicity in the colon cancer cell line HT-29. Bioorg. Med. Chem. 2006, 14, 5625–5631. [Google Scholar] [CrossRef] [PubMed]
- Styers, T.J.; Rodriguez, R.A.; Pan, P.-S.; McAlpine, S.R. High-yielding macrocyclization conditions used in the synthesis of novel Sansalvamide A derivatives. Tetrahedron Lett. 2006, 47, 515–517. [Google Scholar] [CrossRef]
- Rodriguez, R.A.; Pan, P.-S.; Pan, C.-M.; Ravula, S.; Lapera, S.A.; Singh, E.K.; Styers, T.J.; Brown, J.D.; Cajica, J.; Parry, E.; et al. Synthesis of second generation Sansalvamide A derivatives: Novel templates as potent anti-tumor agents. J. Org. Chem. 2007, 72, 1980–2002. [Google Scholar] [CrossRef] [PubMed]
- Singh, E.K.; Ravula, S.; Pan, C.-M.; Pan, P.S.; Vasko, R.C.; Lapera, S.A.; Weerasinghe, S.V.W.; Pflum, M.K.H.; McAlpine, S.R. Synthesis and biological evaluation of histone deacetylase inhibitors that are based on FR235222: A cyclic tetrapeptide scaffold. Bioorg. Med. Chem. Lett. 2008, 18, 2549–2554. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.R.; Styers, T.J.; Rodriguez, R.A.; Pan, P.-S.; Vasko, R.C.; McAlpine, S.R. Synthesis and cytotoxicity of a new class of potent decapeptide macrocycles. Org. Lett. 2008, 10, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Singh, E.K.; Nazarova, L.A.; Lapera, S.A.; Alexander, L.D.; McAlpine, S.R. Histone deacetylace inhibitors: Synthesis of cyclic tetrapeptides and their triazole analogs. Tetrahedron Lett. 2010, 51, 4357–4360. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.R.; Singh, E.K.; Wahyudi, H.; Alexander, L.D.; Kunicki, J.; Nazarova, L.A.; Fairweather, K.A.; Giltrap, A.M.; Jolliffe, K.A.; McAlpine, S.R. Synthesis of Sansalvamide A peptiodmimetics: Trizaole, oxazole, thiazole, and pseudoproline containing compounds. Tetrahedron 2012, 68, 1029–1051. [Google Scholar] [CrossRef] [PubMed]
- Singh, E.; Ramsey, D.M.; McAlpine, S.R. Total synthesis of natural product trans,trans- Sanguinamide B and its structurally related conformational analogs. Org. Lett. 2012, 14, 1198–1201. [Google Scholar] [CrossRef] [PubMed]
- Alexander, L.D.; Sellers, R.P.; Davis, M.E.; Ardi, V.C.; Johnson, V.A.; Vasko, R.C.; McAlpine, S.R. Evaluation of Di-sansalvmide A derivatives: Synthesis, structure-activity relationship, and mechanism of action. J. Med. Chem. 2009, 52, 7927–7930. [Google Scholar] [CrossRef] [PubMed]
- Sellers, R.P.; Alexander, L.D.; Johnson, V.A.; Lin, C.-C.; Savage, J.; Corral, R.; Moss, J.; Slugocki, T.S.; Singh, E.K.; Davis, M.R.; et al. A third generation of Sansalvamide A derivatives: Design and synthesis of Hsp90 Inhibitors. Bioorg. Med. Chem. 2010, 18, 6822–6856. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, D.M.; McConnell, J.R.; Alexander, L.D.; Tanaka, K.W.; Vera, C.M.; Mcalpine, S.R. A new Hsp90 inhibitorthat exhibits a novel biological profile. Bioorg. Med. Chem. Lett. 2012, 22, 3287–3290. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.-M.; Lin, C.-C.; Kim, S.J.; Sellers, R.P.; McAlpine, S.R. Progress toward the synthesis of Urukthapelstatin A and two analogues. Tetrahedron Lett. 2012, 53, 4065–4069. [Google Scholar] [CrossRef] [PubMed]
- Joullié, M.M.; Lassen, K.M. Evolution of amide bond formation. ARKIVOC 2010, 8, 189–250. [Google Scholar]
- Barlos, K.; Gatos, D.; Koutsogianni, S. Fmoc/Trt-amino acids: Comparison to Fmoc/tBu-amino acids in peptide synthesis. J. Pept. Res. 1998, 51, 194–200. [Google Scholar] [CrossRef] [PubMed]
- All AA’s were commercially available from Chem-impex, polypeptide, and GL Biochem.
Sample Availability: Samples of the compounds are available from the authors. |
Figure 1.
Ustat A and related structures: merchercharmycin A (IB-01211) [25,26,27], ascidiacyclamide [20], patellamide D, and telomestatin [25,28].

Scheme 1.
Two synthetic approaches: (A) solution phase and (B) solid phase synthesis of Ustat A.

Scheme 2.
Attempted cyclization conditions for the formation of Ustat A.

Reagents and conditions: (a) O-(Benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TBTU, 0.5 equiv.), 2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate methanaminium (HATU, 0.7 equiv.), 3-(diethoxyphosphoryloxy)-3H-benzo[d][1,2,3] triazin-4-one (DEPBT, 0.5 equiv.), N,N-Diisopropylethylamine (DIPEA, 8 equiv.) in DCM (0.01 M); (b) Pentafluoropropanol (2 equiv.), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, 2.5 equiv.), DIPEA (6 equiv.) in DCM (0.7 mM); (c) FDPP (2 equiv.), HATU (0.6 equiv.), TBTU (0.6 equiv.), DIPEA (6 equiv.) in DCM (0.7 mM).
Scheme 3.
Synthesis of Ustat A using solid phase.

Reagents and conditions: (coupling) Fmoc-Xaa-OH (3 equiv.), HOBt (3 equiv.), DIC (6 equiv.), DMF; (deprotection) 20% piperidine in DMF; (cleavage) TFE/CH2Cl2 (1:1, v/v).
Scheme 4.
Cyclization of the linear precursor.

Reagents and conditions: (a) HATU (0.5 equiv.), TBTU (0.5 equiv.), ethyl 2-cyano-2-(hydroxyimino)acetate (COMU, 0.5 equiv.), 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl morpholinium chloride (DMTMM, 0.5 equiv.), DIPEA (8 equiv.) in CH2Cl2 (0.002 M); (b) TES/CH2Cl2 (1:1, v/v), 1.1% TFA in CH2Cl2 [54]; (c) DAST (3.5 equiv.), pyridine (3 equiv.), K2CO3 (1.5 equiv.) in CH2Cl2 (0.1 M), −70 °C; (d) DBU (6 equiv.), BrCCl3(6 equiv.) in CH2Cl2 (0.1 M), −40 °C.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Kim, S.J.; McAlpine, S.R. Solid Phase versus Solution Phase Synthesis of Heterocyclic Macrocycles. Molecules 2013, 18, 1111-1121. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules18011111
AMA Style
Kim SJ, McAlpine SR. Solid Phase versus Solution Phase Synthesis of Heterocyclic Macrocycles. Molecules. 2013; 18(1):1111-1121. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules18011111
Chicago/Turabian StyleKim, Seong Jong, and Shelli R. McAlpine. 2013. "Solid Phase versus Solution Phase Synthesis of Heterocyclic Macrocycles" Molecules 18, no. 1: 1111-1121. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules18011111