Exploration of Piperidinols as Potential Antitubercular Agents



 ,
,
Abstract
:
1. Introduction

2. Results and Discussion
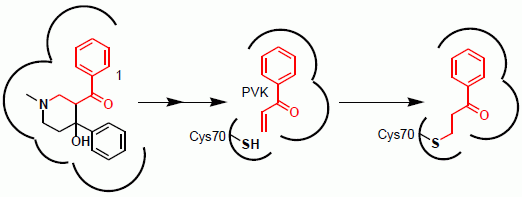
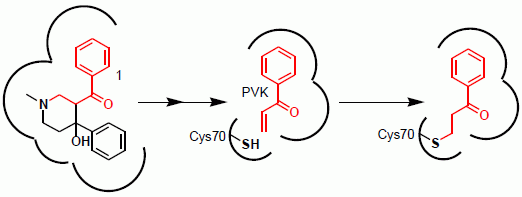
2.1. Mechanism of Inhibition



| Code | R1 | R2 | kobs (10−3 min−1) | t1/2 (min) | Critical Volume (cm3/mol) | cLogP |
|---|---|---|---|---|---|---|
| 1 | H | -CH3 | 9 ± 2 | 81.5 | 864.5 | 2.41 |
| 2 | Cl | -CH3 | 110 ± 2 | 6.3 | 962.5 | 3.53 |
| 3 | Br | -CH3 | 74 ± 7 | 9.4 | 988.5 | 4.07 |
| 4 | H | -CH2CH3 | 15 ± 1 | 45.6 | 920.5 | 2.75 |
| 5 | F | -CH2CH3 | 638 ± 120 | 1.1 | 956.5 | 3.07 |
| 6 | H | -(CH2)3CH3 | 104 ± 8 | 6.6 | 1032.5 | 3.66 |
| 7 | H |  | 573 ± 25 | 1.2 | 1077.5 | 3.96 |
| 8 | H |  | 10 ± 1 | 71.4 | 1092.5 | 4.15 |
| 9 | H |  | 163 ± 39 | 4.2 | 994.5 | 2.84 |
| 10 | H |  | 19 ± 1 | 37.1 | 1017.5 | 0.91 |
| 11 | H |  | 34 ± 1 | 20.2 | 1276.5 | 3.06 |
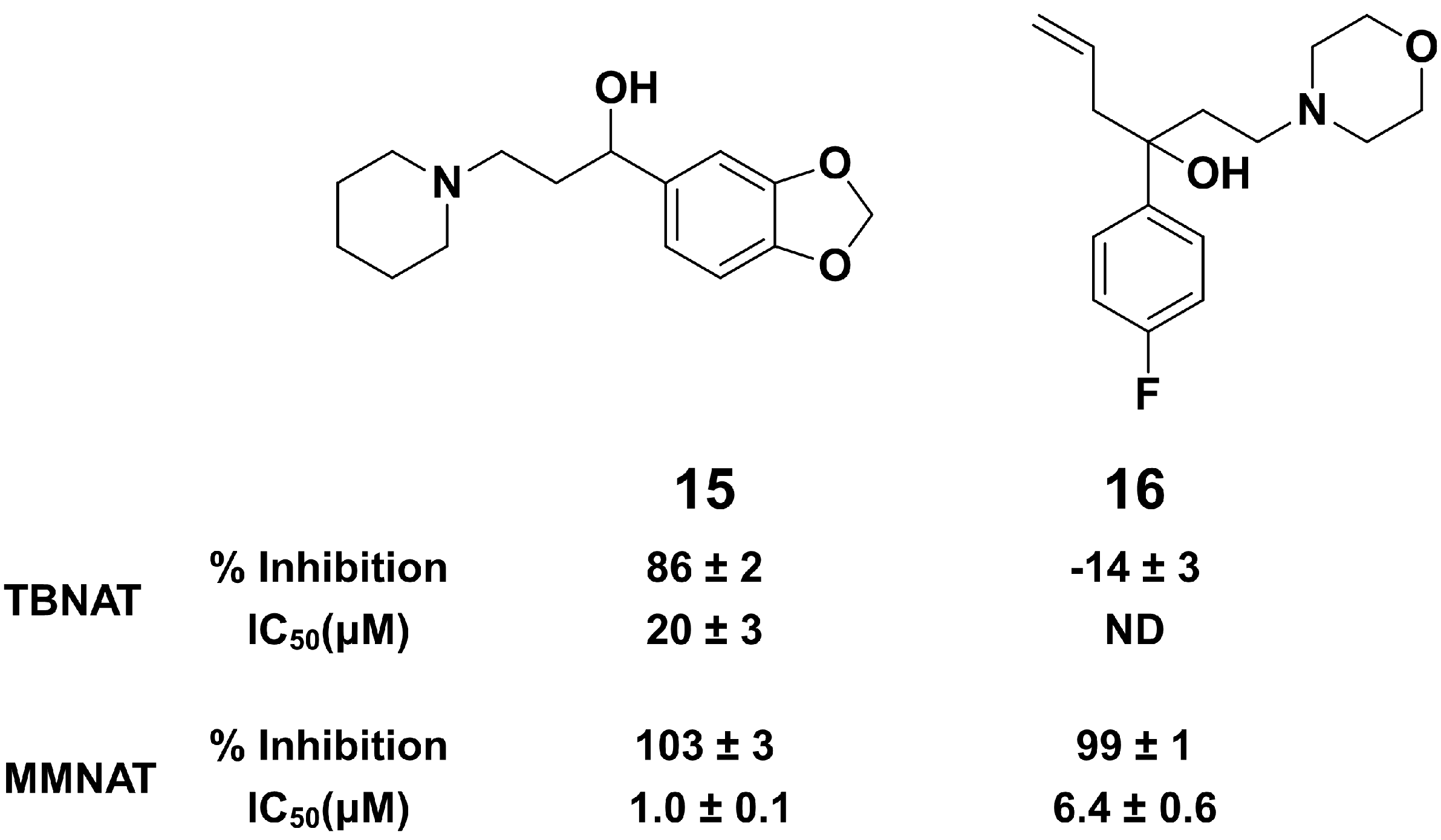
2.2. Effects of Substitutions on the Piperidinol Scaffold
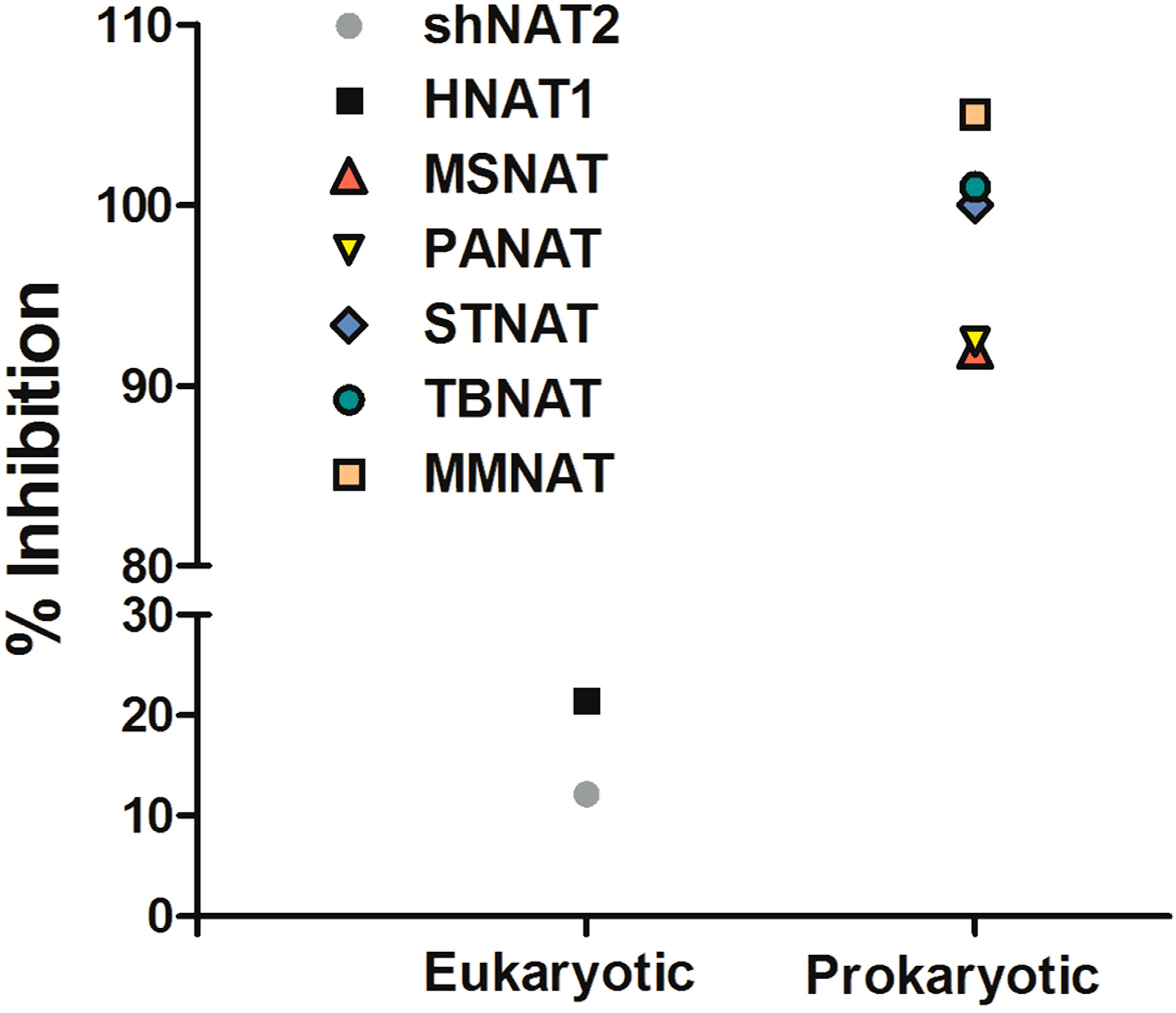
2.3. Comparison of MMNAT and TBNAT

2.4. Effect on Mycobacteria

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | R1 | R2 | TBNAT | MMNAT | MIC (μg/mL) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % Inhibition | IC50 (μM) HLZ | % Inhibition | IC50 (μM) HLZ | IC50 (μM) 5ASA | M. bovis BCG | M. tuberculosis | |||||||||||
| 1 | H | -CH3 | 101 ± 1 | 7.7 ± 0.9 | 105 ± 1 | 1.3 ± 0.0 | 6.0 ±1 | 6.3–12.5 | 1–10 | ||||||||
| 2 | Cl | -CH3 | 98 ± 1 | 1.6 ± 0.1 | 103 ± 2 | 0.16 ± 0.01 | 1.4 ± 0.6 | 6.3–12.5 | ND | ||||||||
| 3 | Br | -CH3 | 98 ± 3 | 2.9 ± 0.4 | 99 ± 1 | 0.08 ± 0.01 | 1.3 ± 0.4 | 6.3–12.5 | 5–10 | ||||||||
| 4 | H | -CH2CH3 | 72 ± 4 | 7.3 ± 0.3 | 126 ± 5 | 1.9 ± 0.0 | 8.0 ± 1 | 6.3–12.5 | 5–10 | ||||||||
| 5 | F | -CH2CH3 | 108 ± 1 | 4.4 ± 0.0 | 102 ± 3 | 0.5 ± 0.0 | 1.3 ± 0.5 | 6.3–12.5 | 5–10 | ||||||||
| 6 | H | -(CH2)3CH3 | 100 ± 3 | 6.9 ± 0.4 | 102 ± 3 | 2.6 ± 1 | 5.0 ± 0.4 | 6.3–12.5 | 1–5 | ||||||||
| 7 | H |  | 72 ± 60 | 4.4 ± 0.1 | 103 ± 1 | ND | 1.7 ± 0.2 | 6.3–12.5 | 1–5 | ||||||||
| 8 | H |  | 58 ± 2 | ND | 101 ± 1 | 4.1 ± 0.4 | ND | 6.3–12.5 | 0–1 | ||||||||
| 9 | H |  | 51 ± 3 | ND | 100.8 ± 0.5 | 2.5 ± 0.3 | 9.0 ± 0.9 | ND | 0–1 | ||||||||
| 10 | H |  | 47 ± 2 | ND | 99 ± 0.7 | 13 ± 1 | >30 | 3.1–6.3 | ND | ||||||||
| 11 | H |  | 67 ± 4 | 1.1 ± 0.3 | 100 ± 2 | 2.7 ± 0.4 | 1.1 ± 0.3 | 6.3–12.5 | 1–5 | ||||||||
2.5. In Silico Screening

3. Experimental Section
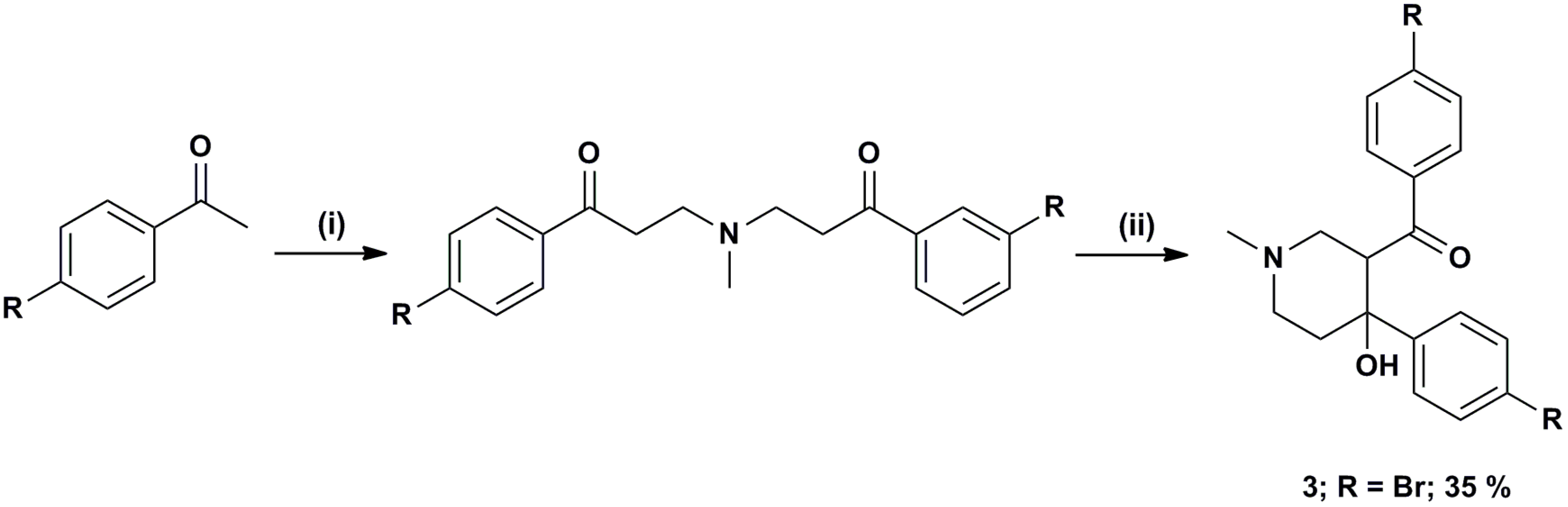
3.1. Range of Inhibitors

3.2. Enzyme Production
3.3. NAT Inhibition Assay
3.4. Mycobacterial Growth Inhibition in Vitro
3.5. Alamar Blue Assay
3.6. In Silico Shape-Based Screening
4. Conclusions
Supplementary Materials
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dye, C.; Williams, B.G. The population dynamics and control of tuberculosis. Science 2010, 328, 856–861. [Google Scholar] [CrossRef]
- WHO. Global Tuberculosis Report 2013. Available online: http://www.who.int/tb/publications/global_report/gtbr13_executive_summary.pdf?ua=1 (accessed on 24 September 2014).
- Sarkar, S.; Suresh, M.R. An overview of tuberculosis chemotherapy—A literature review. J. Pharm. Pharm. Sci. 2011, 14, 148–161. [Google Scholar]
- Shenoi, S.; Friedland, G. Extensively drug-resistant tuberculosis: A new face to an old pathogen. Annu. Rev. Med. 2009, 60, 307–320. [Google Scholar] [CrossRef]
- Udwadia, Z.F.; Amale, R.A.; Ajbani, K.K.; Rodrigues, C. Totally drug-resistant tuberculosis in India. Clin. Infect. Dis. 2012, 54, 579–581. [Google Scholar] [CrossRef]
- Rowland, K. Totally drug-resistant TB emerges in India: Discovery of a deadly form of TB highlights crisis of “mismanagement”. Nat. News 2012. [Google Scholar] [CrossRef]
- Andries, K.; Verhasselt, P.; Guillemont, J.; Gohlmann, H.W.; Neefs, J.M.; Winkler, H.; van Gestel, J.; Timmerman, P.; Zhu, M.; Lee, E.; et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 2005, 307, 223–227. [Google Scholar] [CrossRef]
- Diacon, A.H.; Pym, A.; Grobusch, M.; Patientia, R.; Rustomjee, R.; Page-Shipp, L.; Pistorius, C.; Krause, R.; Bogoshi, M.; Churchyard, G.; et al. The diarylquinoline TMC207 for multidrug-resistant tuberculosis. N. Engl. J. Med. 2009, 360, 2397–2405. [Google Scholar] [CrossRef]
- Osborne, R. First novel anti-tuberculosis drug in 40 years. Nat. Biotechnol. 2013, 31, 89–91. [Google Scholar] [CrossRef]
- Makarov, V.; Manina, G.; Mikusova, K.; Mollmann, U.; Ryabova, O.; Saint-Joanis, B.; Dhar, N.; Pasca, M.R.; Buroni, S.; Lucarelli, A.P.; et al. Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science 2009, 324, 801–804. [Google Scholar] [CrossRef]
- Kaneko, T.; Cooper, C.; Mdluli, K. Challenges and opportunities in developing novel drugs for TB. Future Med. Chem. 2011, 3, 1373–1400. [Google Scholar] [CrossRef]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef]
- Abuhammad, A.; Fullam, E.; Lowe, E.D.; Staunton, D.; Kawamura, A.; Westwood, I.M.; Bhakta, S.; Garner, A.C.; Wilson, D.L.; Seden, P.T.; et al. Piperidinols that show anti-tubercular activity as inhibitors of arylamine N-acetyltransferase: An essential enzyme for mycobacterial survival inside macrophages. PLoS One 2012, 7, e52790. [Google Scholar] [CrossRef]
- Bhakta, S.; Besra, G.S.; Upton, A.M.; Parish, T.; Sholto-Douglas-Vernon, C.; Gibson, K.J.; Knutton, S.; Gordon, S.; DaSilva, R.P.; Anderton, M.C.; et al. Arylamine N-acetyltransferase is required for synthesis of mycolic acids and complex lipids in Mycobacterium bovis BCG and represents a novel drug target. J. Exp. Med. 2004, 199, 1191–1199. [Google Scholar] [CrossRef]
- Anderton, M.C.; Bhakta, S.; Besra, G.S.; Jeavons, P.; Eltis, L.D.; Sim, E. Characterization of the putative operon containing arylamine N-acetyltransferase (nat) in Mycobacterium bovis BCG. Mol. Microbiol. 2006, 59, 181–192. [Google Scholar] [CrossRef]
- Yam, K.C.; D’Angelo, I.; Kalscheuer, R.; Zhu, H.; Wang, J.X.; Snieckus, V.; Ly, L.H.; Converse, P.J.; Jacobs, W.R., Jr.; Strynadka, N.; et al. Studies of a ring-cleaving dioxygenase illuminate the role of cholesterol metabolism in the pathogenesis of Mycobacterium tuberculosis. PLoS Pathog. 2009, 5, e1000344. [Google Scholar] [CrossRef]
- Suleyman, H.; Gul, H.I.; Asoglu, M. Anti-inflammatory activity of 3-benzoyl-1-methyl-4-phenyl-4-piperidinol hydrochloride. Pharmacol. Res. 2003, 47, 471–475. [Google Scholar] [CrossRef]
- Vashishtha, S.C.; Allen, T.M.; Halleran, S.; Szydlowski, J.; Santos, C.L.; De Clercq, E.; Balzarani, J.; Dimmock, J.R. Cytotoxic and anticancer properties of some 4-aryl-3-arylcarbonyl-1-ethyl-4-piperidinols and related compounds. Pharmazie 2001, 56, 390–393. [Google Scholar]
- Gul, H.I.; Calls, U.; Ozturk, Z.; Tutar, E.; Calikiran, L. Evaluation of anticonvulsant activities of bis(3-aryl-3-oxo-propyl) ethylamine hydrochlorides and 4-aryl-3-arylcarbonyl-1-ethyl-4-piperidinol hydrochlorides. Arzneim. Forsch. 2007, 57, 133–136. [Google Scholar]
- Gul, H.I.; Sahin, F.; Gul, M.; Ozturk, S.; Yerdelen, K.O. Evaluation of antimicrobial activities of several Mannich bases and their derivatives. Arch. Pharm. (Weinheim) 2005, 338, 335–338. [Google Scholar] [CrossRef]
- Jeney, E.; Zsolnai, T. Studies in search of new tuberculostatic drugs. I. Hydrazine derivatives, carbolic acid, phenols, quaternary ammonium compounds and their intermediaries. Zentralbl. Bakteriol. Orig. 1956, 167, 55–64. [Google Scholar]
- Sloan, K.B.; Koch, S.A.M.; Siver, K.G. Mannich base derivatives of theophylline and 5-fluorouracil: Syntheses, properties and topical delivery characteristics. Int. J. Pharm. 1984, 21, 251–264. [Google Scholar] [CrossRef]
- Brooke, E.W.; Davies, S.G.; Mulvaney, A.W.; Pompeo, F.; Sim, E.; Vickers, R.J. An approach to identifying novel substrates of bacterial arylamine N-acetyltransferases. Bioorg. Med. Chem. 2003, 11, 1227–1234. [Google Scholar] [CrossRef]
- Westwood, I.; Bhakta, S.; Russell, A.; Fullam, E.; Anderton, M.; Kawamura, A.; Mulvaney, A.; Vickers, R.; Bhowruth, V.; Besra, G.; et al. Identification of aryalmine N-acetyltransferase inhibitors as an approach towards novel anti-tuberculars. Protein Cell 2010, 1, 82–95. [Google Scholar] [CrossRef]
- Westwood, I.M.; Kawamura, A.; Russell, A.J.; Sandy, J.; Davies, S.G.; Sim, E. Novel small-molecule inhibitors of arylamine N-acetyltransferases: Drug discovery by high-throughput screening. Comb. Chem. High Throughput Screen. 2011, 14, 117–124. [Google Scholar] [CrossRef]
- Fullam, E. Arylamine N-Acetyltransferase of Mycobacteria. Ph.D. Thesis, Oxford University, Oxford, UK,, July 2007. [Google Scholar]
- Abuhammad, A.M.; Lowe, E.D.; Fullam, E.; Noble, M.; Garman, E.F.; Sim, E. Probing the architecture of the Mycobacterium marinum arylamine N-acetyltransferase active site. Protein Cell 2010, 1, 384–392. [Google Scholar] [CrossRef]
- Kitz, R.; Wilson, I.B. Esters of methanesulfonic acid as irreversible inhibitors of acetylcholinesterase. J. Biol. Chem. 1962, 237, 3245–3249. [Google Scholar]
- PerkinElmer. Available online: http://www.cambridgesoft.com (accessed on 8 September 2014).
- Fullam, E.; Kawamura, A.; Wilkinson, H.; Abuhammad, A.; Westwood, I.; Sim, E. Comparison of the arylamine N-acetyltransferase from Mycobacterium marinum and Mycobacterium tuberculosis. Protein J. 2009, 28, 281–293. [Google Scholar] [CrossRef]
- Abuhammad, A. Arylamine N-acetyltransferases from Mycobacteria: Investigations of a Potential Target for Anti-Tubercular Therapy. Ph.D. Thesis, University of Oxford, Oxford, UK, April 2013. [Google Scholar]
- Fullam, E.; Westwood, I.M.; Anderton, M.C.; Lowe, E.D.; Sim, E.; Noble, M.E. Divergence of cofactor recognition across evolution: Coenzyme a binding in a prokaryotic arylamine N-acetyltransferase. J. Mol. Biol. 2008, 375, 178–191. [Google Scholar] [CrossRef]
- Pluvinage, B.; Sierra-Gallay, I.L.; Kubiak, X.; Xu, X.; Dairou, J.; Dupret, J.M.; Rodrigues-Lima, F. The Bacillus anthracis arylamine N-acetyltransferase ((BACAN)NAT1) that inactivates sulfamethoxazole, reveals unusual structural features compared with the other NAT isoenzymes. FEBS Lett. 2011, 585, 3947–3952. [Google Scholar] [CrossRef]
- Abuhammad, A.; Lowe, E.D.; McDonough, M.A.; Stewart, P.D.S.; Kolek, S.A.; Sim, E.; Garman, E.F. Structure of arylamine N-acetyltransferase from M. tuberculosis determined by cross-seeding with homologous protein from M. marinum: Triumph over adversity. Acta Crystallogr. D 2013, in press. [Google Scholar]
- Ballester, P.J.; Westwood, I.; Laurieri, N.; Sim, E.; Richards, W.G. Prospective virtual screening with ultrafast shape recognition: The identification of novel inhibitors of arylamine N-acetyltransferases. J. R. Soc. Interface 2010, 7, 335–342. [Google Scholar] [CrossRef]
- Armstrong, M.S.; Finn, P.W.; Morris, G.M.; Richards, W.G. Improving the accuracy of ultrafast ligand-based screening: Incorporating lipophilicity into electroshape as an extra dimension. J. Comput. Aided Mol. Des. 2011, 25, 785–790. [Google Scholar] [CrossRef]
- Armstrong, M.S.; Morris, G.M.; Finn, P.W.; Sharma, R.; Moretti, L.; Cooper, R.I.; Richards, W.G. Electroshape: Fast molecular similarity calculations incorporating shape, chirality and electrostatics. J. Comput. Aided Mol. Des. 2010, 24, 789–801. [Google Scholar] [CrossRef]
- Armstrong, M.S.; Morris, G.M.; Finn, P.W.; Sharma, R.; Richards, W.G. Molecular similarity including chirality. J. Mol. Graph. Model. 2009, 28, 368–370. [Google Scholar] [CrossRef]
- Biovir. Available online: http://accelrys.com/products/discovery-studio/ (accessed on 8 September 2014).
- Cwik, A.; Fuchs, A.; Hell, Z.; Clacens, J.-M. An efficient and environmental-friendly synthesis of 4-hydroxy-arylpiperidines using hydrotalcite catalyst. J. Mol. Catal. A Chem. 2004, 219, 377–381. [Google Scholar] [CrossRef]
- Payton, M.; Auty, R.; Delgoda, R.; Everett, M.; Sim, E. Cloning and characterization of arylamine N-acetyltransferase genes from Mycobacterium smegmatis and Mycobacterium tuberculosis: Increased expression results in isoniazid resistance. J. Bacteriol. 1999, 181, 1343–1347. [Google Scholar]
- Sinclair, J.C.; Sandy, J.; Delgoda, R.; Sim, E.; Noble, M.E. Structure of arylamine N-acetyltransferase reveals a catalytic triad. Nat. Struct. Biol. 2000, 7, 560–564. [Google Scholar] [CrossRef]
- Westwood, I. Structure and Activity of Arylamine N-Acetyltransferase form Pseudomonas aeruginosa; Oxford University: Oxford, UK, 2005. [Google Scholar]
- Abuhammad, A.; Lack, N.; Schweichler, J.; Staunton, D.; Sim, R.B.; Sim, E. Improvement of the expression and purification of Mycobacterium tuberculosis arylamine N-acetyltransferase (TBNAT) a potential target for novel anti-tubercular agents. Protein Expr. Purif. 2011, 80, 246–252. [Google Scholar] [CrossRef]
- Kawamura, A.; Graham, J.; Mushtaq, A.; Tsiftsoglou, S.A.; Vath, G.M.; Hanna, P.E.; Wagner, C.R.; Sim, E. Eukaryotic arylamine N-acetyltransferase. Investigation of substrate specificity by high-throughput screening. Biochem. Pharmacol. 2005, 69, 347–359. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, C.; Marimuthu, A.; Krupka, H.I.; Tabrizizad, M.; Shelloe, R.; Mehra, U.; Eng, K.; Nguyen, H.; Settachatgul, C.; et al. The crystal structures of human steroidogenic factor-1 and liver receptor homologue-1. Proc. Natl. Acad. Sci. USA 2005, 102, 7505–7510. [Google Scholar] [CrossRef]
- Evangelopoulos, D.; Bhakta, S. Rapid methods for testing inhibitors of mycobacterial growth. In Antibiotic Resistance Protocols; Gillespie, S.H., McHugh, T.D., Eds.; Humana Press: New York, NY, USA, 2010; Volume 642, pp. 193–201. [Google Scholar]
- Russell, A.J.; Westwood, I.M.; Crawford, M.H.; Robinson, J.; Kawamura, A.; Redfield, C.; Laurieri, N.; Lowe, E.D.; Davies, S.G.; Sim, E. Selective small molecule inhibitors of the potential breast cancer marker, human arylamine N-acetyltransferase 1, and its murine homologue, mouse arylamine N-acetyltransferase 2. Bioorg. Med. Chem. 2009, 17, 905–918. [Google Scholar] [CrossRef]
- Yajko, D.; Madej, J.; Lancaster, M.; Sanders, C.; Cawthon, V.; Gee, B.; Babst, A.; Hadley, W. Colorimetric method for determining mics of antimicrobial agents for Mycobacterium tuberculosis. J. Clin. Microbiol. 1995, 33, 2324–2327. [Google Scholar]
- Franzblau, S.G.; Witzig, R.S.; McLaughlin, J.C.; Torres, P.; Madico, G.; Hernandez, A.; Degnan, M.T.; Cook, M.B.; Quenzer, V.K.; Ferguson, R.M.; et al. Rapid, low-technology mic determination with clinical Mycobacterium tuberculosis isolates by using the microplate alamar blue assay. J. Clin. Microbiol. 1998, 36, 362–366. [Google Scholar]
- Ebejer, J.-P.; Morris, G.M.; Deane, C.M. Freely available conformer generation methods: How good are they? J. Chem. Inf. Model. 2012, 52, 1146–1158. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abuhammad, A.; Fullam, E.; Bhakta, S.; Russell, A.J.; Morris, G.M.; Finn, P.W.; Sim, E. Exploration of Piperidinols as Potential Antitubercular Agents. Molecules 2014, 19, 16274-16290. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules191016274
Abuhammad A, Fullam E, Bhakta S, Russell AJ, Morris GM, Finn PW, Sim E. Exploration of Piperidinols as Potential Antitubercular Agents. Molecules. 2014; 19(10):16274-16290. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules191016274
Chicago/Turabian StyleAbuhammad, Areej, Elizabeth Fullam, Sanjib Bhakta, Angela J. Russell, Garrett M. Morris, Paul W. Finn, and Edith Sim. 2014. "Exploration of Piperidinols as Potential Antitubercular Agents" Molecules 19, no. 10: 16274-16290. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules191016274