Modern Prodrug Design for Targeted Oral Drug Delivery


{kind=link}
{kind=link}
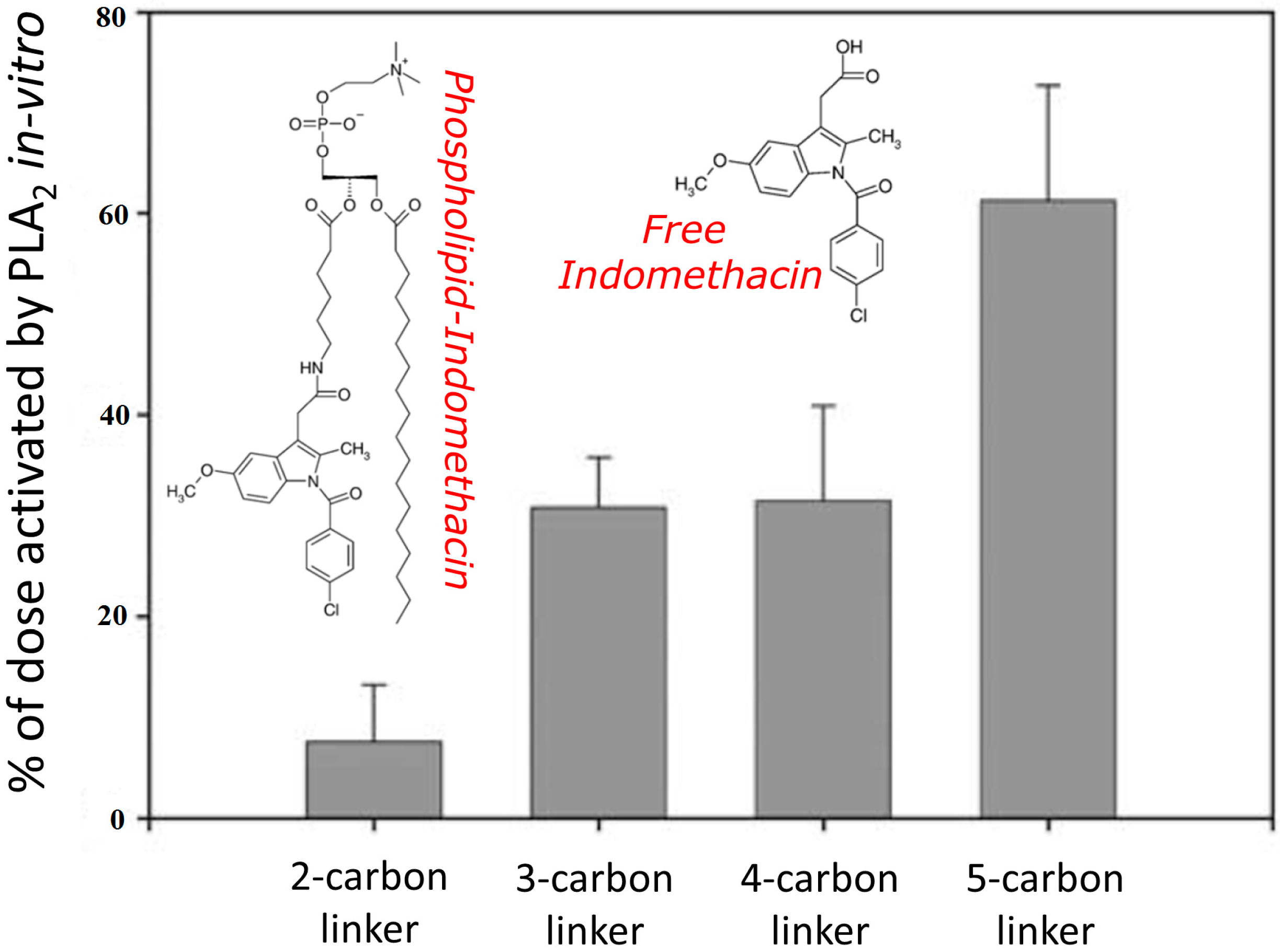{kind=link}
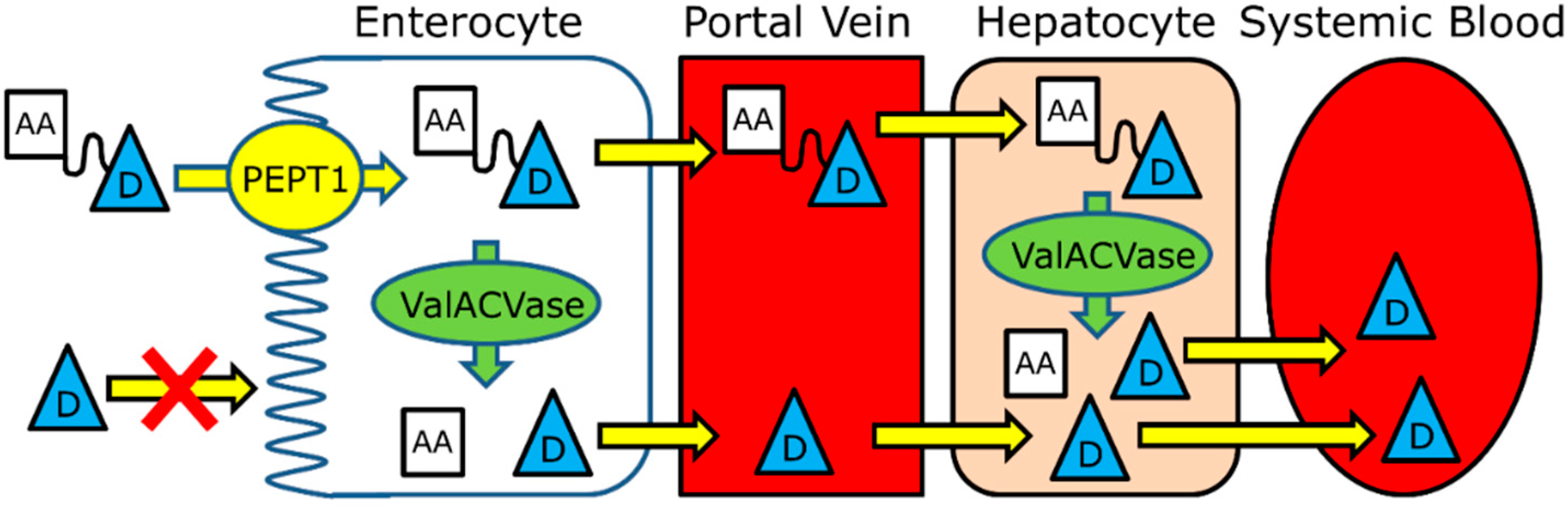{kind=link}
Abstract
:1. Introduction
2. The Classical Prodrug Approach
3. The Modern Prodrug Approach
3.1. Targeting Transporters in Prodrug Design


3.2. Targeting Enzymes in Prodrug Design


4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bildstein, L.; Dubernet, C.; Couvreur, P. Prodrug-based intracellular delivery of anticancer agents. Adv. Drug Deliv. Rev. 2011, 63, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Huttunen, K.M.; Raunio, H.; Rautio, J. Prodrugs—From serendipity to rational design. Pharmacol. Rev. 2011, 63, 750–771. [Google Scholar] [CrossRef] [PubMed]
- Stella, V.J. Prodrugs: Some thoughts and current issues. J. Pharm. Sci. 2010, 99, 4755–4765. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, K.; Webster, R.; Gardner, I.; Dack, K. Design of ester prodrugs to enhance oral absorption of poorly permeable compounds: Challenges to the discovery scientist. Curr. Drug Metab. 2003, 4, 461–485. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Khamis, M.; Agbaria, R.; Karaman, R. Targeted prodrugs in oral drug delivery: The modern molecular biopharmaceutical approach. Expert Opin. Drug Deliv. 2012, 9, 1001–1013. [Google Scholar] [CrossRef] [PubMed]
- Han, H.K.; Amidon, G.L. Targeted prodrug design to optimize drug delivery. AAPS PharmSci. 2000, 2, 48–58. [Google Scholar] [CrossRef]
- Sai, Y.; Tsuji, A. Transporter-mediated drug delivery: Recent progress and experimental approaches. Drug Discov. Today 2004, 9, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Amidon, G.L.; Leesman, G.D.; Elliott, R.L. Improving intestinal absorption of water-insoluble compounds: A membrane metabolism strategy. J. Pharm. Sci. 1980, 69, 1363–1368. [Google Scholar] [CrossRef] [PubMed]
- Fleisher, D.; Bong, R.; Stewart, B.H. Improved oral drug delivery: Solubility limitations overcome by the use of prodrugs. Adv. Drug Deliv. Rev. 1996, 19, 115–130. [Google Scholar] [CrossRef]
- Stella, V.J.; Nti-Addae, K.W. Prodrug strategies to overcome poor water solubility. Adv. Drug Deliv. Rev. 2007, 59, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Bernard, T. Prodrugs: Bridging pharmacodynamic/pharmacokinetic gaps. Curr. Opin. Chem. Biol. 2009, 13, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Duvdevani, R.; Dvir, E.; Elmann, A.; Hoffman, A. A novel mechanism for oral controlled release of drugs by continuous degradation of a phospholipid prodrug along the intestine: In-vivo and in vitro evaluation of an indomethacin—Lecithin conjugate. J. Control. Release 2007, 119, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Duvdevani, R.; Shapiro, I.; Elmann, A.; Finkelstein, E.; Hoffman, A. The oral absorption of phospholipid prodrugs: In vivo and in vitro mechanistic investigation of trafficking of a lecithin-valproic acid conjugate following oral administration. J. Control. Release 2008, 126, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ettmayer, P.; Amidon, G.L.; Clement, B.; Testa, B. Lessons learned from marketed and investigational prodrugs. J. Med. Chem. 2004, 47, 2393–2404. [Google Scholar] [CrossRef] [PubMed]
- Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D.; Jarvinen, T.; Savolainen, J. Prodrugs: Design and clinical applications. Nat. Rev. Drug Discov. 2008, 7, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Zawilska, J.; Wojcieszak, J.; Olejniczak, A. Prodrugs: A challenge for the drug development. Pharmacol. Rep. 2013, 65, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Serafin, A.; Stańczak, A. Different concepts of drug delivery in disease entities. Mini Rev. Med. Chem. 2009, 9, 481–497. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.; Daniel, K.B.; Cohen, S.M. Evaluating prodrug strategies for esterase-triggered release of alcohols. ChemMedChem 2013, 8, 1662–1667. [Google Scholar] [CrossRef]
- Horst, H.; Höltje, W.; Dennis, M.; Coert, A.; Geelen, J.; Voigt, K. Lymphatic absorption and metabolism of orally administered testosterone undecanoate in man. Klin Wochenschr. 1976, 54, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Hoffman, A. Evaluation of a chylomicron flow blocking approach to investigate the intestinal lymphatic transport of lipophilic drugs. Eur. J. Pharm. Sci. 2005, 24, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Hoffman, A. Use of a dynamic in vitro lipolysis model to rationalize oral formulation development for poor water soluble drugs: Correlation with in vivo data and the relationship to intra-enterocyte processes in rats. Pharm. Res. 2006, 23, 2165–2174. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Mendelman, A.; Amsili, S.; Ezov, N.; Hoffman, A. The effect of general anesthesia on the intestinal lymphatic transport of lipophilic drugs: Comparison between anesthetized and freely moving conscious rat models. Eur. J. Pharm. Sci. 2007, 32, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Shackleford, D.M.; Faassen, W.A.; Houwing, N.; Lass, H.; Edwards, G.A.; Porter, C.J.H.; Charman, W.N. Contribution of lymphatically transported testosterone undecanoate to the systemic exposure of testosterone after oral administration of two andriol formulations in conscious lymph duct-cannulated dogs. J. Pharm. Exp. Ther. 2003, 306, 925–933. [Google Scholar] [CrossRef]
- Ring, J.; Kowalzick, L.; Christophers, E.; Schill, W.B.; Schöpf, E.; Ständer, M.; Wolff, H.H.; Altmeyer, P. Calcitriol 3 μg·g−1 ointment in combination with ultraviolet B phototherapy for the treatment of plaque psoriasis: Results of a comparative study. Br. J. Dermatol. 2001, 144, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shabat, S.; Benisty, R.; Wormser, U.; Sintov, A.C. Vitamin D3-based conjugates for topical treatment of psoriasis: Synthesis, antiproliferative activity, and cutaneous penetration studies. Pharm. Res. 2005, 22, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Sintov, A.C.; Yarmolinsky, L.; Dahan, A.; Ben-Shabat, S. Pharmacological effects of vitamin D and its analogs: Recent developments. Drug Discov. Today 2014, in press. [Google Scholar]
- Upadhyay, R.K. Drug delivery systems, CNS protection, and the blood brain barrier. BioMed. Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Qu, B.; Wu, X.; Li, X.; Liu, Q.; Jin, X.; Guo, L.; Hai, L.; Wu, Y. Design, synthesis and biological evaluation of brain targeting l-ascorbic acid prodrugs of ibuprofen with “lock-in” function. Eur. J. Med. Chem. 2014, 82, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, S.; Duvvuri, S.; Mitra, A.K. Membrane transporter/receptor-targeted prodrug design: Strategies for human and veterinary drug development. Adv. Drug Deliv. Rev. 2004, 56, 1437–1452. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Amidon, G.L.; Zimmermann, E.M. Drug targeting strategies for the treatment of inflammatory bowel disease: A mechanistic update. Expert Rev. Clin. Immunol. 2010, 6, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Sabit, H.; Dahan, A.; Sun, J.; Provoda, C.J.; Lee, K.-D.; Hilfinger, J.H.; Amidon, G.L. Cytomegalovirus protease targeted prodrug development. Mol. Pharm. 2013, 10, 1417–1424. [Google Scholar] [CrossRef] [PubMed]
- Wolk, O.; Epstein, S.; Ioffe-Dahan, V.; Ben-Shabat, S.; Dahan, A. New targeting strategies in drug therapy of inflammatory bowel disease: Mechanistic approaches and opportunities. Expert Opin. Drug Deliv. 2013, 10, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Shirasaka, Y.; Kuraoka, E.; Kikuchi, A.; Iguchi, M.; Suzuki, H.; Shibasaki, S.; Kurosawa, T.; Tamai, I. Intestinal absorption mechanism of tebipenem pivoxil, a novel oral carbapenem: Involvement of human OATP family in apical membrane transport. Mol. Pharm. 2010, 7, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Tamai, I. Oral drug delivery utilizing intestinal OATP transporters. Adv. Drug Deliv. Rev. 2012, 64, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Jonker, J.W.; Schinkel, A.H. Pharmacological and physiological functions of the polyspecific organic cation transporters: OCT1, 2, and 3 (SLC22A1–3). J. Pharmacol. Exp. Ther. 2004, 308, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, H.; Lips, K.; Volk, C. Polyspecific organic cation transporters: Structure, function, physiological roles, and biopharmaceutical implications. Pharm. Res. 2007, 24, 1227–1251. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, A.; Polli, J.E. Apical sodium dependent bile acid transporter (ASBT, SLC10A2): A potential prodrug target. Mol. Pharm. 2006, 3, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Kolhatkar, V.; Polli, J.E. Structural requirements of bile acid transporters: C-3 and C-7 modifications of steroidal hydroxyl groups. Eur. J. Pharm. Sci. 2012, 46, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Polli, J.E. Synthesis and in vitro evaluation of potential sustained release prodrugs via targeting ASBT. Int. J. Pharm. 2010, 396, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Gibbs, S.; Fang, L.; Miller, H.; Landowski, C.; Shin, H.-C.; Lennernas, H.; Zhong, Y.; Amidon, G.; Yu, L.; et al. Why is it challenging to predict intestinal drug absorption and oral bioavailability in human using rat model. Pharm. Res. 2006, 23, 1675–1686. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, A. Tissue selective drug delivery utilizing carrier-mediated transport systems. J. Control. Release 1999, 62, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Varma, M.; Ambler, C.; Ullah, M.; Rotter, C.; Sun, H.; Litchfield, J.; Fenner, K.; El-Kattan, A. Targeting intestinal transporters for optimizing oral drug absorption. Curr. Drug Metab. 2010, 11, 730–742. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.M.H.; Grenade, D.S.; Boll, M.; Foltz, M.; Wake, K.A.; Kennedy, D.J.; Munck, L.K.; Miyauchi, S.; Taylor, P.M.; Campbell, F.C.; et al. Thwaites, H+/amino acid transporter 1 (PAT1) is the imino acid carrier: An intestinal nutrient/drug transporter in human and rat. Gastroenterology 2004, 127, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Thwaites, D.T.; Anderson, C.M.H. The SLC36 family of proton-coupled amino acid transporters and their potential role in drug transport. Br. J. Pharmacol. 2011, 164, 1802–1816. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, M.E. Ganapathy, Amino acid transporter ATB0,+ as a delivery system for drugs and prodrugs. Curr. Drug Targets Immune Endocr. Metabol. Disord. 2005, 5, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, T.; Haramura, M.; Fei, Y.-J.; Miyauchi, S.; Bridges, C.C.; Ganapathy, P.S.; Smith, S.B.; Ganapathy, V.; Ganapathy, M.E. Transport of amino acid-based prodrugs by the Na+- and Cl—Coupled amino acid transporter ATB0,+ and expression of the transporter in tissues amenable for drug delivery. J. Pharmacol. Exp. Ther. 2004, 308, 1138–1147. [Google Scholar] [CrossRef] [PubMed]
- Qiu, A.; Jansen, M.; Sakaris, A.; Min, S.H.; Chattopadhyay, S.; Tsai, E.; Sandoval, C.; Zhao, R.; Akabas, M.H.; Goldman, I.D. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell 2006, 127, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Diop-Bove, N.; Visentin, M.; Goldman, I.D. Mechanisms of membrane transport of folates into cells and across epithelia. Ann. Rev. Nutr. 2011, 31, 177–201. [Google Scholar] [CrossRef]
- Bai, J.P.F.; Amidon, G.L. Structural specificity of mucosal-cell transport and metabolism of peptide drugs: Implication for oral peptide drug delivery. Pharm. Res. 1992, 9, 969–978. [Google Scholar] [CrossRef]
- Brandsch, M.; Knütter, I.; Bosse-Doenecke, E. Pharmaceutical and pharmacological importance of peptide transporters. J. Pharm. Pharmacol. 2008, 60, 543–585. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, A.; Tomoyasu, T.; Tanaka, M.; Kanamitsu, K.; Sasabe, H.; Maeda, T.; Odomi, M.; Tamai, I. Peptide derivation of poorly absorbable drug allows intestinal absorption via peptide transporter. J. Pharm. Sci. 2009, 98, 1775–1787. [Google Scholar] [CrossRef]
- Lee, V.H.L.; Chu, C.; Mahlin, E.D.; Basu, S.K.; Ann, D.K.; Bolger, M.B.; Haworth, I.S.; Yeung, A.K.; Wu, S.K.; Hamm-Alvarez, S.; et al. Biopharmaceutics of transmucosal peptide and protein drug administration: Role of transport mechanisms with a focus on the involvement of PepT1. J. Control. Release 1999, 62, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Smith, D.E. Significance of peptide transporter 1 in the intestinal permeability of valacyclovir in wild-type and PepT1 knockout mice. Drug Metab. Dispos. 2013, 41, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, K.; Huang, S.; Tweedie, D.; Benet, L.; Brouwer, K.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.N.; Amidon, G.L. A mechanistic approach to understanding the factors affecting drug absorption: A review of fundamentals. J. Clin. Pharmacol. 2002, 42, 620–643. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, N.; Niwa, T.; Yotsumoto, Y.; Sugiyama, Y. Impact of drug transporter studies on drug discovery and development. Pharmacol. Rev. 2003, 55, 425–461. [Google Scholar] [CrossRef] [PubMed]
- Shugarts, S.; Benet, L. The role of transporters in the pharmacokinetics of orally administered drugs. Pharm. Res. 2009, 26, 2039–2054. [Google Scholar] [CrossRef] [PubMed]
- Posada, M.; Smith, D. Relevance of PepT1 in the intestinal permeability and oral absorption of cefadroxil. Pharm. Res. 2013, 30, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Hu, Y.; Smith, D.E. Impact of peptide transporter 1 on the intestinal absorption and pharmacokinetics of valacyclovir after oral dose escalation in wild-type and PepT1 knockout mice. Drug Metab. Dispos. 2013, 41, 1867–1874. [Google Scholar] [CrossRef] [PubMed]
- Jappar, D.; Hu, Y.; Smith, D.E. Effect of dose escalation on the in vivo oral absorption and disposition of glycylsarcosine in wild-type and pept1 knockout mice. Drug Metab. Dispos. 2011, 39, 2250–2257. [Google Scholar] [CrossRef] [PubMed]
- Jappar, D.; Wu, S.-P.; Hu, Y.; Smith, D.E. Significance and regional dependency of peptide transporter (PEPT) 1 in the intestinal permeability of glycylsarcosine: In situ single-pass perfusion studies in wild-type and Pept1 knockout mice. Drug Metab. Dispos. 2010, 38, 1740–1746. [Google Scholar] [CrossRef] [PubMed]
- Aoki, F.Y.; Doucette, K.E. Oseltamivir: A clinical and pharmacological perspective. Expert Opin. Pharmacother. 2001, 2, 1671–1683. [Google Scholar] [CrossRef] [PubMed]
- Le, Q.M.; Kiso, M.; Someya, K.; Sakai, Y.T.; Nguyen, T.H.; Nguyen, K.H.L.; Pham, N.D.; Ngyen, H.H.; Yamada, S.; Muramoto, Y.; et al. Avian flu: Isolation of drug-resistant H5N1 virus. Nature 2005, 437, 1108. [Google Scholar] [CrossRef] [PubMed]
- Cass, L.; Efthymiopoulos, C.; Bye, A. Pharmacokinetics of zanamivir after intravenous, oral, inhaled or intranasal administration to healthy volunteers. Clin. Pharmacokinet. 1999, 36, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.V.; Gupta, D.; Sun, J.; Dahan, A.; Tsume, Y.; Hilfinger, J.; Lee, K.-D.; Amidon, G.L. Enhancing the intestinal membrane permeability of zanamivir: A carrier mediated prodrug approach. Mol. Pharm. 2011, 8, 2358–2367. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.M.; Dahan, A.; Gupta, D.; Varghese, S.; Amidon, G.L. Enabling the intestinal absorption of highly polar antiviral agents: Ion-pair facilitated membrane permeation of zanamivir heptyl ester and guanidino oseltamivir. Mol. Pharm. 2010, 7, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Dawood, F.; Jain, S.; Finelli, L.; Shaw, M.; Lindstrom, S.; Garten, R.; Gubareva, L.; Xu, X.; Bridges, C.; Uyeki, T. Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N. Engl. J. Med. 2009, 360, 2605–2615. [Google Scholar] [CrossRef] [PubMed]
- Landowski, C.P.; Vig, B.S.; Song, X.; Amidon, G.L. Targeted delivery to PEPT1-overexpressing cells: Acidic, basic, and secondary floxuridine amino acid ester prodrugs. Mol. Cancer Ther. 2005, 4, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Tsume, Y.; Hilfinger, J.; Amidon, G. Potential of amino acid/dipeptide monoester prodrugs of floxuridine in facilitating enhanced delivery of active drug to interior sites of tumors: A two-tier monolayer in vitro study. Pharm. Res. 2011, 28, 2575–2588. [Google Scholar] [CrossRef] [PubMed]
- Tsume, Y.; Hilfinger, J.M.; Amidon, G.L. Enhanced cancer cell growth inhibition by dipeptide prodrugs of floxuridine: Increased transporter affinity and metabolic stability. Mol. Pharm. 2008, 5, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Tsume, Y.; Vig, B.; Sun, J.; Landowski, C.; Hilfinger, J.; Ramachandran, C.; Amidon, G. Enhanced absorption and growth inhibition with amino acid monoester prodrugs of floxuridine by targeting hPEPT1 transporters. Molecules 2008, 13, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Han, H.-k.; de Vrueh, R.L.A.; Rhie, J.K.; Covitz, K.-M.Y.; Smith, P.L.; Lee, C.-P.; Oh, D.-M.; Sadee, W.; Amidon, G.L. 5'-Amino acid Esters of antiviral nucleosides, acyclovir, and AZT are absorbed by the Intestinal PEPT1 peptide transporter. Pharm. Res. 1998, 15, 1154–1159. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Maag, H.; Alfredson, T. Prodrugs of nucleoside analogues for improved oral absorption and tissue targeting. J. Pharm. Sci. 2008, 97, 1109–1134. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Song, X.; Vig, B.S.; Landowski, C.P.; Kim, I.; Hilfinger, J.M.; Amidon, G.L. Prolidase, a potential enzyme target for melanoma: Design of proline-containing dipeptide-like prodrugs. Mol. Pharm. 2005, 2, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Lorenzi, P.L.; Landowski, C.P.; Vig, B.S.; Hilfinger, J.M.; Amidon, G.L. Amino acid ester prodrugs of the anticancer agent gemcitabine: Synthesis, bioconversion, metabolic bioevasion, and hPEPT1-mediated transport. Mol. Pharm. 2005, 2, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Tsume, Y.; Bermejo, B.B.; Amidon, G. The dipeptide monoester prodrugs of floxuridine and gemcitabine-feasibility of orally administrable nucleoside analogs. Pharmaceuticals 2014, 7, 169–191. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, M.; Terada, T.; Irie, M.; Katsura, T.; Niida, A.; Tomita, K.; Fujii, N.; Inui, K.-I. Transport characteristics of a novel peptide transporter 1 substrate, antihypotensive drug midodrine, and its amino acid derivatives. J. Pharmacol. Exp. Ther. 2006, 318, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Ezra, A.; Golomb, G. Administration routes and delivery systems of bisphosphonates for the treatment of bone resorption. Adv. Drug Deliv. Rev. 2000, 42, 175–195. [Google Scholar] [CrossRef] [PubMed]
- Ezra, A.; Hoffman, A.; Breuer, E.; Alferiev, I.S.; Mönkkönen, J.; el Hanany-Rozen, N.; Weiss, G.; Stepensky, D.; Gati, I.; Cohen, H.; et al. A peptide prodrug approach for improving bisphosphonate oral absorption. J. Med. Chem. 2000, 43, 3641–3652. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Gupta, S.V.; Dahan, A.; Tsume, Y.; Hilfinger, J.; Lee, K.-D.; Amidon, G.L. Increasing oral absorption of polar neuraminidase inhibitors: A prodrug transporter approach applied to oseltamivir analogue. Mol. Pharm. 2013, 10, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.; Meredith, D. The SLC16 gene family—From monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflüg. Archiv. Eur. J. Physiol. 2004, 447, 619–628. [Google Scholar] [CrossRef]
- Cundy, K.C.; Annamalai, T.; Bu, L.; de Vera, J.; Estrela, J.; Luo, W.; Shirsat, P.; Torneros, A.; Yao, F.; Zou, J.; et al. XP13512, a novel gabapentin prodrug: II. Improved oral bioavailability, dose proportionality, and colonic absorption compared with gabapentin in rats and monkeys. J. Pharmacol. Exp. Ther. 2004, 311, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Cundy, K.C.; Sastry, S.; Luo, W.; Zou, J.; Moors, T.L.; Canafax, D.M. Clinical pharmacokinetics of XP13512, a novel transported prodrug of gabapentin. J. Clin. Pharmacol. 2008, 48, 1378–1388. [Google Scholar] [CrossRef] [PubMed]
- Cundy, K.C.; Branch, R.; Chernov-Rogan, T.; Dias, T.; Estrada, T.; Hold, K.; Koller, K.; Liu, X.; Mann, A.; Panuwat, M.; et al. XP13512, a novel gabapentin prodrug: I. Design, synthesis, enzymatic conversion to gabapentin, and transport by intestinal solute transporters. J. Pharmacol. Exp. Ther. 2004, 311, 315–323. [Google Scholar] [CrossRef] [PubMed]
- González, P.M.; Lagos, C.F.; Ward, W.C.; Polli, J.E. Structural requirements of the human sodium-dependent bile acid transporter (hASBT): Role of 3- and 7-OH moieties on binding and translocation of bile acids. Mol. Pharm. 2013, 11, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Tolle-Sander, S.; Lentz, K.A.; Maeda, D.Y.; Coop, A.; Polli, J.E. Increased acyclovir oral bioavailability via a bile acid conjugate. Mol. Pharm. 2004, 1, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Rais, R.; Fletcher, S.; Polli, J.E. Synthesis and in vitro evaluation of gabapentin prodrugs that target the human apical sodium-dependent bile acid transporter (hASBT). J. Pharm. Sci. 2011, 100, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Amidon, G.L.; Lennernas, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Lennernäs, H.; Amidon, G.L. The fraction dose absorbed, in humans, and high jejunal human permeability relationship. Mol. Pharm. 2012, 9, 1847–1851. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Miller, J.M.; Amidon, G.L. Prediction of solubility and permeability class membership: Provisional BCS classification of the world’s top oral drugs. AAPS J. 2009, 11, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Miller, J.M.; Hilfinger, J.M.; Yamashita, S.; Yu, L.X.; Lennernas, H.; Amidon, G.L. High-permeability criterion for BCS classification: Segmental/pH dependent permeability considerations. Mol. Pharm. 2010, 7, 1827–1834. [Google Scholar] [CrossRef] [PubMed]
- Fairstein, M.; Swissa, R.; Dahan, A. Regional-dependent intestinal permeability and BCS classification: Elucidation of pH-related complexity in rats using pseudoephedrine. AAPS J. 2013, 15, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Zur, M.; Gasparini, M.; Wolk, O.; Amidon, G.L.; Dahan, A. The low/high BCS permeability class boundary: Physicochemical comparison of metoprolol and labetalol. Mol. Pharm. 2014, 11, 1707–1714. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Wolk, O.; Kim, Y.H.; Ramachandran, C.; Crippen, G.M.; Takagi, T.; Bermejo, M.; Amidon, G.L. Purely in silico BCS classification: Science based quality standards for the world’s drugs. Mol. Pharm. 2013, 10, 4378–4390. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Yu, L.; Hussain, M.; Wall, D.; Smith, R.; Amidon, G. In vitro testing of drug absorption for drug “developability” assessment: Forming an interface between in vitro preclinical data and clinical outcome. Curr. Opin. Drug Discov. Dev. 2004, 7, 75–85. [Google Scholar]
- Patel, M.; Mandava, N.; Gokulgandhi, M.; Pal, D.; Mitra, A. Amino acid prodrugs: An approach to improve the absorption of HIV-1 protease inhibitor, lopinavir. Pharmaceuticals (Basel) 2014, 7, 433–452. [Google Scholar] [CrossRef]
- Landowski, C.P.; Lorenzi, P.L.; Song, X.; Amidon, G.L. Nucleoside ester prodrug substrate specificity of liver carboxylesterase. J. Pharmacol. Exp. Ther. 2006, 316, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Liederer, B.M.; Borchardt, R.T. Enzymes involved in the bioconversion of ester-based prodrugs. J. Pharm. Sci. 2006, 95, 1177–1195. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Hosokawa, M. The mammalian carboxylesterases: From molecules to functions. Ann. Rev. Pharmacol. Toxicol. 1998, 38, 257–288. [Google Scholar] [CrossRef]
- Weller, S.; Blum, M.; Doucette, M.; Burnette, T.; Cederberg, D.; de Miranda, P.; Smiley, M. Pharmacokinetics of the acyclovir pro-drug valaciclovir after escalating single- and multiple-dose administration to normal volunteers. Clin. Pharmacol. Ther. 1993, 54, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Balimane, P.V.; Tamai, I.; Guo, A.; Nakanishi, T.; Kitada, H.; Leibach, F.H.; Tsuji, A.; Sinko, P.J. Direct evidence for peptide transporter (PepT1)-mediated uptake of a nonpeptide prodrug, valacyclovir. Biochem. Biophys. Res. Commun. 1998, 250, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Burnette, T.C.; de Miranda, P. Metabolic disposition of the acyclovir prodrug valaciclovir in the rat. Drug Metab. Dispos. 1994, 22, 60–64. [Google Scholar] [PubMed]
- De Miranda, P.; Burnette, T.C. Metabolic fate and pharmacokinetics of the acyclovir prodrug valaciclovir in cynomolgus monkeys. Drug Metab. Dispos. 1994, 22, 55–59. [Google Scholar] [PubMed]
- Soul-Lawton, J.; Seaber, E.; On, N.; Wootton, R.; Rolan, P.; Posner, J. Absolute bioavailability and metabolic disposition of valaciclovir, the L-valyl ester of acyclovir, following oral administration to humans. Antimicrob. Agents Chemother. 1995, 39, 2759–2764. [Google Scholar] [CrossRef] [PubMed]
- Sinko, P.J.; Balimane, P.V. Carrier-mediated intestinal absorption of valacyclovir, the L-valyl ester prodrug of acyclovir. 1. Interactions with peptides, organic anions and organic cations in rats. Biopharm. Drug Dispos. 1998, 19, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Burnette, T.C.; Harrington, J.A.; Reardon, J.E.; Merrill, B.M.; de Miranda, P. Purification and characterization of a rat liver enzyme that hydrolyzes valaciclovir, the L-valyl ester prodrug of acyclovir. J. Biol. Chem. 1995, 270, 15827–15831. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Chu, X.-y.; Kim, S.; Provoda, C.J.; Lee, K.-D.; Amidon, G.L. Identification of a human valacyclovirase. J. Biol. Chem. 2003, 278, 25348–25356. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.; Xu, Z.; Zhou, J.; Lee, K.-D.; Amidon, G.L. Molecular basis of prodrug activation by human valacyclovirase, an α-amino acid ester hydrolase. J. Biol. Chem. 2008, 283, 9318–9327. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Gupta, S.V.; Lee, K.-D.; Amidon, G.L. Chemical and enzymatic stability of amino acid prodrugs containing methoxy, ethoxy and propylene glycol linkers. Mol. Pharm. 2009, 6, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Crippen, G.M.; Amidon, G.L. Structure and specificity of a human valacyclovir activating enzyme: A homology model of BPHL. Mol. Pharm. 2004, 1, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Song, X.; Vig, B.S.; Mittal, S.; Shin, H.-C.; Lorenzi, P.J.; Amidon, G.L. A novel nucleoside prodrug-activating enzyme: Substrate specificity of biphenyl hydrolase-like protein. Mol. Pharm. 2004, 1, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Dahan, A.; Walls, Z.F.; Lai, L.; Lee, K.-D.; Amidon, G.L. Specificity of a prodrug-activating enzyme hVACVase: The leaving group effect. Mol. Pharm. 2010, 7, 2362–2368. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Hoffman, A. Mode of administration-dependent brain uptake of indomethacin: Sustained systemic input increases brain influx. Drug Metab. Dispos. 2007, 35, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Dvir, E.; Elman, A.; Simmons, D.; Shapiro, I.; Duvdevani, R.; Dahan, A.; Hoffman, A.; Friedman, J.E. DP-155, a lecithin derivative of indomethacin, is a novel nonsteroidal antiinflammatory drug for analgesia and Alzheimer’s disease therapy. CNS Drug Rev. 2007, 13, 260–277. [Google Scholar] [CrossRef] [PubMed]
- Dvir, E.; Friedman, J.E.; Lee, J.Y.; Koh, J.Y.; Younis, F.; Raz, S.; Shapiro, I.; Hoffman, A.; Dahan, A.; Rosenberg, G.; et al. A novel phospholipid derivative of indomethacin, DP-155, shows superior safety and similar efficacy in reducing brain amyloid β in an Alzheimer’s disease model. J. Pharmacol. Exp. Ther. 2006, 318, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Hoffman, A. Rationalizing the selection of oral lipid based drug delivery systems by an in vitro dynamic lipolysis model for improved oral bioavailability of poorly water soluble drugs. J. Control. Release 2008, 129, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kurz, M.; Scriba, G.K.E. Drug–phospholipid conjugates as potential prodrugs: Synthesis, characterization, and degradation by pancreatic phospholipase A2. Chem. Phys. Lipids 2000, 107, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Dahan, A.; Amidon, G.L. Enhancing the intestinal absorption of molecules containing the polar guanidino functionality: A double-targeted prodrug approach. J. Med. Chem. 2010, 53, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Miller, J.M.; Beig, A.; Rozen, L.; Amidon, G.L.; Dahan, A. Mechanistic enhancement of the intestinal absorption of drugs containing the polar guanidino functionality. Expert Opin. Drug Metab. Toxicol. 2011, 7, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Amidon, G.L. Segmental dependent transport of low permeability compounds along the small intestine due to P-glycoprotein: The role of efflux transport in the oral absorption of BCS class III drugs. Mol. Pharm. 2009, 6, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Sabit, H.; Amidon, G.L. Multiple efflux pumps are involved in the transepithelial transport of colchicine: Combined effect of p-glycoprotein and multidrug resistance-associated protein 2 leads to decreased intestinal absorption throughout the entire small intestine. Drug Metab. Dispos. 2009, 37, 2028–2036. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Amidon, G.L. Small intestinal efflux mediated by MRP2 and BCRP shifts sulfasalazine intestinal permeability from high to low, enabling its colonic targeting. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G371–G377. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Amidon, G.L. MRP2 mediated drug—Drug interaction: Indomethacin increases sulfasalazine absorption in the small intestine, potentially decreasing its colonic targeting. Int. J. Pharm. 2010, 386, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Sabit, H.; Amidon, G. The H2 receptor antagonist nizatidine is a P-glycoprotein substrate: Characterization of its intestinal epithelial cell efflux transport. AAPS J. 2009, 11, 205–213. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dahan, A.; Zimmermann, E.M.; Ben-Shabat, S. Modern Prodrug Design for Targeted Oral Drug Delivery. Molecules 2014, 19, 16489-16505. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules191016489
Dahan A, Zimmermann EM, Ben-Shabat S. Modern Prodrug Design for Targeted Oral Drug Delivery. Molecules. 2014; 19(10):16489-16505. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules191016489
Chicago/Turabian StyleDahan, Arik, Ellen M. Zimmermann, and Shimon Ben-Shabat. 2014. "Modern Prodrug Design for Targeted Oral Drug Delivery" Molecules 19, no. 10: 16489-16505. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules191016489