Therapeutic Targeting the Cell Division Cycle 25 (CDC25) Phosphatases in Human Acute Myeloid Leukemia — The Possibility to Target Several Kinases through Inhibition of the Various CDC25 Isoforms



Abstract
:1. Introduction
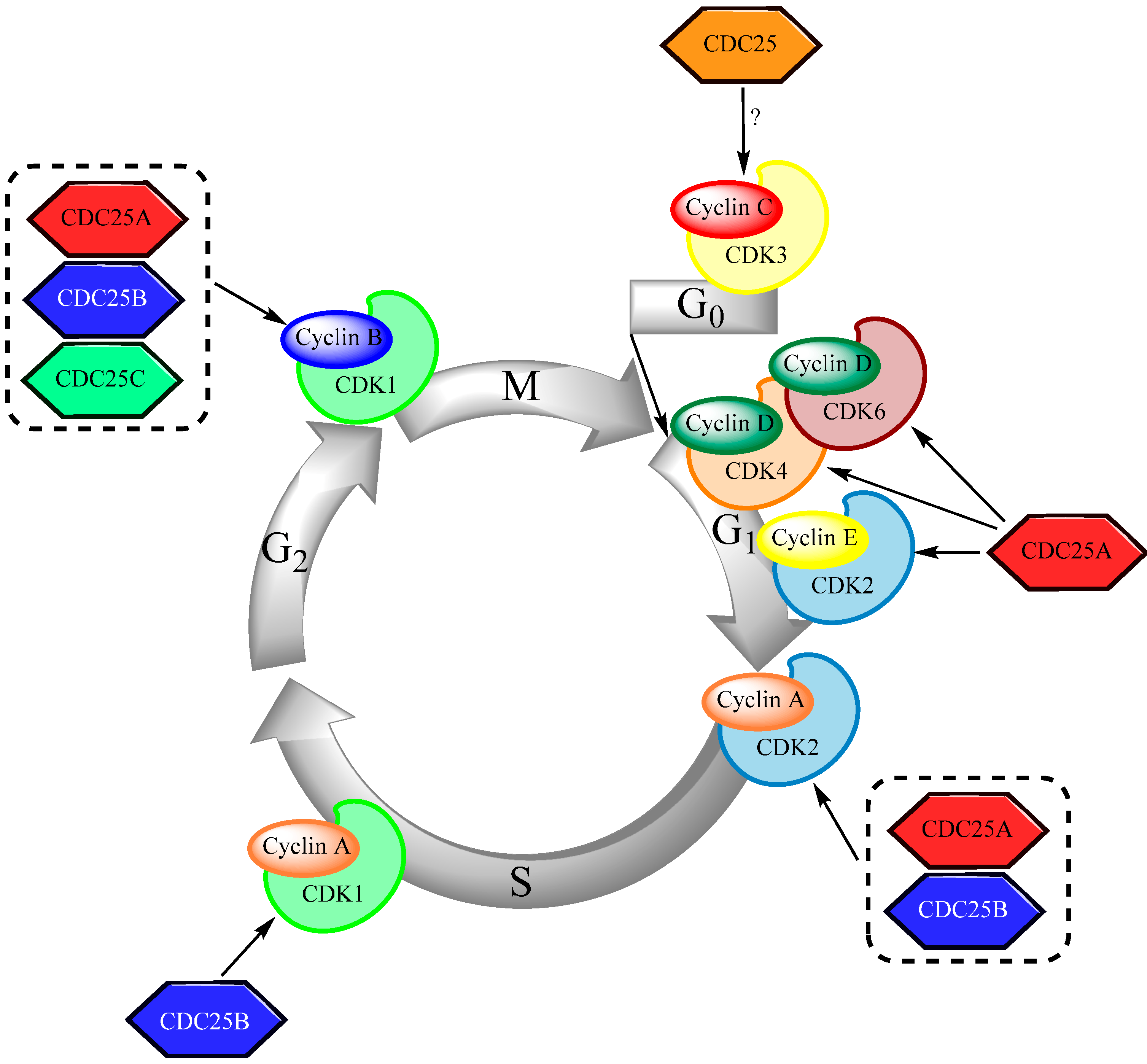
2. CDC25 and the Regulation of Cell Cycle Progression
2.1. The Structure of CDC25

2.2. CDC25 in Cell Cycle Regulation

2.3. Control of CDC25 Expression and Activity

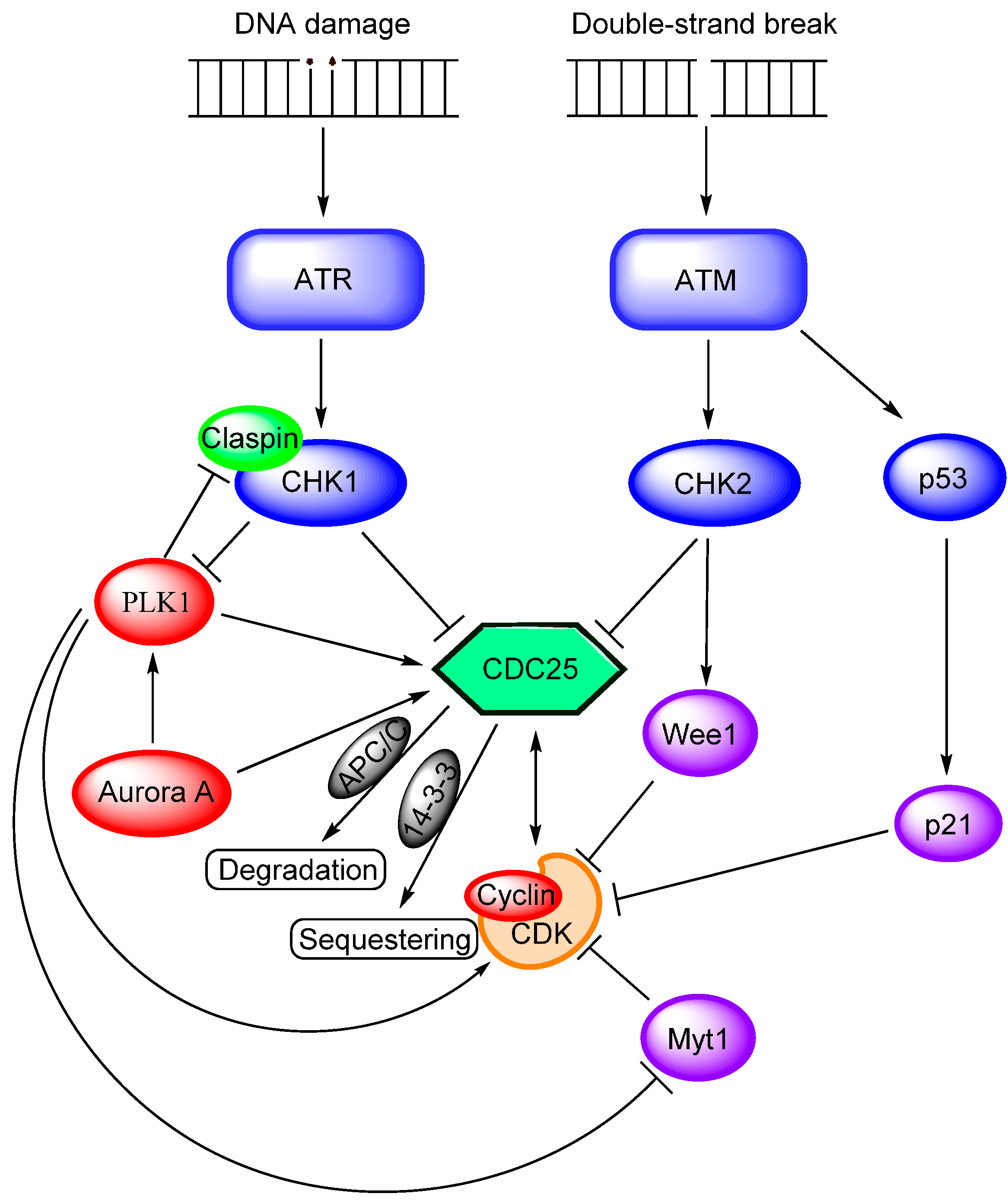
2.4. Cell Cycle Arrest and CDC25 Inhibition
3. Small Molecule CDC25 Inhibitors

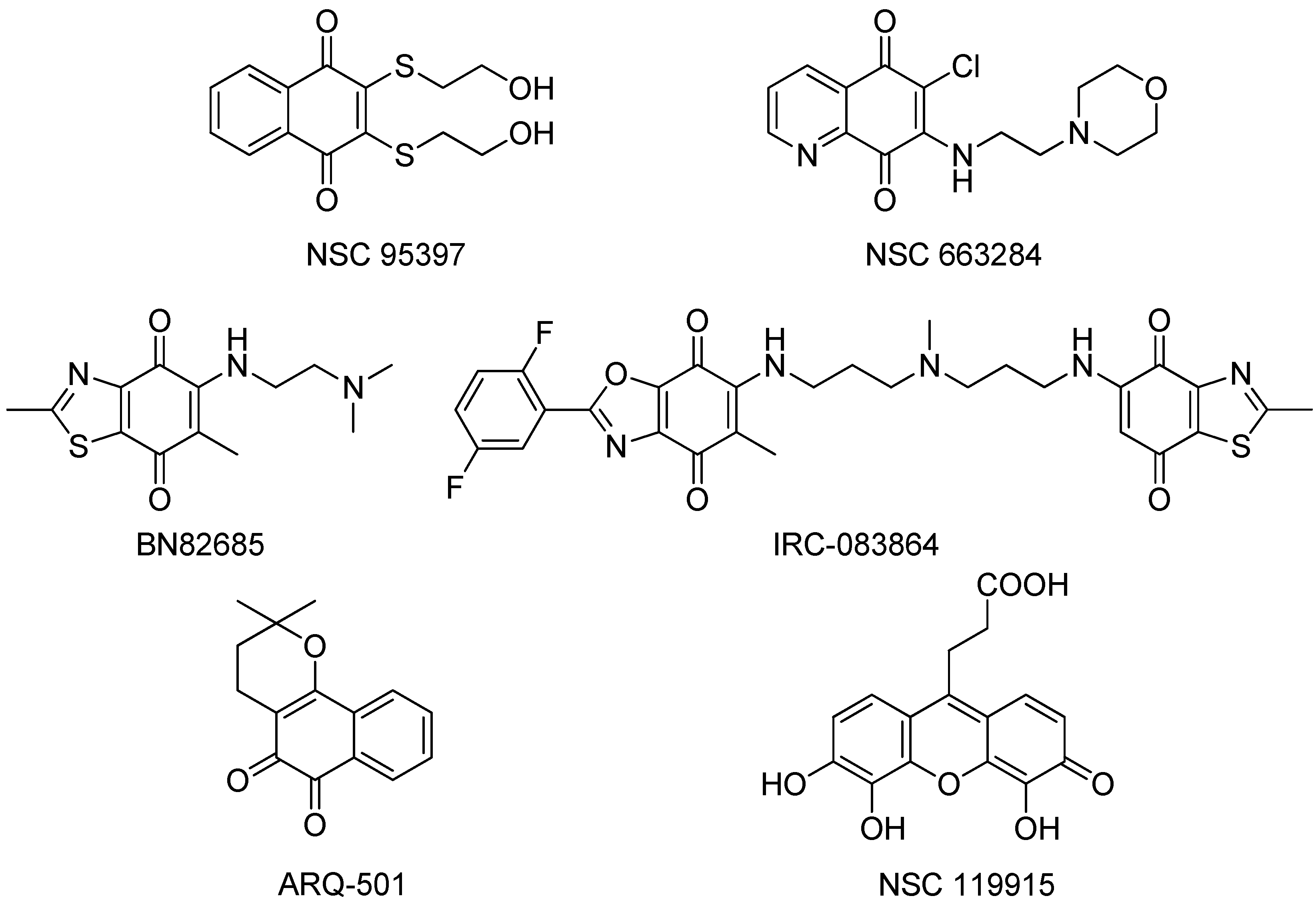
- NSC95397, an unspecific naphthoquinone isoform shows IC50 values in the mid nM range [72], significantly inhibits the proliferation of human and murine carcinoma cells and blocks the G2/M phase transition of the cell cycle.
- NSC 663284 is a potent, cell-permeable, and unspecific irreversible CDC25 inhibitor, arrests cells in the G1 and G2/M phases and induces significant growth inhibition of human breast cancer cell lines [75].
- The quinone-based compound BN 82685 inhibits all three mammalian CDC25 phosphatases in the high nM range in biochemical assays (IC50 = 250, 250, and 170 nM, respectively) and blocks the growth of human pancreatic tumor Mia PaCa-2 cells xenografted into athymic nude mice [78]. BN 82685 was further reported to retain its activity when taken orally. Furthermore, combining low concentrations of BN 82685 and paclitaxel (Taxol®), inhibits the proliferation of colon cancer cells [79], suggesting that combinations of CDC25 inhibitors with microtubule-targeting agents may be of therapeutic interest.
- The bis-quinonoid IRC-083864 is the most potent CDC25 inhibitor described thus far with an IC50 value of ~20 nM. It has activity in pancreatic and prostate cancer xenografts [80] and has entered into clinical trial under the name of Debio 0931, but no data are available yet. The two quinone groups of this large molecule are thought to deactivate the enzyme by covalent bond formation or oxidation of the critical active site thiolate anion.
- Another CDC25 inhibitor, ARQ-501, entered phase I clinical trials in patients with advanced and chemotherapy-unresponsive solid tumors, and is undergoing a phase II trial in patients with leiomyosarcoma and head and neck cancer, having completed an additional trial in combination with the nucleoside analog gemcitabine [81,82]. It is probable, however, that ARQ-501 is not directly a CDC25 inhibitor, but rather functions by some other mechanism.
- Recently, Lavecchia et al. discovered a new quinonoid CDC25 inhibitor (NSC 119915) with micromolar activity by means of a structure-based high-throughput virtual screening [24]. Mechanistically, NSC 119915 displays irreversible inhibition kinetics with in vitro Ki values for CDC25A and CDC25B of 70 and 80 nM, respectively, generates intracellular ROS in cells, arrests cells in the G0/G1 and G2/M phase transitions of the cell cycle, and significantly suppresses the growth of human MCF-7 breast, PC-3 prostate, and K562 leukemia cancer cell lines.
4. Biological Functions of CDC25 in Human AML

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CDC25 Isoform [Reference] Experimental Model | Observations (Scientific Background, Observations, Conclusion) |
|---|---|
| CDC25A [83] AML cell lines (KBM3/Bu250 and OCI-AML3) | Background: The combination of fludarabine, clofarabine and busulfan has a cytotoxic effect on AML cells; the authors investigated the effect of adding the HDAC inhibitor suberoylanilide hydroxamic acid (SAHA) to the triple combination. |
| Observations: | |
| 1. HDAC inhibition increased the antiproliferative effect together with caspase activation and induction of apoptosis. | |
| 2. Additional HDAC inhibition induced an ATM-initiated DNA damage response with activation of p53 and CHK2. | |
| 3. The four-drug combination activated the ATM-CHK2-CDC25-CDK1 pathway with increased CHK2 phosphorylation, decreased CDC25A levels and thereby increased phosphorylation and deactivation of the downstream target CDK1. | |
| 4. Similar observations could be seen in primary human AML cells (only 3 patients examined). | |
| Conclusion: Decreased CDC25A expression is seen in AML cells following exposure to antiproliferative and cytotoxic treatment. | |
| CDC25A [84] Various cell lines, including the HL60 AML cell line | Background: The authors investigated the antiproliferative effects of extracts from the plant Acalypha alopecuroidea. |
| Observations: | |
| 1. The extract had an antiproliferative and cytotoxic effect. | |
| 2. A DNA damage response was seen with CHK2 phosphorylation/activation subsequently leading to inactivation of CDC25A through phosphorylation and finally CDK1 Y15 hyperphosphorylation with cell cycle arrest. | |
| Conclusion: Antiproliferative and cytotoxic effects with cell cycle arrest are associated with CDC25A inactivation. | |
| CDC25A [85] HL60 AML cells | Background: The anticancer-effect of extracts from the plant Scutellaria orientalis was investigated. |
| Observations: | |
| 1. The extract had a dose-dependent proapoptotic effect and induced genotoxic stress. | |
| 2. Dose-dependent p21 induction together with dose-dependent CDC25A/cyclin D1 downregulation was also observed. | |
| Conclusion: Genotoxic stress and antiproliferative/proapoptotic effects are associated with downregulation of CDC25A. | |
| CDC25A [86] Various cell lines, including AML lines | Background: The PI3K-Akt-mTOR pathway can be constitutively activated in human AML cells; the authors investigated a possible functional link between this pathway and CDC25A expression. |
| Observations: | |
| 1. Alk-dependent proliferation was inhibited by RNA interference-mediated downregulation of CDC25A. | |
| 2. Pharmacological CDC25 inhibition reduced ALK (anaplastic lymphoma kinase)-dependent proliferation. | |
| 3. PI3K-Akt-CDC25 mediated intracellular signaling downstream to the FLT3 growth factor receptor. | |
| Conclusion: High CDC25 protein levels are important for PI3K-Akt-mediated growth enhancement. | |
| CDC25A [87] AML cell lines (KG1a, U937) | Background: Interactions with the microenvironment are important for regulation of proliferation for many different cells. |
| Observations: | |
| 1. Adhesion to fibronectin increased AML cell proliferation through increased S-phase entry; differentiated normal progenitors showed a similar effect whereas normal CD34+ cells showed decreased proliferation. | |
| 2. The AML cells showed accumulation of CDC25A; pharmacological inhibition or siRNA-mediated downregulation of CDC25A impaired adhesion-dependent proliferation. CDC25A accumulation was CDH1 dependent and due to modified proteasomal degradation. | |
| 3. The adhesion-induced proliferation and CDC25A upregulation was mediated through activation of the PI3K-Akt-mTOR pathway. | |
| Conclusion: CDC25A upregulation can be mediated by integrin-initiated signaling through PI3K-Akt-mTOR. | |
| CDC25B [88] AML cell lines but also an immature subset of primary human AML cells (CD34+, high aldehyde dehydrogenase activity) | Background: The immediate-early response gene 5 (IER5) can function as a regulator of cell proliferation in cancer cells; the authors investigated whether IER5 is a regulator of AML cell proliferation. |
| Observations: | |
| 1. IER5 was constitutively expressed in human AML cells. | |
| 2. Overexpression of IER5 caused a decrease in CDC25B associated with an antiproliferative effect and G2/M arrest. | |
| 3. Downregulation of CDC25B mRNA expression was caused by IER5 binding to the promoter and mediated through NF-YB and p300. | |
| Conclusion: IER5-induced growth inhibition of immature AML cells is associated with downregulation of CDC25 expression. | |
| CDC25B [49] AML cell lines (KG1a, U937) and primary human AML cells | Background: The importance of the G2/M checkpoint for the sensitivity of AML cells to genotoxic stress was examined; exposure to etoposide was used as the genotoxic stress. |
| Observations: | |
| 1. G2/M checkpoint stringency varied between AML cell lines; U937 cells showed an earlier exit from the checkpoint than KG1a cells after exposure to etoposide. Etoposide caused increased phosphorylation/activation of CHK1 in both cell lines, but the earlier exit by U937 cells was associated with an earlier decrease in CHK1 activation. | |
| 2. CDC25B protein levels increased after etoposide exposure. CDC25B is important for checkpoint recovery, and pharmacological as well as siRNA inhibition of CDC25B caused an inhibition of the checkpoint recovery and entry into mitosis. | |
| 3. Combination of etoposide + CHK1 inhibition increased the number of mitotic and apoptotic cells. Increased cytotoxicity was also seen for primary AML cells when combining etoposide with CHK1 inhibition, but this potentiating effect differed between patients. | |
| Conclusion: CDC25 is important for the sensitivity of human AML cells for genotoxic stress. | |
| CDC25B [89] Primary human AML cells | Background: Is CDC25 important for the antiproliferative effect of PI3K/mTOR inhibitors in primary human AML cells? |
| Observations: | |
| 1. Resistance to PI3K/mTOR inhibition was associated with increased expression of CDC25B. | |
| 2. Pharmacological CDC25 inhibition has an additive antiproliferative effect to PI3K/mTOR inhibition only for certain cell lines and for a subset of patients. | |
| Conclusion: The effect of CDC25 inhibition differs between patients, additive antiproliferative effects to PI3K-mTOR inhibition are seen only for a subset. | |
| CDC25C [90] AML cell line U937 | Background: Does CDC25C associate with cyclin A in human AML cells? |
| Observations: | |
| CDC25C coprecipitated with cyclin A in proliferating cells, this was not seen after apoptosis induction through the extrinsic and intrinsic pathways. | |
| Conclusion: CDC25C contribute to cell cycle regulation in human AML cell lines through complexing with cyclin A. | |
| CDC25C [91] Primary human AML cells | Background: Normal cells show different CDC25C splice variants, is this also true for primary human AML cells? |
| Observations: | |
| Several splice variants were detected in primary human AML cells, and the splicing pattern differed between AML cells and normal CD34+ hematopoietic cells. | |
| Conclusion: Aberrant splicing may contribute to the AML cell phenotype and to functional differences in cell cycle regulation between patients. |
- CDC25A. Upregulation of CDC25A enhances lymphoma cell proliferation [87]. Its expression seems to be controlled through PI3K-Akt-mTOR signaling [87]. Loss of CDC25A is also important for cell cycle arrest during the differentiation of malignant hematopoietic cells [92]. This isoform seems to be important for the DNA damage response during exposure of human AML cells to cytotoxic drugs. It also seems important for Akt-dependent proliferation in AML cells because inhibition or knock-down of CDC25A reduces the Akt-dependent AML cell proliferation. The FLT3 gene is frequently mutated and thereby encodes a constitutively activated kinase in human AML and is an adverse prognostic factor [93]; this FLT3-initiated signaling is mediated through different pathways (including PI3K-Akt-mTOR) and CDC25 may thereby mediate or contribute to the adverse prognostic impact of this genetic abnormality.
- CDC25B. High expression is associated with resistance against the antiproliferative effect of PI3K-Akt-mTOR inhibitors [89], but the molecular mechanisms behind this effect are not known. Inhibition of this isoform seems to reduce AML cell line proliferation through effects on NF-YB and p300 [88]; whereas in other cells growth inhibition is seen only with combined Cyclin D1 and CDC25B inhibition [84]. These regulatory events thus represent a link between CDC25 and the NF-YB system that consist of the three subunits NF-YA, NF-YB and NF-YC; this system seems to contribute to carcinogenesis for several human malignancies [94]. Finally, activation of the NFκB system is also important for transcriptional regulation, and inhibition of NFκB at G2/M phase transition delays mitotic entry and inhibits transcription of G2/M-specific genes, including cyclin B, PLK1, and CDC25B [95]. NFκB is important for leukemic stem cell functions and their chemosensitivity [96], but it is not known whether this importance of NFκB on AML stem cells is mediated through its effects on CDC25B transcription.
5. The Molecular Context of CDC25 in Human AML Cells
5.1. Regulation of the G1 Phase of the Cell Cycle in Human AML Cells
5.2. Regulation of the S-Phase
5.3. Regulation of the G2 Phase
5.4. Regulation of Mitosis
| Signaling Cascade | Effects on Cell Cycle Regulation—Effects of Genetic Abnormalities | |
|---|---|---|
| Receptor ligation or mutations | ← RTK | Receptor ligation and mutations of growth factor receptor tyrosine kinases: Proliferation of primary AML cells is increased by receptor ligation [103]; receptor tyrosine kinase (RTK) mutations (e.g., c-kit mutations, FLT3-mutations) can cause constitutive signaling [98]. High CXCR4 levels is seen for a minority of patients and is associated with adverse prognosis [105]. |
| ↓ | CDK4/6: These mediators stimulate G1 progression and bind/sequester CDK inhibitors (CKI), e.g., p14, p16, p27 [98]. p14 and p27 can show increased expression in AML, p16 may show low expression [106]. | |
| STATs | ||
| MAP kinases | ↓ | PIM kinases: These kinases are activated by the upstream receptor ligation together with STAT/MAP kinases and thereafter increase CDK4 activation [98]. They also decrease the effects of the CKIs, and PIM1 activates CDC25 directly (see Table 4). |
| PLC | ||
| ↓ | CKI (CDK inhibitors): High p27 levels seem to be predictive of complete remission after intensive induction chemotherapy [102]. | |
| CDK4/6 | ← PIM kinases | Phospholipase Cβ 1 (PLC): The nuclear form of this enzyme is an important checkpoint that controls progression through the G1 phase, its activation depends on type 1 insulin-like receptor (IGF-R) and monoallelic deletions seem to be associated with aggressive disease both for patients with AML and patients with preleukemic myelodysplastic syndromes [108]. |
| Cyclin D | ||
| ↓ | ┴ | |
| ├ CKIs | CDK2 activation: The upstream receptor-initiated signaling activates CDK2 through CDK4/6 dephosphorylation. | |
| ↓ | Cyclin E: The expression varies between AML patients, high expression is seen in one third of the patients [99]. | |
| CDK2 | ├ p53/p21 | p53 and p21—different p53 isoforms associated with FLT3-ITD and NPM1 mutations: p53 inhibits G1 progression through p2 induction. p53 is mutated only in a minority of AML patients; but the balance between various isoforms differ between patients, e.g., between patients with adverse prognosis FLT3-ITD and NPM1 mutations [107]. |
| Cyclin E | ← E2F | |
| ↓ | E2F: Inactivation of the Rb gene by CDK4/6 releases the E2F transcription factor that increase transcription of cyclin E; aberrant expression of a variant E2F can be seen in human AML [100,101]. | |
| ↓ | TET2 mutations: These loss-of-function mutations are detected in 10-20% of AML patients, loss of TET activity leads to increased proliferation and is associated with adverse prognosis [104]. | |
| Entering of S-phase | MLL fusion proteins—11q23 translocations: The wild-type MLL protein cause histone methylation; its expression normally peaks at the G1/S boundary but the fusion protein shows stable expression and therefore seems to abrogate checkpoint control and increase the expression of homeobox transcription factors as described in detail by [98]. | |
| Signaling Pathway | Additional Regulator | Normal Molecular Function | Abnormality in Human AML |
|---|---|---|---|
| DNA damage Replication checkpoint | Claspin | ATR: a serine-threonine kinase activated by DNA damage [98]. | ATR: stabilizes the chromatin-remodeller MLL (see below), this ability is lost for MLL-fusion proteins encoded by 11q23 translocations [98]. |
| Claspin: activated by ATR through phosphorylation, initiates DNA repair and enhances this DNA damage response through stimulation of CHK1 [120,123]. | Claspin: differentially expressed in AML cell lines [49] | ||
| CHK1: experimental and clinical studies suggest that CHK1 inhibition has antileukemic effects [112,114]. | |||
| Chemotherapy: cytarabine and clofarabine cause S-phase/replication arrest and CHK1 and CHK2-mediated inhibitory phosphorylation of CDC25 [98]. | |||
| ↓ | CHK1: a serine/threonine kinase that inhibits CDC25 through addition of inhibitory phosphate groups. Differentially expressed in AML cell lines [111]. | ||
| ATR | |||
| ↓ | |||
| CHK1 | |||
| Clinical relevance: MLL-fusion proteins caused by 11q23 translocations can inhibit the MLL-ATR interaction, causing MLL degradation and continued replication [115]. | |||
| ┴ | |||
| CDC25 | |||
| Double-strand break | RAD51C PIM kinase 1 FLT3-ITD | ATM: a serine/threonine kinase activated by DNA double strand breaks. | ATM: genetic variants of ATM influence treatment outcome, heterozygous ATM 4138C>T is associated with an inferior treatment outcome after intensive chemotherapy [121]. Missense mutations have been detected in AML [118]. |
| CHK2: a serine/threonine kinase that adds inhibitory phosphate to CDC25 and thereby inhibits CDK2. | |||
| Intra-S phase | |||
| Checkpoint | CHK2: can be mutated in human AML, but this is uncommon [111]. | ||
| ↓ | |||
| ATM | RAD15C: Involved in ATM-dependent binding to DNA double-strand breaks required for CHK2 phosphorylation [122]. | RAD51C: polymorphisms do not influence risk of AML or outcome after chemotherapy [116]. | |
| ↓ | |||
| CHK2 | |||
| ┴ | FLT3-ITD: this is a common AML mutation associated with chemoresistance and reduced AML-free survival. It shows constitutive signaling and causes activation of CDC25. Normal myeloid differentiation requires activation of the transcription factor C/EBPα; constitutively activated FLT3-ITD causes an inhibitory phosphorylation and thereby a block of differentiation mediated either by ERK1/2 or by CDK1 (see description of the G2 phase, Table 4) [127,128,129]. | ||
| CDC25 | PIM kinase 1: this serine/threonine kinase transduces cytokine-initiated mitogenic signals; it phosphorylates CDC25A directly and this phosphorylation increases the phosphatase activity of CDC25A [126]. | ||
| Chemotherapy: topoisomerase inhibitors (etoposide, anthracyclines) favor DNA strand breaks and provoke a checkpoint response involving ATM [98]. | |||
| CDC25 | CDK2: a serine/threonine kinase activated by cyclin A and cyclin C and inactivated by p21 and p27. | CDK2: high activity with fast progression through S-phase has been described in AML [102]. | |
| ↓ | |||
| ↓ | |||
| CDK2 | Cyclin A: high activity with fast progression through S-phase has been described [113,117,124]. | ||
| Cyclin A | Cyclin A: regulates S-phase progression, interact with c-Myb [113,117,124]. | ||
| Cyclin C | |||
| ↓ | Cyclin E: high activity with fast progression through S-phase has been described in human AML [99]. | ||
| ↓ | |||
| CDC45 | Cyclin E: regulates G1/S transition | CDC45: altered regulation in 11q23 AML [102]. | |
| CDC45: regulator of replication, regulated by wild-type MLL [102]. | p21: deficiency cooperates in t(8;21) variants of AML [119]. | ||
| p27: downregulated in AML, faster S-phase progression [125]. | |||
| p21: inhibitor of CDK2 and CDK4 | |||
| p27: inhibitor of CDK2 and CDK4 |
| Signaling Cascade | Effects on Cell Cycle Regulation—Effects of AML-Associated Abnormalities | |
|---|---|---|
| DNA-damage ↓ ATR-ATM CHK1-CHK2 ┴ CDC25 ↓ CDK1 Cyclin B ↓ Mitosis | γ-H2Ax, BRAC1 SET ┴ ← PP2A ← PLK1 ← FOXM1 ├ p53/p21 | NFκB and cell cycle regulation: |
| Activation of the NFκB system is important for transcriptional regulation, and inhibition of NFκB delays mitotic entry and inhibits transcription of G2/M-specific genes, including cyclin B, PLK1, and CDC25B [95]. NFκB is important for AML stem cell functions and their chemosensitivity [96], but it is not known whether its effect on leukemic stem cells is mediated through altered CDC25B expression. | ||
| The DNA damage response: | ||
| γ-H2Ax: Phosphorylation of this histone is an indication of a DNA damage response, the AML cell level after treatment shows considerable variation between patients [135]. | ||
| BRCA1/2: DNA repair is closely linked to cell cycle regulation; reduced BRCA1 gene expression is seen for a minority of de novo AML but for a majority of secondary AMLs [138]. | ||
| SET: an endogenous inhibitor of PP2A [134]. | ||
| PP2A: Inactivation through phosphorylation is seen in primary AML cells for a majority of patients; this inactivation can be caused by either deregulated expression of endogenous PP2A inhibitors (e.g., SET, see above), overexpression of SETBP1 or downregulation of PP2A subunits [134]. | ||
| PLK1: This kinase is overexpressed in primary AML cells for a majority of patients [137,139]. | ||
| FOXM1: This transcription factor seems to be overexpressed in primary AML cells for most patients; decreased expression is associated with G2-arrest and reduced levels of Aurora kinase B, cyclin B1 and CDC25B together with increased levels of p21 and p27 at the protein level [136]. | ||
| Cyclin B: Cyclin B1 is commonly expressed in primary AML cells, but the expression pattern (cytoplasmic versus nuclear) varies between patients [140]. The expression is controlled by FOXM1, see above [136]. | ||
| p53/p21: See Table 2 and Table 3 | ||
| CDK1 and FLT3 mutations: | ||
| An alternative pathway for activation of CDK1 is the constitutive activation by FLT3-ITD; this signaling can activate both ERK1/2 and CDK1 and these kinases can then phosphorylate the transcription factor C/EBPα; this phosphorylation will inhibit its transcriptional activity and thereby contribute to the differentiation block in these AML cells [127,128,129]. | ||
- The mitotic checkpoint. Several regulators of the mitotic checkpoint show decreased levels in primary human AML cells and this weakens the mitotic checkpoint response [141,142,143]. Blinkin can also be a fusion partner with MLL in 11q23 translocations; this fusion molecule seems to cause a dominant negative effect over the wild type protein.
- The t(8;21) abnormality. This AML-associated abnormality can also disturb this step through downregulation of securin. Furthermore, cyclin B is involved in regulation of mitosis and can be downregulated by the fusion protein encoded by the t(8;21) fusion gene [141].
- CDK1 and FLT3 mutations. An alternative pathway for activation of CDK1 is constitutive activation of FLT3-initiated signaling by FLT3-ITD; this signaling can activate both ERK1/2 and CDK1 and these kinases then phosphorylate the transcription factor C/EBPα; this phosphorylation will inhibit its transcriptional activity and thereby contribute to the differentiation block in these AML cells [127,128,129]. Whether this phosphorylation is mediated by ERK1/2 or CDK1 seems to differ between patients [127,151], and this difference between FLT3-ITD patients may depend on the large variation of the ITDs between patients [152]. Thus, the FLT3-ITD abnormalities can alter both differentiation and cell cycle regulation through their effects on CDK1.
5.5. Conclusions—Patients are Heterogeneous with Regard to Cell Cycle Regulation of the AML Cells
6. CDC25 Inhibition in the Treatment of Human AML
6.1. CDC25 and the Anticancer Effects of mTOR Inhibition
6.2. CDC25 and Induction of Differentiation in AML Cells
7. The Possible Use of CDC25 Inhibitors in the Treatment of human AML
7.1. AML is a Heterogeneous Disease—the Possible Use of CDC25 Inhibitors Only in Subsets of Patients
7.2. Combined Targeting of Cell Cycle Regulation—the Possible Combination of CDC25 Inhibition with Inhibitors of PLKs, AURORA Kinases or CDK Inhibitors
7.3. Combination of CDC25 Inhibitors with Conventional Chemotherapy
7.4. Toxicity versus Antileukemic Efficiency
7.5. Inhibition of Several Therapeutic Targets through the Use of a Single CDC25 Inhibitor
| Upstream Events Affecting CDC25 Activation (See Also Figure 3) | |
|---|---|
| Akt/PKB [60,62,63,64,65] | This pathway can be an upstream inhibitor of CDC25 through sequestering it in the cytoplasm, but at the same time CDC25A is necessary for Akt-initiated proliferation. |
| MAP kinases [30] | Negative regulator during cellular stress. |
| CDK1/cyclin B [44] | Activates CDC25B and CDC25C in a positive feedback loop. |
| CDK2/cyclin E [21,29] | Activates CDC25A in a positive feedback loop. |
| PLK1 [44] | Activates CDC25 and promotes mitosis; both direct and indirect activating effects. |
| Aurora kinases [47,48] | Activate CDC25s and promote mitosis; both direct and indirect activating effects via activation of PLK1. |
| Wee1 and Myt1 kinases [33] | Inhibition of CDC25s. |
| CHK1 [50,172] | Inactivates all three isoforms of CDC25; it also inhibits CDC25s indirectly, e.g., through PLK1 inhibition. |
| CHK2 [50,172] | Inactivates all three isoforms of CDC25; it also inhibits CDC25s indirectly, e.g., through PLK1 inhibition. |
| CDK1 [30,32] | CDK1/cyclin A is dephosphorylated by CDC25B in late S-phase. Dephosphorylation of CDK1/cyclin B by CDC25 is a rate-limiting step for transition from G2 to mitosis. |
| CDK2 [25,30] | Activated during several steps in the cell cycle. Associates with cyclin E during G1/S transition and cyclin A during S-phase. |
| CDK4 [25] | Important for entry into S phase. Associates with cyclin D. |
| CDK6 [25] | Important for entry into S phase. Associates with cyclin D. |
| Akt/protein kinase B [61] | Its phosphorylation can be regulated by CDC25B. |
8. Final Comments
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hunter, T. Protein kinases and phosphatases: The yin and yang of protein phosphorylation and signaling. Cell 1995, 80, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; di Giovanni, C.; Novellino, E. Inhibitors of Cdc25 phosphatases as anticancer agents: A patent review. Expert Opin. Ther. Pat. 2010, 20, 405–425. [Google Scholar] [CrossRef] [PubMed]
- Galaktionov, K.; Beach, D. Specific activation of cdc25 tyrosine phosphatases by B-type cyclins: Evidence for multiple roles of mitotic cyclins. Cell 1991, 67, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Kristjansdottir, K.; Rudolph, J. Cdc25 phosphatases and cancer. Chem. Biol. 2004, 11, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; Di Giovanni, C.; Novellino, E. CDC25 phosphatase inhibitors: An update. Mini Rev. Med. Chem. 2012, 12, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Johnston, P.A.; Foster, C.A.; Tierno, M.B.; Shun, T.Y.; Shinde, S.N.; Paquette, W.D.; Brummond, K.M.; Wipf, P.; Lazo, J.S. Cdc25B dual-specificity phosphatase inhibitors identified in a high-throughput screen of the NIH compound library. Assay Drug Dev. Technol. 2009, 7, 250–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudolf, V.; Inze, D.; de Veylder, L. What if higher plants lack a CDC25 phosphatase? Trends Plant. Sci. 2006, 11, 474–479. [Google Scholar]
- Obtained from the NCBI protein database. Available online: http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/protein (accessed on 7 November 2014).
- Keyse, S.M.; Ginsburg, M. Amino acid sequence similarity between CL100, a dual-specificity MAP kinase phosphatase and cdc25. Trends Biochem. Sci. 1993, 18, 377–378. [Google Scholar] [CrossRef]
- Dillet, V.; van Etten, R.L.; Bashford, D. Stabilization of charges and protonation states in the active site of the protein tyrosine phosphatases: A computational study. J. Phys. Chem. B 2000, 104, 11321–11333. [Google Scholar] [CrossRef]
- Fauman, E.B.; Cogswell, J.P.; Lovejoy, B.; Rocque, W.J.; Holmes, W.; Montana, V.G.; Piwnica-Worms, H.; Rink, M.J.; Saper, M.A. Crystal structure of the catalytic domain of the human cell cycle control phosphatase, Cdc25A. Cell 1998, 93, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, R.A.; Yem, A.W.; Wolfe, C.L.; Deibel, M.R., Jr.; Chidester, C.G.; Watenpaugh, K.D. Crystal structure of the catalytic subunit of Cdc25B required for G2/M phase transition of the cell cycle. J. Mol. Biol. 1999, 293, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Gabrielli, B.G.; Clark, J.M.; McCormack, A.K.; Ellem, K.A. Hyperphosphorylation of the N-terminal domain of Cdc25 regulates activity toward cyclin B1/Cdc2 but not cyclin A/Cdk2. J. Biol. Chem. 1997, 272, 28607–28614. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, J. Targeting the neighbor’s pool. Mol. Pharmacol. 2004, 66, 780–782. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, J. Cdc25 phosphatases: Structure, specificity, and mechanism. Biochemistry 2007, 46, 3595–3604. [Google Scholar] [CrossRef] [PubMed]
- Sohn, J.; Kristjansdottir, K.; Safi, A.; Parker, B.; Kiburz, B.; Rudolph, J. Remote hot spots mediate protein substrate recognition for the Cdc25 phosphatase. Proc. Natl. Acad. Sci. USA 2004, 101, 16437–16441. [Google Scholar] [CrossRef] [PubMed]
- Boutros, R.; Dozier, C.; Ducommun, B. The when and wheres of CDC25 phosphatases. Curr. Opin. Cell Biol. 2006, 18, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Kallstrøm, H.; Lindqvist, A.; Pospisil, V.; Lundgren, A.; Rosenthal, C.K. Cdc25A localisation and shuttling: Characterisation of sequences mediating nuclear export and import. Exp. Cell Res. 2005, 303, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Muslin, A.J.; Xing, H. 14–3-3 proteins: Regulation of subcellular localization by molecular interference. Cell Signal. 2000, 12, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Mammalian cyclin-dependent kinases. Trends Biochem. Sci. 2005, 30, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, I.; Draetta, G.; Karsenti, E. Activation of the phosphatase activity of human cdc25A by a cdk2-cyclin E dependent phosphorylation at the G1/S transition. EMBO J. 1994, 13, 4302–4310. [Google Scholar] [PubMed]
- Chen, M.S.; Hurov, J.; White, L.S.; Woodford-Thomas, T.; Piwnica-Worms, H. Absence of apparent phenotype in mice lacking Cdc25C protein phosphatase. Mol. Cell. Biol. 2001, 21, 3853–3861. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, A.M.; White, L.S.; Donovan, P.J.; Piwnica-Worms, H. Normal cell cycle and checkpoint responses in mice and cells lacking Cdc25B and Cdc25C protein phosphatases. Mol. Cell. Biol. 2005, 25, 2853–2860. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; di Giovanni, C.; Pesapane, A.; Montuori, N.; Ragno, P.; Martucci, N.M.; Masullo, M.; de Vendittis, E.; Novellino, E. Discovery of new inhibitors of Cdc25B dual specificity phosphatases by structure-based virtual screening. J. Med. Chem. 2012, 55, 4142–4158. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Rollins, B.J. Cyclin C/cdk3 promotes Rb-dependent G0 exit. Cell 2004, 117, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Resnitzky, D.; Reed, S.I. Different roles for cyclins D1 and E in regulation of the G1-to-S transition. Mol. Cell. Biol. 1995, 15, 3463–3469. [Google Scholar] [PubMed]
- Shen, T.; Huang, S. The role of Cdc25A in the regulation of cell proliferation and apoptosis. Anticancer Agents Med. Chem. 2012, 12, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Blomberg, I.; Hoffmann, I. Ectopic expression of Cdc25A accelerates the G(1)/S transition and leads to premature activation of cyclin E- and cyclin A-dependent kinases. Mol. Cell. Biol. 1999, 19, 6183–6194. [Google Scholar] [PubMed]
- Frazer, C.; Young, P.G. Phosphorylation mediated regulation of Cdc25 activity, localization and stability. In Protein Phosphorylation in Human Health; Huang, C., Ed.; InTech: Rijeka, Croatia, 2012; pp. 395–436. [Google Scholar]
- Turowski, P.; Franckhauser, C.; Morris, M.C.; Vaglio, P.; Fernandez, A.; Lamb, N.J. Functional cdc25C dual-specificity phosphatase is required for S-phase entry in human cells. Mol. Biol. Cell 2003, 14, 2984–2998. [Google Scholar] [CrossRef] [PubMed]
- Mailand, N.; Podtelejnikov, A.V.; Groth, A.; Mann, M.; Bartek, J.; Lukas, J. Regulation of G(2)/M events by Cdc25A through phosphorylation-dependent modulation of its stability. EMBO J. 2002, 21, 5911–5920. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ji, P.; Liu, J.; Broaddus, R.R.; Xue, F.; Zhang, W. Centrosome-associated regulators of the G(2)/M checkpoint as targets for cancer therapy. Mol. Cancer 2009, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Sananes, M.R.; Kapuy, O.; Hunt, T.; Novak, B. Switches and latches: A biochemical tug-of-war between the kinases and phosphatases that control mitosis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 3584–3594. [Google Scholar] [CrossRef] [PubMed]
- Gabrielli, B.G.; de Souza, C.P.; Tonks, I.D.; Clark, J.M.; Hayward, N.K.; Ellem, K.A. Cytoplasmic accumulation of cdc25B phosphatase in mitosis triggers centrosomal microtubule nucleation in HeLa cells. J. Cell Sci. 1996, 109, 1081–1093. [Google Scholar] [PubMed]
- Lindqvist, A.; Kallstrøm, H.; Lundgren, A.; Barsoum, E.; Rosenthal, C.K. Cdc25B cooperates with Cdc25A to induce mitosis but has a unique role in activating cyclin B1-Cdk1 at the centrosome. J. Cell Biol. 2005, 171, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, E.; Boutros, R.; Froment, C.; Monsarrat, B.; Ducommun, B.; Dozier, C. CHK1 phosphorylates CDC25B during the cell cycle in the absence of DNA damage. J. Cell Sci. 2006, 119, 4269–4275. [Google Scholar] [CrossRef] [PubMed]
- Donzelli, M.; Squatrito, M.; Ganoth, D.; Hershko, A.; Pagano, M.; Draetta, G.F. Dual mode of degradation of Cdc25 A phosphatase. EMBO J. 2002, 21, 4875–4884. [Google Scholar] [CrossRef] [PubMed]
- Tumurbaatar, I.; Cizmecioglu, O.; Hoffmann, I.; Grummt, I.; Voit, R. Human Cdc14B promotes progression through mitosis by dephosphorylating Cdc25 and regulating Cdk1/cyclin B activity. PLoS One 2011, 6, e14711. [Google Scholar] [CrossRef] [PubMed]
- Lincoln, A.J.; Wickramasinghe, D.; Stein, P.; Schultz, R.M.; Palko, M.E.; de Miguel, M.P.; Tessarollo, L.; Donovan, P.J. Cdc25b phosphatase is required for resumption of meiosis during oocyte maturation. Nat. Genet. 2002, 30, 446–449. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Wang, L.N.; Zhou, Y.Y.; Yu, H.P.; Shen, Q.; Zang, Y.; Zhou, Y.B.; Li, J.Y.; Zhang, H. X.; Li, J. Discovery and characterization of a novel inhibitor of CDC25B, LGH00045. Acta. Pharmacol. Sin. 2008, 29, 1268–1274. [Google Scholar] [CrossRef] [PubMed]
- Bugler, B.; Quaranta, M.; Aressy, B.; Brezak, M.C.; Prevost, G.; Ducommun, B. Genotoxic-activated G2-M checkpoint exit is dependent on CDC25B phosphatase expression. Mol. Cancer Ther. 2006, 5, 1446–1451. [Google Scholar] [CrossRef] [PubMed]
- Barre, B.; Vigneron, A.; Coqueret, O. The STAT3 transcription factor is a target for the Myc and riboblastoma proteins on the Cdc25A promoter. J. Biol. Chem. 2005, 280, 15673–15681. [Google Scholar] [CrossRef] [PubMed]
- Salaun, P.; Rannou, Y.; Prigent, C. Cdk1, Plks, Auroras, and Neks: The mitotic bodyguards. Adv. Exp. Med. Biol. 2008, 617, 41–56. [Google Scholar] [PubMed]
- Boutros, R.; Lobjois, V.; Ducommun, B. CDC25 phosphatases in cancer cells: Key players? Good targets? Nat. Rev. Cancer 2007, 7, 495–507. [Google Scholar] [CrossRef]
- Macůrek, L.; Lindqvist, A.; Medema, R.H. Aurora-A and hBora join the game of Polo. Cancer Res. 2009, 69, 4555–4558. [Google Scholar] [CrossRef] [PubMed]
- Seki, A.; Coppinger, J.A.; Jang, C.Y.; Yates, J.R.; Fang, G. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science 2008, 320, 1655–1658. [Google Scholar] [CrossRef] [PubMed]
- Kotani, S.; Tugendreich, S.; Fujii, M.; Jørgensen, P.M.; Watanabe, N.; Hoog, C.; Hieter, P.; Todokoro, K. PKA and MPF-activated polo-like kinase regulate anaphase-promoting complex activity and mitosis progression. Mol. Cell 1998, 1, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Didier, C.; Cavelier, C.; Quaranta, M.; Galcera, M.O.; Demur, C.; Laurent, G.; Manenti, S.; Ducommun, B. G2/M checkpoint stringency is a key parameter in the sensitivity of AML cells to genotoxic stress. Oncogene 2008, 27, 3811–3820. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, H.C.; Yaffe, M.B. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr. Opin. Cell Biol. 2009, 21, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Niida, H.; Nakanishi, M. DNA damage checkpoints in mammals. Mutagenesis 2006, 21, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Falck, J.; Mailand, N.; Syljuåsen, R.G.; Bartek, J.; Lukas, J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 2001, 410, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Uto, K.; Inoue, D.; Shimuta, K.; Nakajo, N.; Sagata, N. Chk1, but not Chk2, inhibits Cdc25 phosphatases by a novel common mechanism. EMBO J. 2004, 23, 3386–3396. [Google Scholar] [CrossRef]
- Krämer, A.; Mailand, N.; Lukas, C.; Syljuåsen, R.G.; Wilkinson, C.J.; Nigg, E.A.; Bartek, J.; Lukas, J. Centrosome-associated Chk1 prevents premature activation of cyclin-B-Cdk1 kinase. Nat. Cell Biol. 2004, 6, 884–891. [Google Scholar] [PubMed]
- Yarden, R.I.; Pardo-Reoyo, S.; Sgagias, M.; Cowan, K.H.; Brody, L.C. BRCA1 regulates the G2/M checkpoint by activating Chk1 kinase upon DNA damage. Nat. Genet. 2002, 30, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Busino, L.; Chiesa, M.; Draetta, G.F.; Donzelli, M. Cdc25A phosphatase: Combinatorial phosphorylation, ubiquitylation and proteolysis. Oncogene 2004, 23, 2050–2056. [Google Scholar] [CrossRef] [PubMed]
- Smits, V.A.; Klompmaker, R.; Arnaud, L.; Rijksen, G.; Nigg, E.A.; Medema, R.H. Polo-like kinase-1 is a target of the DNA damage checkpoint. Nat. Cell Biol. 2000, 2, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Dunphy, W.G. Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts. Mol. Cell 2000, 6, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, H.C.; Aslanian, A.S.; Lees, J.A.; Yaffe, M.B. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 2007, 11, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Piwnica-Worms, H. DNA damage and replication checkpoints in fission yeast require nuclear exclusion of the Cdc25 phosphatase via 14–3-3 binding. Mol. Cell. Biol. 1999, 19, 7410–7419. [Google Scholar]
- Chen, R.-Q.; Yang, Q.-K.; Lu, B.-W.; Yi, W.; Cantin, G.; Chen, Y.-L.; Fearns, C.; Yates, J.R., III; Lee, J.-D. CDC25B Mediates Rapamycin-induced Oncogenic Responses in Cancer Cells. Cancer Res. 2009, 69, 2663–2668. [Google Scholar] [PubMed]
- Li, G.Y.; Jung, K.H.; Lee, H.; Son, M.K.; Seo, J.; Hong, S.W.; Jeong, Y.; Hong, S.; Hong, S.S. A novel imidazopyridine derivative, HS-106, induces apoptosis of breast cancer cells and represses angiogenesis by targeting the PI3K/mTOR pathway. Cancer Lett. 2013, 329, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Slingerland, J.M. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle 2003, 2, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Puc, J.; Keniry, M.; Li, H.S.; Pandita, T.K.; Choudhury, A.D.; Memeo, L.; Mansukhani, M.; Murty, V.V.; Gaciong, Z.; Meek, S.E.; et al. Lack of PTEN sequesters CHK1 and initiates genetic instability. Cancer Cell 2005, 7, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Hirose, Y.; Katayama, M.; Mirzoeva, O.K.; Berger, M.S.; Pieper, R.O. Akt activation suppresses Chk2-mediated, methylating agent-induced G2 arrest and protects from temozolomide-induced mitotic catastrophe and cellular senescence. Cancer Res. 2005, 65, 4861–4869. [Google Scholar] [CrossRef] [PubMed]
- Tonic, I.; Yu, W.N.; Park, Y.; Chen, C.C.; Hay, N. Akt activation emulates Chk1 inhibition and Bcl2 overexpression and abrogates G2 cell cycle checkpoint by inhibiting BRCA1 foci. J. Biol. Chem. 2010, 285, 23790–23798. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Hegarat, N.; Black, E.J.; Scott, M.T.; Hochegger, H.; Gillespie, D.A. Akt/PKB suppresses DNA damage processing and checkpoint activation in late G2. J. Cell Biol. 2010, 190, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; Coluccia, A.; di Giovanni, C.; Novellino, E. Cdc25B phosphatase inhibitors in cancer therapy: Latest developments, trends and medicinal chemistry perspective. Anticancer Agents Med. Chem. 2008, 8, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; di Giovanni, C.; Novellino, E. CDC25A and B dual-specificity phosphatase inhibitors: Potential agents for cancer therapy. Curr. Med. Chem. 2009, 16, 1831–1849. [Google Scholar] [CrossRef] [PubMed]
- Kolb, S.; Mondesert, O.; Goddard, M.L.; Jullien, D.; Villoutreix, B.O.; Ducommun, B.; Garbay, C.; Braud, E. Development of novel thiazolopyrimidines as CDC25B phosphatase inhibitors. ChemMedChem 2009, 4, 633–648. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; Cosconati, S.; Limongelli, V.; Novellino, E. Modeling of Cdc25B dual specifity protein phosphatase inhibitors: Docking of ligands and enzymatic inhibition mechanism. ChemMedChem 2006, 1, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Lazo, J.S.; Nemoto, K.; Pestell, K.E.; Cooley, K.; Southwick, E.C.; Mitchell, D.A.; Furey, W.; Gussio, R.; Zaharevitz, D.W.; Joo, B.; et al. Identification of a potent and selective pharmacophore for Cdc25 dual specificity phosphatase inhibitors. Mol. Pharmacol. 2002, 61, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Li, M.; Choi, J.; Cho, H.; Ham, S.W. Structure-based virtual screening approach to identify novel classes of Cdc25B phosphatase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 4372–4375. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Lefterov, I.M.; Wang, M.; Lazo, J.S.; Scott, C.N.; Wilcox, C.S.; Carr, B.I. Binding and inhibition of Cdc25 phosphatases by vitamin K analogues. Biochemistry 2003, 42, 10490–10497. [Google Scholar] [CrossRef] [PubMed]
- Pu, L.; Amoscato, A.A.; Bier, M.E.; Lazo, J.S. Dual G1 and G2 phase inhibition by a novel, selective Cdc25 inhibitor 6-chloro-7-[corrected](2-morpholin-4-ylethylamino)-quinoline-5,8-dione. J. Biol. Chem. 2002, 277, 46877–46885. [Google Scholar] [CrossRef] [PubMed]
- Brisson, M.; Nguyen, T.; Wipf, P.; Joo, B.; Day, B.W.; Skoko, J.S.; Schreiber, E.M.; Foster, C.; Bansal, P.; Lazo, J.S. Redox regulation of Cdc25B by cell-active quinolinediones. Mol. Pharmacol. 2005, 68, 1810–1820. [Google Scholar] [PubMed]
- Zhou, Y.B.; Feng, X.; Wang, L.N.; Du, J.Q.; Zhou, Y.Y.; Yu, H.P.; Zang, Y.; Li, J.Y.; Li, J. LGH00031, a novel ortho-quinonoid inhibitor of cell division cycle 25B, inhibits human cancer cells via ROS generation. Acta. Pharmacol. Sin. 2009, 30, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Brezak, M.C.; Quaranta, M.; Contour-Galcera, M.O.; Lavergne, O.; Mondesert, O.; Auvray, P.; Kasprzyk, P.G.; Prevost, G.P.; Ducommun, B. Inhibition of human tumor cell growth in vivo by an orally bioavailable inhibitor of CDC25 phosphatases. Mol. Cancer Ther. 2005, 4, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Cazales, M.; Boutros, R.; Brezak, M.C.; Chaumeron, S.; Prevost, G.; Ducommun, B. Pharmacologic inhibition of CDC25 phosphatases impairs interphase microtubule dynamics and mitotic spindle assembly. Mol. Cancer Ther. 2007, 6, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Brezak, M.C.; Valette, A.; Quaranta, M.; Contour-Galcera, M.O.; Julien, D.; Lavergne, O.; Frongia, C.; Bigg, D.; Kasprzyk, P.G.; Prevost, G.P.; et al. IRC-083864, a novel bis quinone inhibitor of CDC25 phosphatases active against human cancer cells. Int. J. Cancer 2009, 124, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Hartner, L.P.; Rosen, L.; Hensley, M.; Mendelson, D.; Staddon, A.P.; Chow, W.; Kovalyov, O.; Ruka, W.; Skladowski, K.; Jagiello-Gruszfeld, A.; et al. Phase 2 dose multi-center, open-label study of ARQ 501, a checkpoint activator, in adult patients with persistent, recurrent or metastatic leiomyosarcoma (LMS). ASCO Meeting Abstracts 2007, 25 (Suppl. 18), 20521. [Google Scholar]
- Khong, H.T.; Dreisbach, L.; Kindler, H.L.; Trent, D.F.; Jeziorski, K.G.; Bonderenko, I.; Popiela, T.; Yagovane, D.M.; Dombal, G. A phase 2 study of ARQ 501 in combination with gemcitabine in adult patients with treatment naive, unresectable pancreatic adenocarcinoma. ASCO Meeting Abstracts 2007, 25 (Suppl. 18), 15017. [Google Scholar]
- Song, G.; Valdez, B.C.; Li, Y.; Dominguez, J.R.; Corn, P.; Champlin, R.E.; Andersson, B.S. The histone deacetylase inhibitor SAHA sensitizes acute myeloid leukemia cells to a combination of nucleoside analogs and the DNA-alkylating agent busulfan. Leuk. Lymphoma 2014, 55, 1625–1634. [Google Scholar] [CrossRef]
- Madlener, S.; Svacinova, J.; Kitner, M.; Kopecky, J.; Eytner, R.; Lackner, A.; Vo, T.P.; Frisch, R.; Grusch, M.; de Martin, R.; et al. In vitro anti-inflammatory and anticancer activities of extracts of Acalypha alopecuroidea (Euphorbiaceae). Int. J. Oncol. 2009, 35, 881–891. [Google Scholar] [PubMed]
- Ozmen, A.; Madlener, S.; Bauer, S.; Krasteva, S.; Vonach, C.; Giessrigl, B.; Gridling, M.; Viola, K.; Stark, N.; Saiko, P.; et al. In vitro anti-leukemic activity of the ethno-pharmacological plant Scutellaria orientalis ssp. carica endemic to western Turkey. Phytomedicine 2010, 17, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Vidal, A.; Mazars, A.; Gautier, E.F.; Prevost, G.; Payrastre, B.; Manenti, S. Upregulation of the CDC25A phosphatase down-stream of the NPM/ALK oncogene participates to anaplastic large cell lymphoma enhanced proliferation. Cell Cycle 2009, 8, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Vidal, A.; Ysebaert, L.; Didier, C.; Betous, R.; de Toni, F.; Prade-Houdellier, N.; Demur, C.; Contour-Galcera, M.O.; Prevost, G.P.; Ducommun, B.; et al. Cell adhesion regulates CDC25A expression and proliferation in acute myeloid leukemia. Cancer Res. 2006, 66, 7128–7135. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Nagata, Y.; Tan, L.; Takemura, T.; Shibata, K.; Fujie, M.; Fujisawa, S.; Tanaka, Y.; Toda, M.; Makita, R.; et al. Transcriptional repression of Cdc25B by IER5 inhibits the proliferation of leukemic progenitor cells through NF-YB and p300 in acute myeloid leukemia. PLoS One 2011, 6, e28011. [Google Scholar] [CrossRef] [PubMed]
- Reikvam, H.; Tamburini, J.; Skrede, S.; Holdhus, R.; Poulain, L.; Ersvær, E.; Hatfield, K.J.; Bruserud, Ø. Antileukaemic effect of PI3K-mTOR inhibitors in acute myeloid leukaemia-gene expression profiles reveal CDC25B expression as determinate of pharmacological effect. Br. J. Haematol. 2014, 164, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Ekberg, J.; Persson, J.L. Post-translational modification of cyclin A1 is associated with staurosporine and TNFalpha induced apoptosis in leukemic cells. Mol. Cell. Biochem. 2009, 320, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Caudill, J.S.; Porcher, J.C.; Steensma, D.P. Aberrant pre-mRNA splicing of a highly conserved cell cycle regulator, CDC25C, in myelodysplastic syndromes. Leuk. Lymphoma 2008, 49, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Tamir, A.; Petrocelli, T.; Stetler, K.; Chu, W.; Howard, J.; Croix, B.S.; Slingerland, J.; Ben-David, Y. Stem cell factor inhibits erythroid differentiation by modulating the activity of G1-cyclin-dependent kinase complexes: A role for p27 in erythroid differentiation coupled G1 arrest. Cell Growth Differ. 2000, 11, 269–277. [Google Scholar] [PubMed]
- Levis, M.; Small, D. FLT3: ITDoes matter in leukemia. Leukemia 2003, 17, 1738–1752. [Google Scholar] [CrossRef] [PubMed]
- Dolfini, D.; Gatta, R.; Mantovani, R. NF-Y and the transcriptional activation of CCAAT promoters. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 29–49. [Google Scholar] [CrossRef] [PubMed]
- Cude, K.; Wang, Y.; Choi, H. J.; Hsuan, S.L.; Zhang, H.; Wang, C.Y.; Xia, Z. Regulation of the G2-M cell cycle progression by the ERK5-NFkappaB signaling pathway. J. Cell Biol. 2007, 177, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Reikvam, H.; Olsnes, A.M.; Gjertsen, B.T.; Ersvar, E.; Bruserud, Ø. Nuclear factor-kappaB signaling: A contributor in leukemogenesis and a target for pharmacological intervention in human acute myelogenous leukemia. Crit. Rev. Oncog. 2009, 15, 1–41. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Chen, X.; Rocha, K.; Epling-Burnette, P.K.; Djeu, J.Y.; Liu, Q.; Byrd, J.; Sokol, L.; Lawrence, N.; Pireddu, R.; et al. A critical role for phosphatase haplodeficiency in the selective suppression of deletion 5q MDS by lenalidomide. Proc. Natl. Acad. Sci. USA 2009, 106, 12974–12979. [Google Scholar] [CrossRef] [PubMed]
- Schnerch, D.; Yalcintepe, J.; Schmidts, A.; Becker, H.; Follo, M.; Engelhardt, M.; Wasch, R. Cell cycle control in acute myeloid leukemia. Am. J. Cancer Res. 2012, 2, 508–528. [Google Scholar] [PubMed]
- Iida, H.; Towatari, M.; Tanimoto, M.; Morishita, Y.; Kodera, Y.; Saito, H. Overexpression of cyclin E in acute myelogenous leukemia. Blood 1997, 90, 3707–3713. [Google Scholar] [PubMed]
- Paggi, M.G.; de Fabritiis, P.; Bonetto, F.; Amadio, L.; Santarelli, G.; Spadea, A.; Gentile, F.P.; Floridi, A.; Felsani, A. The retinoblastoma gene product in acute myeloid leukemia: A possible involvement in promyelocytic leukemia. Cancer Res. 1995, 55, 4552–4556. [Google Scholar] [PubMed]
- Pulikkan, J.A.; Dengler, V.; Peramangalam, P.S.; Peer Zada, A.A.; Müller-Tidow, C.; Bohlander, S.K.; Tenen, D.G.; Behre, G. Cell-cycle regulator E2F1 and microRNA-223 comprise an autoregulatory negative feedback loop in acute myeloid leukemia. Blood 2010, 115, 1768–1778. [Google Scholar] [CrossRef] [PubMed]
- Radosevic, N.; Delmer, A.; Tang, R.; Marie, J.P.; Ajchenbaum-Cymbalista, F. Cell cycle regulatory protein expression in fresh acute myeloid leukemia cells and after drug exposure. Leukemia 2001, 15, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Reikvam, H.; Oyan, A.M.; Kalland, K.H.; Hovland, R.; Hatfield, K.J.; Bruserud, Ø. Differences in proliferative capacity of primary human acute myelogenous leukaemia cells are associated with altered gene expression profiles and can be used for subclassification of patients. Cell Prolif. 2013, 46, 554–562. [Google Scholar] [PubMed]
- Sanders, M.A.; Valk, P.J. The evolving molecular genetic landscape in acute myeloid leukaemia. Curr. Opin. Hematol. 2013, 20, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Spoo, A.C.; Lubbert, M.; Wierda, W.G.; Burger, J.A. CXCR4 is a prognostic marker in acute myelogenous leukemia. Blood 2007, 109, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Zolota, V.; Sirinian, C.; Melachrinou, M.; Symeonidis, A.; Bonikos, D.S. Expression of the regulatory cell cycle proteins p21, p27, p14, p16, p53, mdm2, and cyclin E in bone marrow biopsies with acute myeloid leukemia. Correlation with patients’ survival. Pathol. Res. Pract. 2007, 203, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Ånensen, N.; Haaland, I.; D’Santos, C.; van Belle, W.; Gjertsen, B.T. Proteomics of p53 in diagnostics and therapy of acute myeloid leukemia. Curr. Pharm. Biotechnol. 2006, 7, 199–207. [Google Scholar] [CrossRef]
- Cocco, L.; Manzoli, L.; Palka, G.; Martelli, A.M. Nuclear phospholipase C beta1, regulation of the cell cycle and progression of acute myeloid leukemia. Adv. Enzym. Regul. 2005, 45, 126–135. [Google Scholar] [CrossRef]
- Haaland, I.; Opsahl, J.A.; Berven, F.S.; Reikvam, H.; Fredly, H.K.; Haugse, R.; Thiede, B.; McCormack, E.; Lain, S.; Bruserud, Ø.; et al. Molecular mechanisms of nutlin-3 involve acetylation of p53, histones and heat shock proteins in acute myeloid leukemia. Mol. Cancer 2014, 13, 116. [Google Scholar] [CrossRef] [PubMed]
- McCormack, E.; Haaland, I.; Venas, G.; Forthun, R.B.; Huseby, S.; Gausdal, G.; Knappskog, S.; Micklem, D.R.; Lorens, J.B.; Bruserud, Ø.; et al. Synergistic induction of p53 mediated apoptosis by valproic acid and nutlin-3 in acute myeloid leukemia. Leukemia 2012, 26, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Aktas, D.; Arno, M.J.; Rassool, F.; Mufti, G.J. Analysis of CHK2 in patients with myelodysplastic syndromes. Leuk. Res. 2002, 26, 985–987. [Google Scholar] [CrossRef] [PubMed]
- Cavelier, C.; Didier, C.; Prade, N.; Mansat-De Mas, V.; Manenti, S.; Recher, C.; Demur, C.; Ducommun, B. Constitutive activation of the DNA damage signaling pathway in acute myeloid leukemia with complex karyotype: Potential importance for checkpoint targeting therapy. Cancer Res. 2009, 69, 8652–8661. [Google Scholar] [CrossRef] [PubMed]
- Ekberg, J.; Landberg, G.; Holm, C.; Richter, J.; Wolgemuth, D.J.; Persson, J.L. Regulation of the cyclin A1 protein is associated with its differential subcellular localization in hematopoietic and leukemic cells. Oncogene 2004, 23, 9082–9089. [Google Scholar] [CrossRef] [PubMed]
- Karp, J.E.; Thomas, B.M.; Greer, J.M.; Sorge, C.; Gore, S.D.; Pratz, K.W.; Smith, B.D.; Flatten, K.S.; Peterson, K.; Schneider, P.; et al. Phase I and pharmacologic trial of cytosine arabinoside with the selective checkpoint 1 inhibitor Sch 900776 in refractory acute leukemias. Clin. Cancer Res. 2012, 18, 6723–6731. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Takeda, S.; Kumar, R.; Westergard, T.D.; Brown, E.J.; Pandita, T.K.; Cheng, E.H.; Hsieh, J.J. Phosphorylation of MLL by ATR is required for execution of mammalian S-phase checkpoint. Nature 2010, 467, 343–346. [Google Scholar] [CrossRef]
- Liu, L.; Yang, L.; Mi, Y.; Wang, J.; Li, J.; Zhang, Y.; Ma, X.; Qin, T.; Xu, Z.; Xiao, Z. RAD51 and XRCC3 polymorphisms: Impact on the risk and treatment outcomes of de novo inv(16) or t(16;16)/CBFbeta-MYH11(+) acute myeloid leukemia. Leuk. Res. 2011, 35, 1020–1026. [Google Scholar] [CrossRef] [PubMed]
- Müller-Tidow, C.; Wang, W.; Idos, G.E.; Diederichs, S.; Yang, R.; Readhead, C.; Berdel, W.E.; Serve, H.; Saville, M.; Watson, R.; et al. Cyclin A1 directly interacts with B-myb and cyclin A1/cdk2 phosphorylate B-myb at functionally important serine and threonine residues: Tissue-specific regulation of B-myb function. Blood 2001, 97, 2091–2097. [Google Scholar] [PubMed]
- Oguchi, K.; Takagi, M.; Tsuchida, R.; Taya, Y.; Ito, E.; Isoyama, K.; Ishii, E.; Zannini, L.; Delia, D.; Mizutani, S. Missense mutation and defective function of ATM in a childhood acute leukemia patient with MLL gene rearrangement. Blood 2003, 101, 3622–3627. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.F.; Yan, M.; Zhang, D.E. The p21Waf1 pathway is involved in blocking leukemogenesis by the t(8;21) fusion protein AML1-ETO. Blood 2007, 109, 4392–4398. [Google Scholar] [CrossRef] [PubMed]
- Segurado, M.; Tercero, J. A. The S-phase checkpoint: Targeting the replication fork. Biol. Cell 2009, 101, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.Y.; Ren, Z.H.; Jiao, B.; Xiao, R.; Yun, H.Y.; Chen, B.; Zhao, W.L.; Zhu, Q.; Chen, Z.; Chen, S.J. Genetic variations of DNA repair genes and their prognostic significance in patients with acute myeloid leukemia. Int. J. Cancer 2011, 128, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Somyajit, K.; Subramanya, S.; Nagaraju, G. RAD51C: A novel cancer susceptibility gene is linked to Fanconi anemia and breast cancer. Carcinogenesis 2010, 31, 2031–2038. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K. Multiple functions of the S-phase checkpoint mediator. Biosci. Biotechnol. Biochem. 2010, 74, 2367–2373. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Nakamaki, T.; Lubbert, M.; Said, J.; Sakashita, A.; Freyaldenhoven, B.S.; Spira, S.; Huynh, V.; Muller, C.; Koeffler, H.P. Cyclin A1 expression in leukemia and normal hematopoietic cells. Blood 1999, 93, 2067–2074. [Google Scholar] [PubMed]
- Yokozawa, T.; Towatari, M.; Iida, H.; Takeyama, K.; Tanimoto, M.; Kiyoi, H.; Motoji, T.; Asou, N.; Saito, K.; Takeuchi, M.; et al. Prognostic significance of the cell cycle inhibitor p27Kip1 in acute myeloid leukemia. Leukemia 2000, 14, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, T.; Kitanaka, C.; Noguchi, K.; Muramatsu, T.; Asai, A.; Kuchino, Y. Physical and functional interactions between Pim-1 kinase and Cdc25A phosphatase. Implications for the Pim-1-mediated activation of the c-Myc signaling pathway. J. Biol. Chem. 1999, 274, 18659–18666. [Google Scholar] [CrossRef] [PubMed]
- Radomska, H.S.; Alberich-Jorda, M.; Will, B.; Gonzalez, D.; Delwel, R.; Tenen, D.G. Targeting CDK1 promotes FLT3-activated acute myeloid leukemia differentiation through C/EBPalpha. J. Clin. Investig. 2012, 122, 2955–2966. [Google Scholar] [CrossRef] [PubMed]
- Radomska, H.S.; Basseres, D.S.; Zheng, R.; Zhang, P.; Dayaram, T.; Yamamoto, Y.; Sternberg, D.W.; Lokker, N.; Giese, N.A.; Bohlander, S.K.; et al. Block of C/EBP alpha function by phosphorylation in acute myeloid leukemia with FLT3 activating mutations. J. Exp. Med. 2006, 203, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Friedman, A.D.; Levis, M.; Li, L.; Weir, E.G.; Small, D. Internal tandem duplication mutation of FLT3 blocks myeloid differentiation through suppression of C/EBPalpha expression. Blood 2004, 103, 1883–1890. [Google Scholar] [CrossRef] [PubMed]
- Mesa, R.A.; Loegering, D.; Powell, H.L.; Flatten, K.; Arlander, S.J.; Dai, N.T.; Heldebrandt, M.P.; Vroman, B.T.; Smith, B.D.; Karp, J.E.; et al. Heat shock protein 90 inhibition sensitizes acute myelogenous leukemia cells to cytarabine. Blood 2005, 106, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, A.; Biamonti, G. Cellular response to etoposide treatment. Cancer Lett. 2007, 252, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Rossi, R.; Lidonnici, M.R.; Soza, S.; Biamonti, G.; Montecucco, A. The dispersal of replication proteins after Etoposide treatment requires the cooperation of Nbs1 with the ataxia telangiectasia Rad3-related/Chk1 pathway. Cancer Res. 2006, 66, 1675–1683. [Google Scholar] [CrossRef] [PubMed]
- Seedhouse, C.; Grundy, M.; Shang, S.; Ronan, J.; Pimblett, H.; Russell, N.; Pallis, M. Impaired S-phase arrest in acute myeloid leukemia cells with a FLT3 internal tandem duplication treated with clofarabine. Clin. Cancer Res. 2009, 15, 7291–7298. [Google Scholar] [CrossRef] [PubMed]
- Cristobal, I.; Garcia-Orti, L.; Cirauqui, C.; Alonso, M.M.; Calasanz, M.J.; Odero, M.D. PP2A impaired activity is a common event in acute myeloid leukemia and its activation by forskolin has a potent anti-leukemic effect. Leukemia 2011, 25, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Halicka, H.D.; Ozkaynak, M.F.; Levendoglu-Tugal, O.; Sandoval, C.; Seiter, K.; Kajstura, M.; Traganos, F.; Jayabose, S.; Darzynkiewicz, Z. DNA damage response as a biomarker in treatment of leukemias. Cell Cycle 2009, 8, 1720–1724. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Hirano, I.; Okinaka, K.; Takemura, T.; Yokota, D.; Ono, T.; Shigeno, K.; Shibata, K.; Fujisawa, S.; Ohnishi, K. The FOXM1 transcriptional factor promotes the proliferation of leukemia cells through modulation of cell cycle progression in acute myeloid leukemia. Carcinogenesis 2010, 31, 2012–2021. [Google Scholar] [CrossRef] [PubMed]
- Renner, A.G.; Dos Santos, C.; Recher, C.; Bailly, C.; Creancier, L.; Kruczynski, A.; Payrastre, B.; Manenti, S. Polo-like kinase 1 is overexpressed in acute myeloid leukemia and its inhibition preferentially targets the proliferation of leukemic cells. Blood 2009, 114, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Scardocci, A.; Guidi, F.; D'Alo, F.; Gumiero, D.; Fabiani, E.; Diruscio, A.; Martini, M.; Larocca, L.M.; Zollino, M.; Hohaus, S.; et al. Reduced BRCA1 expression due to promoter hypermethylation in therapy-related acute myeloid leukaemia. Br. J. Cancer 2006, 95, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Tsykunova, G.; Reikvam, H.; Ahmed, A.B.; Nepstad, I.; Gjertsen, B.T.; Bruserud, Ø. Targeting of polo-like kinases and their cross talk with Aurora kinases--possible therapeutic strategies in human acute myeloid leukemia? Expert Opin. Investig. Drugs 2012, 21, 587–603. [Google Scholar]
- Ersvær, E.; Zhang, J.Y.; McCormack, E.; Olsnes, A.; Ånensen, N.; Tan, E.M.; Gjertsen, B.T.; Bruserud, Ø. Cyclin B1 is commonly expressed in the cytoplasm of primary human acute myelogenous leukemia cells and serves as a leukemia-associated antigen associated with autoantibody response in a subset of patients. Eur. J. Haematol. 2007, 79, 210–225. [Google Scholar] [CrossRef] [PubMed]
- Boyapati, A.; Yan, M.; Peterson, L.F.; Biggs, J.R.; le Beau, M.M.; Zhang, D.E. A leukemia fusion protein attenuates the spindle checkpoint and promotes aneuploidy. Blood 2007, 109, 3963–3971. [Google Scholar] [CrossRef] [PubMed]
- Kiyomitsu, T.; Obuse, C.; Yanagida, M. Human Blinkin/AF15q14 is required for chromosome alignment and the mitotic checkpoint through direct interaction with Bub1 and BubR1. Dev. Cell 2007, 13, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.F.; Lin, P.M.; Yang, M.C.; Liu, T.C.; Chang, J.G.; Sue, Y.C.; Chen, T.P. Expression of hBUB1 in acute myeloid leukemia. Leuk. Lymphoma 2002, 43, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Cerveira, N.; Correia, C.; Bizarro, S.; Pinto, C.; Lisboa, S.; Mariz, J.M.; Marques, M.; Teixeira, M.R. SEPT2 is a new fusion partner of MLL in acute myeloid leukemia with t(2;11)(q37;q23). Oncogene 2006, 25, 6147–6152. [Google Scholar] [CrossRef] [PubMed]
- Damm, F.; Oberacker, T.; Thol, F.; Surdziel, E.; Wagner, K.; Chaturvedi, A.; Morgan, M.; Bomm, K.; Gohring, G.; Lubbert, M.; et al. Prognostic importance of histone methyltransferase MLL5 expression in acute myeloid leukemia. J. Clin. Oncol. 2011, 29, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, X.N.; Cheng, F.; Liou, Y.C.; Deng, L.W. Phosphorylation of mixed lineage leukemia 5 by CDC2 affects its cellular distribution and is required for mitotic entry. J. Biol. Chem. 2010, 285, 20904–20914. [Google Scholar] [CrossRef] [PubMed]
- Lucena-Araujo, A.R.; de Oliveira, F.M.; Leite-Cueva, S.D.; dos Santos, G.A.; Falcao, R.P.; Rego, E.M. High expression of AURKA and AURKB is associated with unfavorable cytogenetic abnormalities and high white blood cell count in patients with acute myeloid leukemia. Leuk. Res. 2011, 35, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Ricke, R.M.; Jeganathan, K.B.; van Deursen, J.M. Bub1 overexpression induces aneuploidy and tumor formation through Aurora B kinase hyperactivation. J. Cell Biol. 2011, 193, 1049–1064. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Garcia-Manero, G.; Kantarjian, H.M.; Xiao, L.; Vadhan-Raj, S.; Fernandez, M.H.; Nguyen, M.H.; Medeiros, L.J.; Bueso-Ramos, C.E. Analysis of Aurora kinase A expression in CD34(+) blast cells isolated from patients with myelodysplastic syndromes and acute myeloid leukemia. J. Hematop. 2009, 2, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Mundt, K.E.; Golsteyn, R.M.; Lane, H.A.; Nigg, E.A. On the regulation and function of human polo-like kinase 1 (PLK1): Effects of overexpression on cell cycle progression. Biochem. Biophys. Res. Commun. 1997, 239, 377–385. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, A.M.; Foran, J.M.; Fiedler, W.; Serve, H.; Paquette, R.L.; Cooper, M.A.; Yuen, H.A.; Louie, S.G.; Kim, H.; Nicholas, S.; et al. An innovative phase I clinical study demonstrates inhibition of FLT3 phosphorylation by SU11248 in acute myeloid leukemia patients. Clin. Cancer Res. 2003, 9, 5465–5476. [Google Scholar] [PubMed]
- Hatfield, K.J.; Hovland, R.; Oyan, A.M.; Kalland, K.H.; Ryningen, A.; Gjertsen, B.T.; Bruserud, Ø. Release of angiopoietin-1 by primary human acute myelogenous leukemia cells is associated with mutations of nucleophosmin, increased by bone marrow stromal cells and possibly antagonized by high systemic angiopoietin-2 levels. Leukemia 2008, 22, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.Y.; Rosenberg, L.M.; Wang, X.R.; Zhou, Z.M.; Yue, P.; Fu, H.; Khuri, F.R. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005, 65, 7052–7058. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. WHO classification of tumours of haemotopoietic and lymphoid tissues, 4th ed.; IARC Press: Lyon, France, 2008. [Google Scholar]
- Shimizu, T.; Oka, Y.; Awai, N.; Takeda, K. Hypophosphorylation of pRB and repression of cyclin D3 and cdc25A during the granulocytic differentiation of human myeloblastic leukemia ML-1 cells. Leukemia Res. 1999, 23, 901–907. [Google Scholar] [CrossRef]
- Irish, J.M.; Ånensen, N.; Hovland, R.; Skavland, J.; Borresen-Dale, A.L.; Bruserud, Ø.; Nolan, G.P.; Gjertsen, B.T. Flt3 Y591 duplication and Bcl-2 overexpression are detected in acute myeloid leukemia cells with high levels of phosphorylated wild-type p53. Blood 2007, 109, 2589–2596. [Google Scholar] [CrossRef] [PubMed]
- Irish, J.M.; Hovland, R.; Krutzik, P.O.; Perez, O.D.; Bruserud, Ø.; Gjertsen, B.T.; Nolan, G.P. Single cell profiling of potentiated phospho-protein networks in cancer cells. Cell 2004, 118, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Placke, T.; Faber, K.; Nonami, A.; Putwain, S.L.; Salih, H.R.; Heidel, F.H.; Kramer, A.; Root, D.E.; Barbie, D.A.; Kvitsov, A.V.; et al. Requirement for CDK6 in MLL-rearranged acute myeloid leukemia. Blood 2014, 124, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Van der Linden, M.H.; Willekes, M.; van Roon, E.; Seslija, L.; Schneider, P.; Pieters, R.; Stam, R.W. MLL fusion-driven activation of CDK6 potentiates proliferation in MLL-rearranged infant ALL. Cell Cycle 2014, 13, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Abbate, F.; Casini, A.; Scozzafava, A.; Supuran, C.T. Carbonic anhydrase inhibitors: X-ray crystallographic structure of the adduct of human isozyme II with a topically acting antiglaucoma sulfonamide. Bioorg. Med. Chem. Lett. 2004, 14, 2357–2361. [Google Scholar] [CrossRef] [PubMed]
- Owa, T.; Yoshino, H.; Okauchi, T.; Yoshimatsu, K.; Ozawa, Y.; Sugi, N.H.; Nagasu, T.; Koyanagi, N.; Kitoh, K. Discovery of novel antitumor sulfonamides targeting G1 phase of the cell cycle. J. Med. Chem. 1999, 42, 3789–3799. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, Y.; Kusano, K.; Owa, T.; Yokoi, A.; Asada, M.; Yoshimatsu, K. Therapeutic potential and molecular mechanism of a novel sulfonamide anticancer drug, indisulam (E7070) in combination with CPT-11 for cancer treatment. Cancer Chemother. Pharmacol. 2012, 69, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Chow, D.C.; Wenning, L.A.; Miller, W.M.; Papoutsakis, E.T. Modeling pO(2) distributions in the bone marrow hematopoietic compartment. II. Modified Kroghian models. Biophys. J. 2001, 81, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Chow, D.C.; Wenning, L.A.; Miller, W.M.; Papoutsakis, E.T. Modeling pO(2) distributions in the bone marrow hematopoietic compartment. I. Krogh’s model. Biophys. J. 2001, 81, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, K.J.; Bedringsaas, S.L.; Ryningen, A.; Gjertsen, B.T.; Bruserud, Ø. Hypoxia increases HIF-1alpha expression and constitutive cytokine release by primary human acute myeloid leukaemia cells. Eur. Cytokine Netw. 2010, 21, 154–164. [Google Scholar] [PubMed]
- Borthakur, G.; Cortes, J.E.; Kadia, T.M.; Pemmaraju, N.; Jabbour, E.; Jain, N.; Daver, N.; Kantarjian, H. Phase 2, open-label study of E7070, idarubicin and cytarabine in patients with relapsed or refractory acute myeloid leukemia and high-risk myelodisplastic syndrome. Haematologica 2014, 99, 34. [Google Scholar]
- Fredly, H.; Gjertsen, B.T.; Bruserud, Ø. Histone deacetylase inhibition in the treatment of acute myeloid leukemia: The effects of valproic acid on leukemic cells, and the clinical and experimental evidence for combining valproic acid with other antileukemic agents. Clin. Epigenetics 2013, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Fredly, H.; Ersvær, E.; Kittang, A.O.; Tsykunova, G.; Gjertsen, B.T.; Bruserud, Ø. The combination of valproic acid, all-trans retinoic acid and low-dose cytarabine as disease-stabilizing treatment in acute myeloid leukemia. Clin. Epigenetics 2013, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Fredly, H.; Stapnes Bjørnsen, C.; Gjertsen, B.T.; Bruserud, Ø. Combination of the histone deacetylase inhibitor valproic acid with oral hydroxyurea or 6-mercaptopurin can be safe and effective in patients with advanced acute myeloid leukaemia-a report of five cases. Hematology 2010, 15, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Bruserud, Ø.; Liseth, K.; Stamnesfet, S.; Cacic, D.L.; Melve, G.; Kristoffersen, E.; Hervig, T.; Reikvam, H. Hyperleukocytosis and leukocytapheresis in acute leukaemias: Experience from a single centre and review of the literature of leukocytapheresis in acute myeloid leukaemia. Transfus Med. 2013, 23, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Petti, M.C.; Tafuri, A.; Latagliata, R.; Aloe Spiriti, M.A.; Montefusco, E.; Mancini, M.; Meloni, G.; Petrucci, M.T.; Spaeda, A.; Redi, R.; et al. High-dose hydroxyurea in the treatment of poor-risk myeloid leukemias. Ann. Hematol 2003, 82, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.T.; Poon, R.Y. How protein kinases co-ordinate mitosis in animal cells. Biochem. J. 2011, 435, 17–31. [Google Scholar] [CrossRef] [PubMed]
- McCormack, E.; Bruserud, Ø.; Gjertsen, B.T. Animal models of acute myelogenous leukaemia-development, application and future perspectives. Leukemia 2005, 19, 687–706. [Google Scholar] [CrossRef] [PubMed]
- McCormack, E.; Bruserud, Ø.; Gjertsen, B.T. Review: Genetic models of acute myeloid leukaemia. Oncogene 2008, 27, 3765–3779. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brenner, A.K.; Reikvam, H.; Lavecchia, A.; Bruserud, Ø. Therapeutic Targeting the Cell Division Cycle 25 (CDC25) Phosphatases in Human Acute Myeloid Leukemia — The Possibility to Target Several Kinases through Inhibition of the Various CDC25 Isoforms. Molecules 2014, 19, 18414-18447. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules191118414
Brenner AK, Reikvam H, Lavecchia A, Bruserud Ø. Therapeutic Targeting the Cell Division Cycle 25 (CDC25) Phosphatases in Human Acute Myeloid Leukemia — The Possibility to Target Several Kinases through Inhibition of the Various CDC25 Isoforms. Molecules. 2014; 19(11):18414-18447. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules191118414
Chicago/Turabian StyleBrenner, Annette K., Håkon Reikvam, Antonio Lavecchia, and Øystein Bruserud. 2014. "Therapeutic Targeting the Cell Division Cycle 25 (CDC25) Phosphatases in Human Acute Myeloid Leukemia — The Possibility to Target Several Kinases through Inhibition of the Various CDC25 Isoforms" Molecules 19, no. 11: 18414-18447. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules191118414