Determination and Occurrence of Phenoxyacetic Acid Herbicides and Their Transformation Products in Groundwater Using Ultra High Performance Liquid Chromatography Coupled to Tandem Mass Spectrometry

 ,
,
Abstract
:1. Introduction

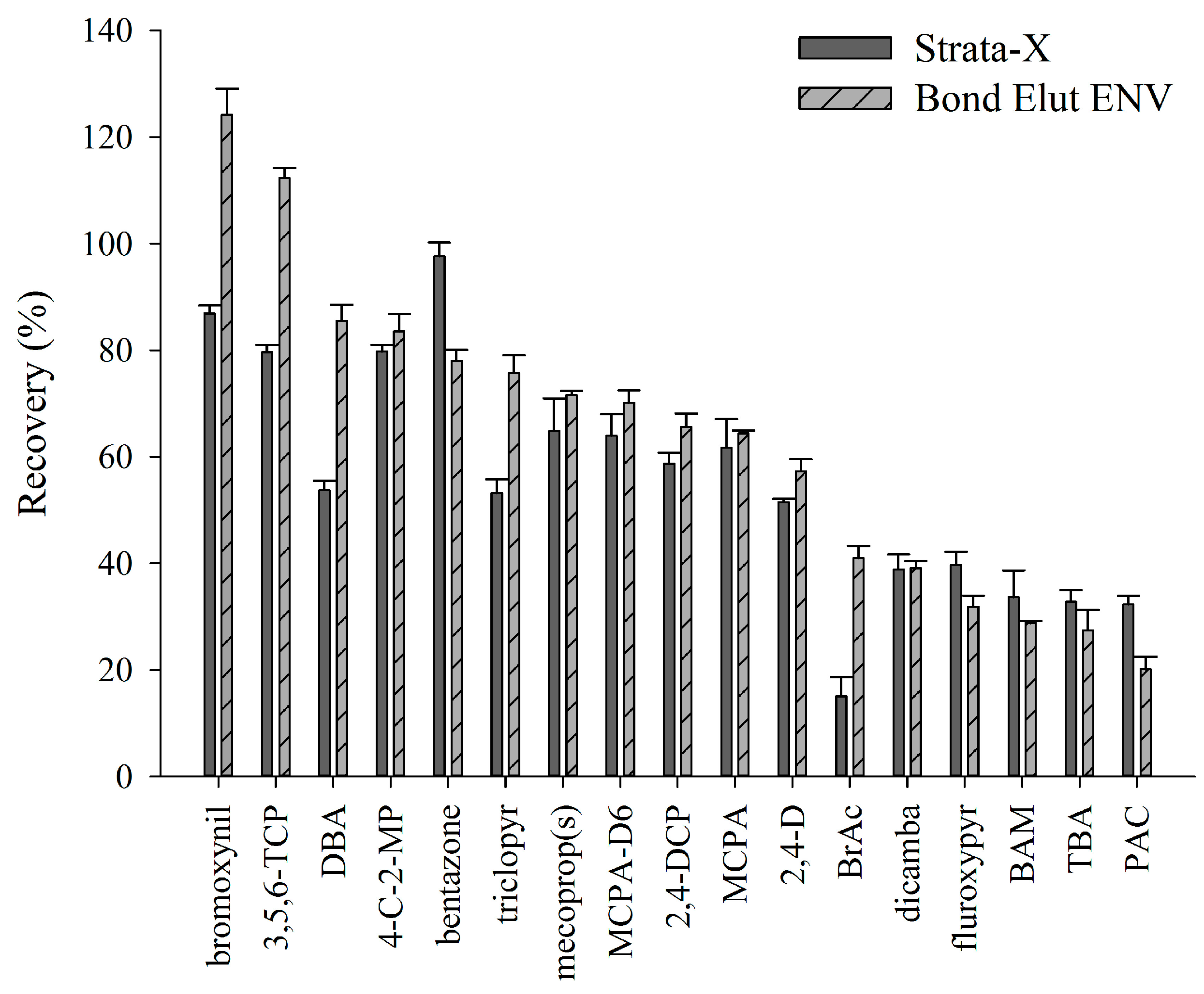{kind=link}
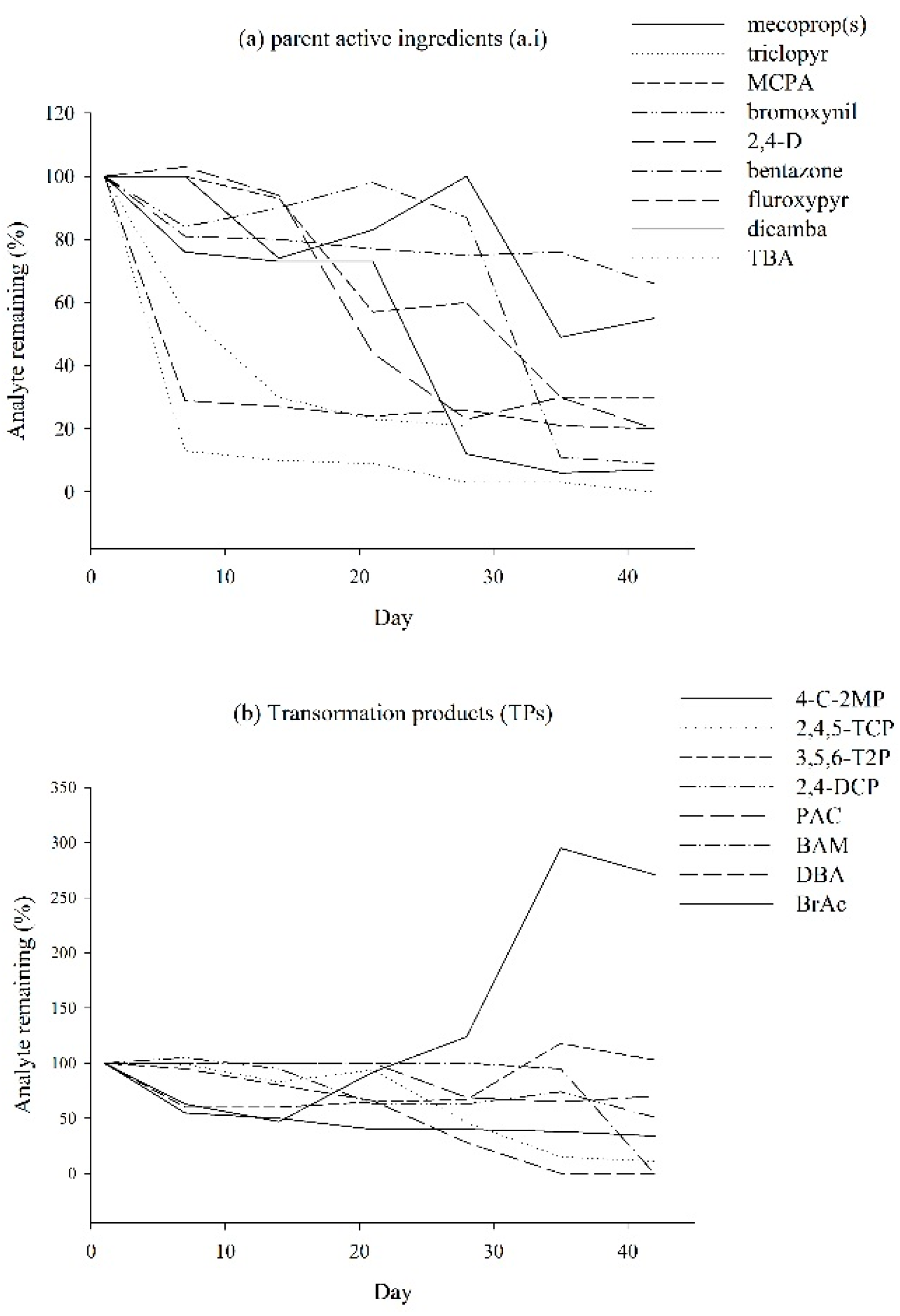{kind=link}
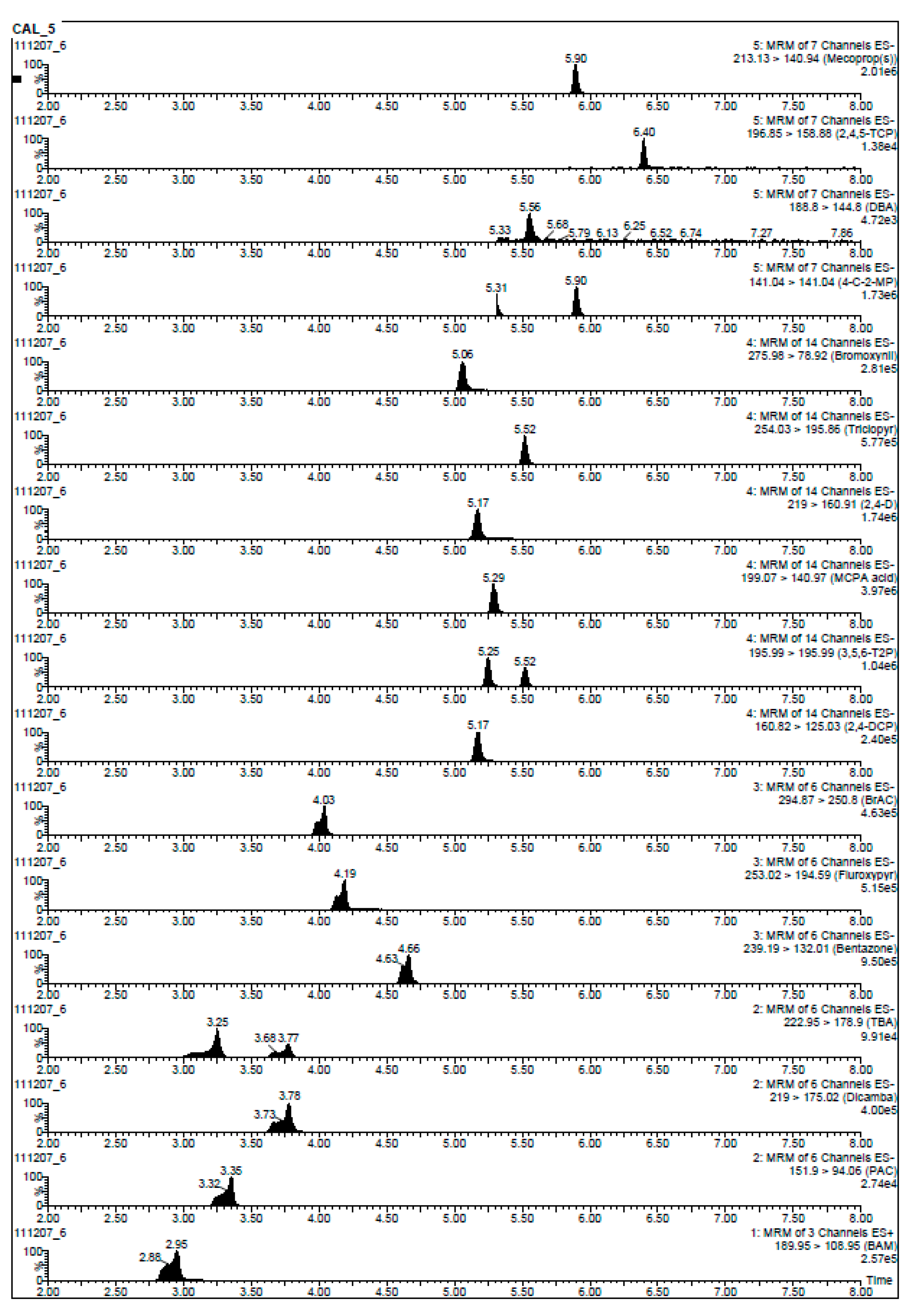{kind=link}
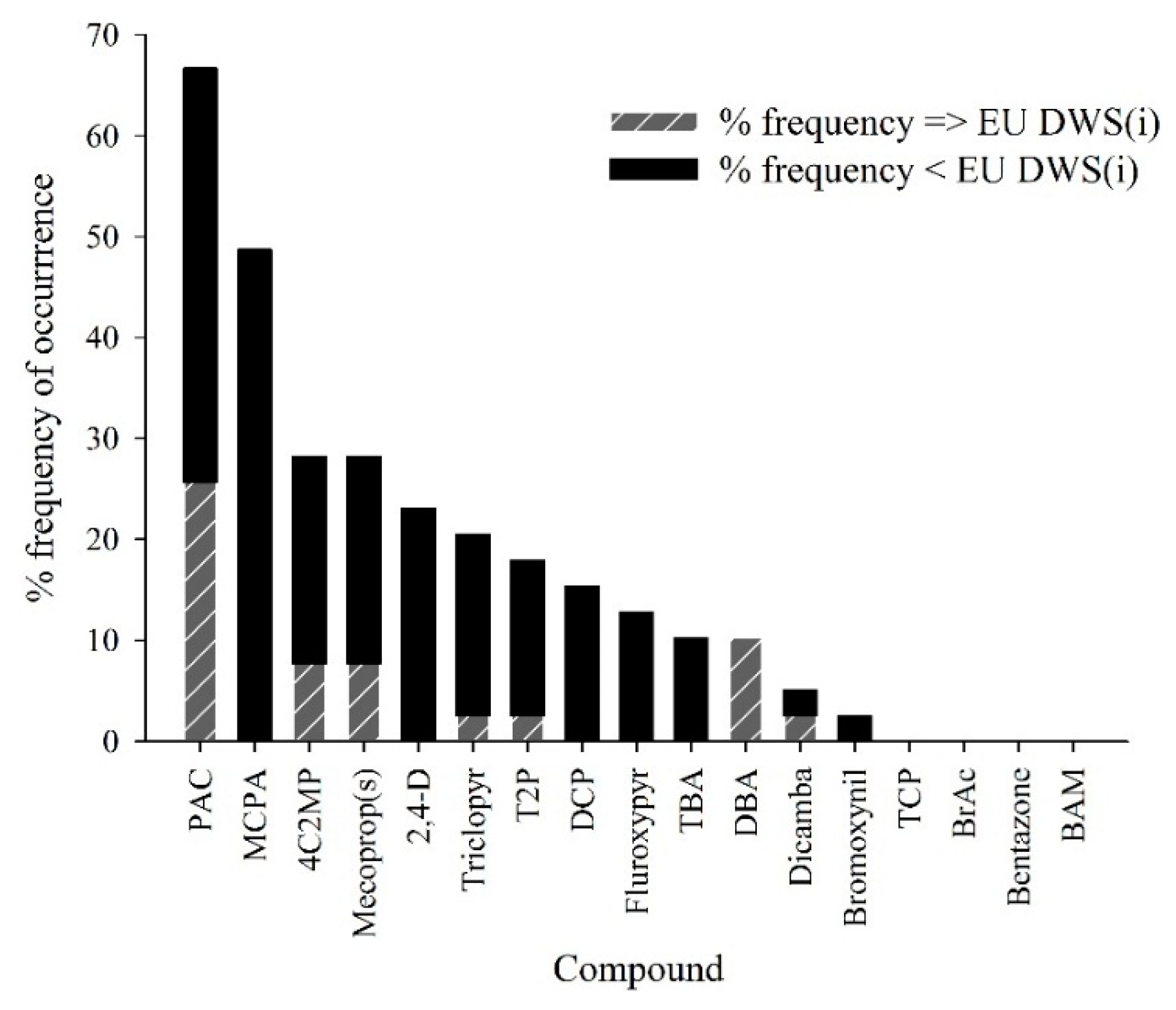{kind=link}
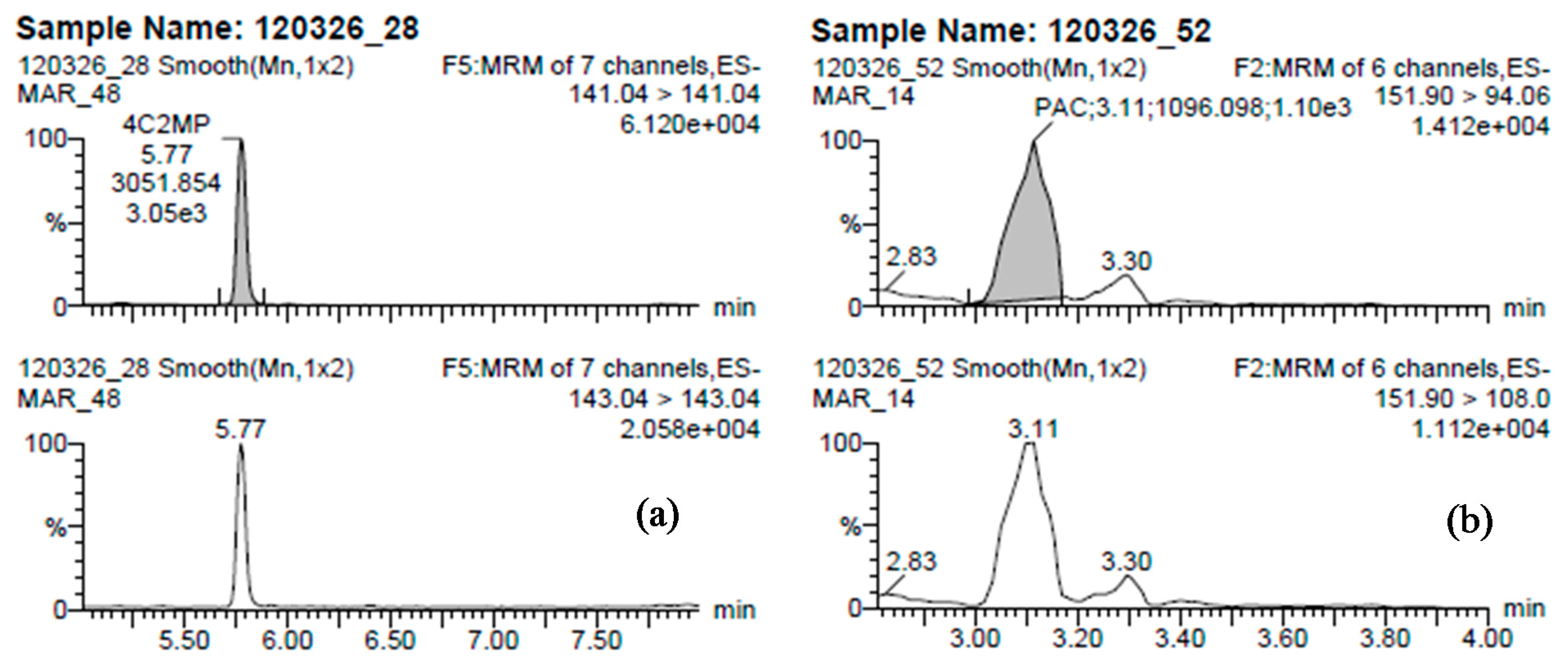{kind=link}
| Parent Active Ingredient (a.i.) | CAS Number | Transformation Product(s) (TP) | CAS Number |
|---|---|---|---|
| MCPA (4-chloro-2-methylphenoxy acetic acid) | 94-74-6 | Phenoxyacetic acid (PAC) | 122-59-8 |
 |  | ||
| 4-chloro-2-methylphenol (4C2MP) | 1570-64-5 | ||
 | |||
| Mecoprop (2-(4-chloro-2-methylphenoxy) propanoic acid) | 7085-19-0 | 4-chloro-2-methylphenol (4C2MP) | 1570-64-5 |
 | | ||
| Mecoprop-p ((2-R)-2-(4-chloro-2- methylphenoxy)propanoic acid) | 16484-77-8 | 4-chloro-2-methylphenol (4C2MP) | 1570-64-5 |
 | | ||
| 2,4-D ((2,4-dichlorophenoxy)acetic acid) | 94-75-7 | Phenoxyacetic acid (PAC) | 122-59-8 |
 | | ||
| 2,4-Dichlorophenol (DCP) | 120-83-2 | ||
 | |||
| Bromoxynil (3,5-dibromo-4-hydroxy-benzonitrile) | 1689-84-5 | 3,5-Dibromo-4-hydroxybenzoic acid (BrAc) | 3337-62-0 |
 |  | ||
| Triclopyr ([3,5,6-trichloropyridinyl)oxy] acetic acid) | 55335-06-3 | 3,5,6-Trichloro-2-pyridinol (T2P) | 6515-38-4 |
 |  | ||
| TBA (2,3,6-trichlorobenzoic acid) | 50-31-7 | 2,4,5-Trichlorophenol (TCP) | 95-95-4 |
 |  | ||
| Dichlobenil (2,6-dichlorobenzonitrile) * | 1194-65-6 | 2,6-Dichlorobenzamide (BAM) | 2008-58-4 |
 |  | ||
| 3,5-dichlorobenzoic acid (DBA) | 50-30-5 | ||
 | |||
| Bentazone (3-(1-methylethyl)-1H-2,1,3-benzothiadiazin-4(3H)-one 2,2-dioxide † | 25057-89-0 | ||
 | |||
| Dicamba (3,6-dichloro-2-methoxybenzoic acid) † | 1918-00-9 | ||
 | |||
| Fluroxypyr [(4-amino-3,5-dichloro-6-fluoro-2-pyridinyl)oxy]acetic acid) † | 69377-81-7 | ||
 | |||
2. Results and Discussion
2.1. Method Development
2.1.1. UHPLC-MS/MS Conditions
| Compound | Empirical Formula | MW 1 (g/mole) | Transition (m/z) | Cone (V) | CE 2 (V) | Dwell Time(s) | ESI Polarity | SRM Window 3 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phenoxyacetic acid herbicides | ||||||||||||||||
| PAC | C8H8O3 | 152.2 | 151.9 → 94.1 | 23 | 14 | 0.07 | Neg. | 2 | ||||||||
| 151.9 → 108.0 | 23 | 9 | 0.07 | |||||||||||||
| Dicamba | C8H6Cl2O3 | 221.0 | 219.0 → 175.0 | 15 | 8 | 0.07 | Neg. | 2 | ||||||||
| 221.1 → 176.8 | 15 | 7 | 0.07 | |||||||||||||
| TBA | C7H3Cl3O2 | 225.5 | 223.0 → 178.9 | 14 | 7 | 0.07 | Neg. | 2 | ||||||||
| 224.9 → 180.9 | 14 | 7 | 0.07 | |||||||||||||
| Bentazone | C10H12N2O3S | 240.3 | 239.2 → 132.0 | 26 | 27 | 0.07 | Neg. | 3 | ||||||||
| 239.2 → 175.0 | 26 | 20 | 0.07 | |||||||||||||
| 239.2 → 196.9 | 26 | 21 | 0.07 | |||||||||||||
| Fluroxypyr | C7H5Cl2FN2O3 | 255.0 | 253.0 → 194.6 | 22 | 13 | 0.07 | Neg. | 3 | ||||||||
| 253.0 → 232.9 | 22 | 4 | 0.07 | |||||||||||||
| BrAc | C7H4Br2O3 | 295.9 | 294.9 → 250.8 | 35 | 18 | 0.005 | Neg. | 3 | ||||||||
| DCP | C6H4Cl2O | 162.9 | 160.8 → 125.0 | 36 | 16 | 0.02 | Neg. | 4 | ||||||||
| 162.9 → 127.0 | 36 | 19 | 0.02 | |||||||||||||
| T2P | C5H2Cl3NO | 198.4 | 196.0 → 196.0 | 28 | 1 | 0.02 | Neg. | 4 | ||||||||
| 197.9 → 197.9 | 28 | 1 | 0.02 | |||||||||||||
| 199.9 → 199.9 | 28 | 1 | 0.02 | |||||||||||||
| MCPA | C9H9ClO3 | 200.6 | 199.1 → 141.0 | 26 | 15 | 0.015 | Neg. | 4 | ||||||||
| 201.1 → 143.0 | 26 | 15 | 0.015 | |||||||||||||
| MCPA D6 (methyl-D3, phenoxy-D3) | C9H9ClO3D6 | 206.6 | 204.9 → 146.9 | 25 | 15 | 0.01 | Neg. | 4 | ||||||||
| 2,4-D | C8H6Cl2O3 | 221.0 | 219.0 → 160.9 | 24 | 16 | 0.015 | Neg. | 4 | ||||||||
| 220.9 → 162.9 | 24 | 15 | 0.015 | |||||||||||||
| Phenoxyacetic acid herbicides | ||||||||||||||||
| Triclopyr | C7H4Cl3NO3 | 256.5 | 254.0 → 195.9 | 19 | 14 | 0.015 | Neg. | 4 | ||||||||
| 254.0 → 218.1 | 19 | 6 | 0.015 | |||||||||||||
| Bromoxynil | C7H3Br2NO | 276.9 | 276.0 → 78.9 | 38 | 26 | 0.015 | Neg. | 4 | ||||||||
| 275.9 → 80.9 | 38 | 26 | 0.015 | |||||||||||||
| 4C2MP | C7H7ClO | 142.6 | 141.0 → 141.0 | 36 | 5 | 0.1 | Neg. | 4 | ||||||||
| 143.0 → 143.0 | 36 | 5 | 0.1 | |||||||||||||
| TCP | C6H3Cl3O | 197.5 | 196.9 → 159.0 | 25 | 18 | 0.08 | Neg. | 5 | ||||||||
| 196.9 → 160.9 | 25 | 18 | 0.08 | |||||||||||||
| Mecoprop | C10H11ClO3 | 214.7 | 213.1 → 140.9 | 25 | 16 | 0.1 | Neg. | 5 | ||||||||
| Mecoprop-p | C10H11ClO3 | 214.7 | 213.1 → 140.9 | 25 | 16 | 0.1 | Neg. | 5 | ||||||||
| Benzonitrile herbicides | ||||||||||||||||
| BAM | C7H5Cl2NO | 190.0 | 190.0 → 109.0 | 34 | 34 | 0.15 | Pos. | 1 | ||||||||
| 190.0 → 144.9 | 34 | 27 | 0.15 | |||||||||||||
| 190.0 → 172.9 | 34 | 18 | 0.15 | |||||||||||||
| DBA | C7H4Cl2O2 | 190.0 | 188.8 → 144.8 | 25 | 11 | 0.015 | Neg. | 5 | ||||||||
| 190.8 → 147.0 | 25 | 11 | 0.015 | |||||||||||||
2.1.2. Sample Preparation Procedure

2.2. Method Validation
2.2.1. Recovery, Precision, Limit of Detection and Calibration
| Analyte | Validation Level (μg·L−1) | LOQ (µg·L−1) | LOD (µg·L−1) | |||||
|---|---|---|---|---|---|---|---|---|
| Accuracy (%) | Precision (%RSD) | |||||||
| 0.02 | 0.04 | 0.06 | 0.02 | 0.04 | 0.06 | |||
| BAM | 95 | 105 | 88 | 18 | 12 | 22 | 0.0009 | 0.0006 |
| PAC | 110 | 105 | 109 | 18 | 2 | 13 | 0.0063 | 0.0015 |
| Dicamba | 106 | 116 | 108 | 13 | 11 | 10 | 0.0004 | 0.0003 |
| TBA | 97 | 111 | 94 | 18 | 9 | 16 | 0.0517 | 0.0047 |
| Bentazone | 112 | 101 | 97 | 23 | 8 | 10 | 0.001 | 0.00009 |
| Fluroxypyr | 92 | 118 | 99 | 30 | 5 | 15 | 0.0007 | 0.0002 |
| BrAc | 101 | 99 | 79 | 16 | 13 | 15 | 0.0023 | 0.0018 |
| DCP | 109 | 103 | 99 | 16 | 8 | 5 | 0.0014 | 0.0007 |
| T2P | 113 | 109 | 94 | 8 | 9 | 10 | 0.0306 | 0.0025 |
| MCPA | 109 | 90 | 71 | 32 | 20 | 18 | 0.0003 | 0.0001 |
| 2,4-D | 112 | 107 | 98 | 14 | 8 | 5 | 0.0005 | 0.0003 |
| Triclopyr | 113 | 108 | 102 | 15 | 8 | 8 | 0.0023 | 0.0004 |
| Bromoxynil | 112 | 106 | 94 | 16 | 15 | 17 | 0.0015 | 0.0002 |
| 4C2MP | 100 | 109 | 103 | 46 | 5 | 25 | 0.0002 | 0.0001 |
| DBA | 103 | 96 | 88 | 45 | 42 | 41 | 0.0625 | 0.0036 |
| TCP | 105 | 108 | 97 | 12 | 7 | 9 | 0.0049 | 0.0012 |
| Mecoprop(s) | 115 | 96 | 102 | 19 | 16 | 11 | 0.0002 | 0.00008 |
2.2.2. Stability of Analytes in Water Samples

2.2.3. Comparison with Published Methods

| Author(s) | Separation | Detection | Extraction | Range of Analytes | Number of Phenoxyacetic Acid Herbicides | Separation Time (min) | Sample Size (mL) | Recovery Range (%) | Detection Limit (µg·L−1) |
|---|---|---|---|---|---|---|---|---|---|
| LOD from this method | UHPLC | MS/MS | SPE | 18 | 16 | 11.9 | 500 | 88–118 | 0.00008–0.0047 (refer to Table 3) |
| Rodil et al. [21] | LC | MS/MS | SPE | 53 | 6 | 41 | 200 | 22–146 | 0.0006 (2,4-D)0.0025 (mecoprop) |
| Marin et al. [25] | UHPLC | MS/MS | SPE | 37 | 3 | 10 | 100 | 70–120 | 0.025 |
| Marchese et al. [17] | LC | MS/MS | Graphite cartridge | 8 | 4 | c.15 | 500 | 85–103 | 0.0001 |
| Solymosné Majzik et al. [26] | LC | MS/MS | SPE | 6 | 4 | 15 | 500 | 76–108 | 0.0011 (dicamba)0.0017 (2,4-D)0.0029 (MCPA)0.0015 (mecoprop) |
| Sklivagou et al. [3] | LC | MS/MS | SPE | 6 | 3 | 15 | 500 | 61–120 | 0.03 (2,4-D)0.003 (MCPA)0.003 (bentazone) |
| Tabani et al. [23] | CE † | UV ‡ | SPE-EME * | 3 | 3 | 20 | 100 | 75–77 | 1.0 (mecoprop)1.5 (MCPA) |
2.3. Application to Environmental Groundwater Samples

| Compound | Minimum | Maximum | Median | Average |
|---|---|---|---|---|
| 2,4-D | 0.002 | 0.007 | 0 | 0.001 |
| 4C2MP | 0.005 | 1.364 | 0.005 | 0.076 |
| BAM | <LOD | <LOD | 0 | - |
| Bentazone | <LOD | <LOD | 0 | - |
| BrAc | <LOD | <LOD | 0 | - |
| Bromoxynil | 0.008 | 0.08 | 0 | 0.003 |
| DBA | 3.019 | 14.218 * | 0 | 1.172 |
| DCP | 0.001 | 0.004 | 0 | 0.001 |
| Dicamba | 0.003 | 0.126 | 0 | 0.005 |
| Fluroxypyr | 0.003 | 0.004 | 0 | 0.001 |
| MCPA | 0.005 | 0.01 | 0.006 | 0.004 |
| Mecoprop(s) | 0.006 | 1.461 | 0.006 | 0.079 |
| PAC | 0.015 | 4.148 * | 0.216 | 0.456 |
| T2P | 0.037 | 0.146 | 0 | 0.012 |
| TBA | 0.005 | 0.026 | 0 | 0.002 |
| TCP | <LOD | <LOD | 0 | - |
| Triclopyr | 0.023 | 0.15 | 0.001 | 0.013 |

| Site Name | Number of Samples Analysed | Number of Replicates across Site Collected and Analysed | Sample Date | pH | Redox (mV) | Conductivity (µS/cm @ 25 °C) | Turbidity (NTU) | Temperature (°C) |
|---|---|---|---|---|---|---|---|---|
| KWDg | 2 | 2 | 13th March | 7.2 | 189 | 595 | 0 | 8.4 |
| KWDa | 2 | 2 | 21st March | 7.9 | 113 | 470 | 0 | 10.2 |
| I/KWDa | 7 | 2 | 13th March | 7.9 | 180 | 406 | 128 | 9.6 |
| FvWDa | 3 | 2 | 14th March | 6.1 | 164 | 127 | 167 | 9.5 |
| FvPDa | 9 | 2 | 14th March | 7.3 | -40 | 263 | 194 | 9.8 |
| FmWDa1 | 10 | 2 | 21st March | 7.1 | 117 | 220 | 28 | 9.7 |
| FmWDa2 | 9 | 2 | 20th March | 6.5 | 120 | 196 | 53 | 10.6 |
3. Experimental Section
3.1. Reagents and Materials
3.2. Standard Solutions and Calibration
3.3. Quality Control
3.4. Sample Preparation
3.5. UHPLC-MS/MS Conditions
3.6. Validation Procedure
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- European Commission. Council Directive 98/83/EC. Off. J. Eur. Commun 1998, L330, 32–54. [Google Scholar]
- Nadin, P. The Use of Plant Protection Products in the European Union Data 1992–2003; European Commission: Luxembourg, France, 2007. [Google Scholar]
- Sklivagou, E.; Papadopoulou, K.; Bakoulis, A. Determination of acid herbicides in water by LC/MS/MS. Desalin. Water Treat. 2010, 13, 320–327. [Google Scholar] [CrossRef]
- Kolpin, D.W.; Barbash, J.E.; Gilliom, R.J. Occurrence of Pesticides in Shallow Groundwater of the United States: Initial Results from the National Water-Quality Assessment Program. Environ. Sci. Technol. 1998, 32, 558–566. [Google Scholar] [CrossRef]
- Arias-Estévez, M.; López-Periago, E.; Martínez-Carballo, E.; Sinal-Gándara, J.; Mejuto, J.-C.; García-Río, L. The mobility and degradation of pesticides in soils and the pollution of groundwater resources. Agric. Ecosyst. Environ. 2008, 123, 247–260. [Google Scholar] [CrossRef]
- Hildebrandt, A.; Guillamón, M.; Lacorte, S.; Tauler, R.; Barceló, D. Impact of pesticides used in agriculture and vineyards to surface and groundwater quality (North Spain). Water Res. 2008, 42, 3315–3326. [Google Scholar] [CrossRef] [PubMed]
- Lanças, F.M.; Olivares, I.R.B.; Alves, P.M. Development, validation and application of a method to analyze phenols in water samples by solid phase microextraction-gas chromatography-flame ionization detector. J. Environ. Sci. Heal. B 2007, 42, 491–498. [Google Scholar] [CrossRef]
- Clausen, L.; Arildskov, N.P.; Larsen, F.; Aamand, J.; Albrechtsen, H.-J. Degradation of the herbicide dichlobenil and its metabolite BAM in soils and subsurface sediments. J. Contam. Hydrol. 2007, 89, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Holtze, M.S.; Sorensen, S.R.; Sorensen, J.; Aamand, J. Microbial degradation of the benzonitrile herbicides dichlobenil, bromoxynil and ioxynil in soil and subsurface environments—Insights into degradation pathways, persistent metabolites and involved degrader organisms. Environ. Pollut. 2008, 154, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Jensen, G.G.; Björklund, E.; Simonsen, A.; Halling-Sørensen, B. Determination of 2,6-dichlorobenzamide and its degradation products in water samples using solid-phase extraction followed by liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2009, 1216, 5199–5206. [Google Scholar] [CrossRef] [PubMed]
- Eades, J.F. A Note on the Persistence of the Herbicide Dichlobenil in Groundwater. Irish J. Agric. Food Res. 1992, 31, 81–83. [Google Scholar]
- European Commission. EC no. 689/2008. Available online: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2008:204:0001:0035:EN:PDF (accessed on 3 December 2014).
- European Parliament. Directive 2000/60/EC. Available online: http://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex:32000L0060 (accessed on 3 December 2014).
- European Commission. 2006/118/EC. Available online: http://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:32006L0118 (accessed on 3 December 2014).
- Cappiello, A.; Famiglini, G.; Palma, P.; Pierini, E.; Termopoli, V.; Trufelli, H. Overcoming Matrix Effects in Liquid Chromatography Mass Spectrometry. Anal. Chem. 2008, 80, 9343–9348. [Google Scholar] [CrossRef] [PubMed]
- Laganà, A.; Bacaloni, A.; de Leva, I.; Faberi, A.; Fago, G.; Marino, A. Occurrence and determination of herbicides and their major transformation products in environmental waters. Anal. Chim. Acta 2002, 462, 187–198. [Google Scholar] [CrossRef]
- Marchese, S.; Perret, D.; Gentili, A.; D’Ascenzo, G.; Faberi, A. Determination of phenoxyacetic acid herbicides and their phenolic metabolites in surface and drinking water. Rapid Commun. Mass Spectrom. 2002, 16, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Gervais, G.; Brosillon, S.; Laplanche, A.; Helen, C. Ultra-pressure liquid chromatography-electrosray tandem mass spectrometry for multiresidue determination of pesticides in water. J. Chromatogr. A 2008, 1902, 163–172. [Google Scholar] [CrossRef]
- Jordan, T.B.; Nichols, D.S.; Kerr, N.I. Selection of SPE cartridge for automated solid-phase extraction of pesticides from water followed by liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2009, 394, 2257–2266. [Google Scholar] [CrossRef] [PubMed]
- Matamoros, V.; Jover, E.; Bayona, J.M. Part-per-Trillion Determination of Pharmaceuticals, Pesticides, and Related Organic Contaminants in River Water by Solid-Phase Extraction Followed by Comprehensive Two-Dimensional Gas Chromatography Time-of-Flight Mass Spectrometry. Anal. Chem. 2009, 82, 699–706. [Google Scholar] [CrossRef]
- Rodil, R.; Quintana, J.B.; López-Mahía, P.; Muniategui-Lorenzo, S.; Prada-Rodríguez, D. Multi-residue analytical method for the determination of emerging pollutants in water by solid-phase extraction and liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2009, 1216, 2958–2969. [Google Scholar] [CrossRef] [PubMed]
- Chiron, S.; Comoretto, L.; Rinaldi, E.; Maurino, V.; Minero, C.; Vione, D. Pesticide by-products in the Rhône delta (Southern France). The case of 4-chloro-2-methylphenol and of its nitroderivative. Chemosphere 2009, 74, 599–604. [Google Scholar]
- Tabani, H.; Fakhari, A.R.; Shahsavani, A.; Behbahani, M.; Salarian, M.; Bagheri, A.; Nojavan, S. Combination of graphene oxide-based solid phase extraction and electro membrane extraction for the preconcentration of chlorophenoxy acid herbicides in environmental samples. J. Chromatogr. A 2013, 1300, 227–235. [Google Scholar] [CrossRef] [PubMed]
- European Commission. SANCO/10232/2006. Available online: http://ec.europa.eu/food/plant/resources/qualcontrol_en.pdf (accessed on 3 December 2014).
- Marín, J.M.; Gracia-Lor, E.; Sancho, J.V.; López, F.J.; Hernández, F. Application of ultra-high-pressure liquid chromatography–tandem mass spectrometry to the determination of multi-class pesticides in environmental and wastewater samples: Study of matrix effects. J. Chromatogr. A 2009, 1216, 1410–1420. [Google Scholar] [CrossRef] [PubMed]
- SolymosnéMajzik, E.; Tóth, F.; Bneke, L.; Kiss, Z. SPE-LC-MS-MS Determination of Phenoxy Acid Herbicides in Surface and Ground Water. Chromatographia 2006, 63, 105–109. [Google Scholar] [CrossRef]
- Fava, L.; Orru, M.A.; Crobe, A.; Caracciolo, A.B.; Bottoni, P.; Funari, E. Pesticide metabolites as contaminants of groundwater resources: assessment of the leaching potential of endosulfan sulfate, 2,6-dichlorobenzoic acid, 3,4-dichloroaniline, 2,4-dichlorophenol and 4-chloro-2-methylphenol. Microchem. J. 2005, 79, 207–211. [Google Scholar] [CrossRef]
- Roberts, T.R. Metabolic pathways of Agrochemicals: Part.2. Insecticides and Fungicides. R. Soc. Chem. 1999, 1–1500. [Google Scholar]
- Horgan, E. Waterways Ireland, Carrick-on-Shannon, Ireland. Personal communication. 2011. [Google Scholar]
- Conroy, K. Pesticide Control Service, Department of Agriculture, Celbridge, Ireland. Personal communication. 2011. [Google Scholar]
- Onneby, K.; Jonsson, A.; Stenstrom, J. A new concept for reduction of diffuse contamination by simultaneous application of pesticide and pesticide-degrading microorganisms. Biodegradation 2010, 21, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Moody, A. Pesticide Control Service, Department of Agriculture, Celbridge, Ireland. Personal communication. 2011. [Google Scholar]
- Vroumsia, T.; Steiman, R.; Seigle-Murandi, F.; Benoit-Guyod, J.L. Groupe pour l’Étude du Devenir des Xénobiotiques dans l’Environnement (GEDEXE). Fungal bioconversion of 2,4-dichlorophenoxyacetic acid (2,4-D) and 2,4-dichlorophenol (2,4-DCP). Chemosphere 2005, 60, 1471–1480. [Google Scholar] [PubMed]
- Zertal, A.; Jacquet, M.; Lave’drine, B.; Sehili, T. Photodegradation of chlorinated pesticides dispersed on sand. Chemosphere 2005, 58, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Zertal, A.; Sehili, T.; Boule, P. Photochemical behaviour of 4-chloro-2-methylphenoxyacetic acid: Influence of pH and irridiation wavelength. J. Photochem. Photobiol. A 2001, 146, 37–48. [Google Scholar] [CrossRef]
- Vione, D.; Khanra, S.; Das, R.; Minero, C.; Maurino, V.; Brigante, M.; Mailhot, G. Effect of dissolved organic compounds on the photodegradation of the herbicide MCPA in aqueous solution. Water Res. 2010, 44, 6053–6062. [Google Scholar] [CrossRef] [PubMed]
- Buss, S.R.; Thrasher, J.; Morgan, P.; Smith, J.W.N. A review of mecoprop attenuation in the subsurface. Q. J. Eng. Geol. Hydrog. 2006, 39, 283–292. [Google Scholar] [CrossRef]
- Tett, V.A.; Willetts, A.J.; Lappin-Scott, H.M. Enantioselective degradation of the herbicide mecoprop by mixed and pure bacterial cultures. FEMS Microbiol. Ecol. 1994, 14, 191–200. [Google Scholar] [CrossRef]
- Klint, M.; Arvin, E.; Jfnsen, B.K. Degradation of the pesticides mecoprop and atrazine in unpolluted sandy aquifers. J. Environ. Qual. 1993, 22, 262–266. [Google Scholar] [CrossRef]
- Agertved, J.; Rügge, K.; Barker, J.F. Transformation of the Herbicides MCPP and Atrazine under Natural Aquifer Conditions. Ground Water 1992, 30, 500–506. [Google Scholar] [CrossRef]
- Harrison, I.; Williams, G.M.; Carlick, C.A. Enantioselective biodegradation of mecoprop in aerobic and anaerobic microcosms. Chemosphere 2003, 53, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McManus, S.-L.; Moloney, M.; Richards, K.G.; Coxon, C.E.; Danaher, M. Determination and Occurrence of Phenoxyacetic Acid Herbicides and Their Transformation Products in Groundwater Using Ultra High Performance Liquid Chromatography Coupled to Tandem Mass Spectrometry. Molecules 2014, 19, 20627-20649. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules191220627
McManus S-L, Moloney M, Richards KG, Coxon CE, Danaher M. Determination and Occurrence of Phenoxyacetic Acid Herbicides and Their Transformation Products in Groundwater Using Ultra High Performance Liquid Chromatography Coupled to Tandem Mass Spectrometry. Molecules. 2014; 19(12):20627-20649. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules191220627
Chicago/Turabian StyleMcManus, Sarah-Louise, Mary Moloney, Karl G. Richards, Catherine E. Coxon, and Martin Danaher. 2014. "Determination and Occurrence of Phenoxyacetic Acid Herbicides and Their Transformation Products in Groundwater Using Ultra High Performance Liquid Chromatography Coupled to Tandem Mass Spectrometry" Molecules 19, no. 12: 20627-20649. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules191220627