Tyrosine Kinase Inhibitors as Reversal Agents for ABC Transporter Mediated Drug Resistance


Abstract
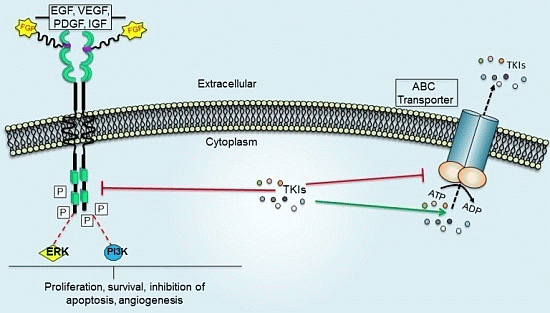
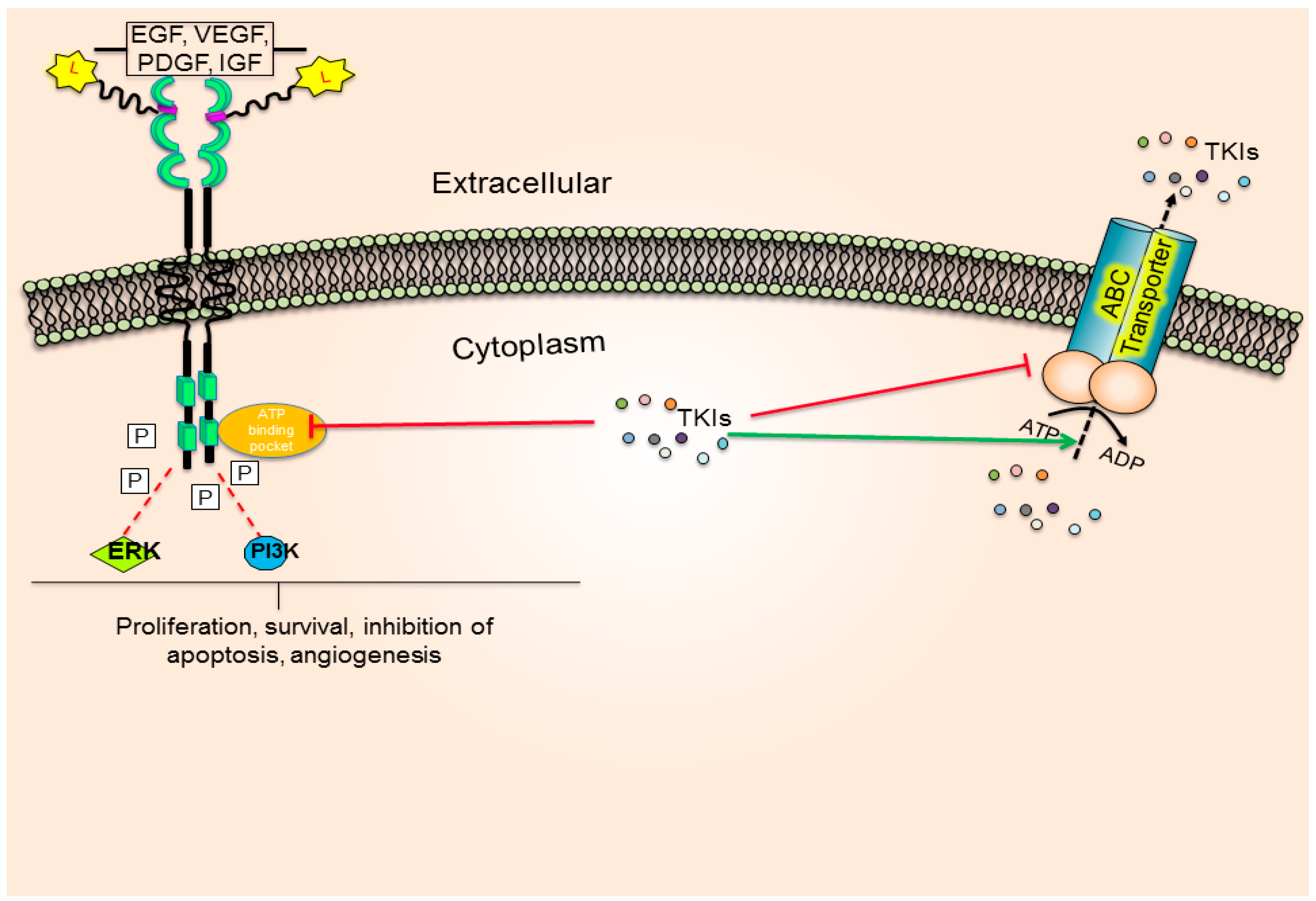
:
1. Tyrosine Kinases and Their Role in Cancer
2. TKIs and Their Role in Cancer Chemotherapy
3. Multidrug Resistance

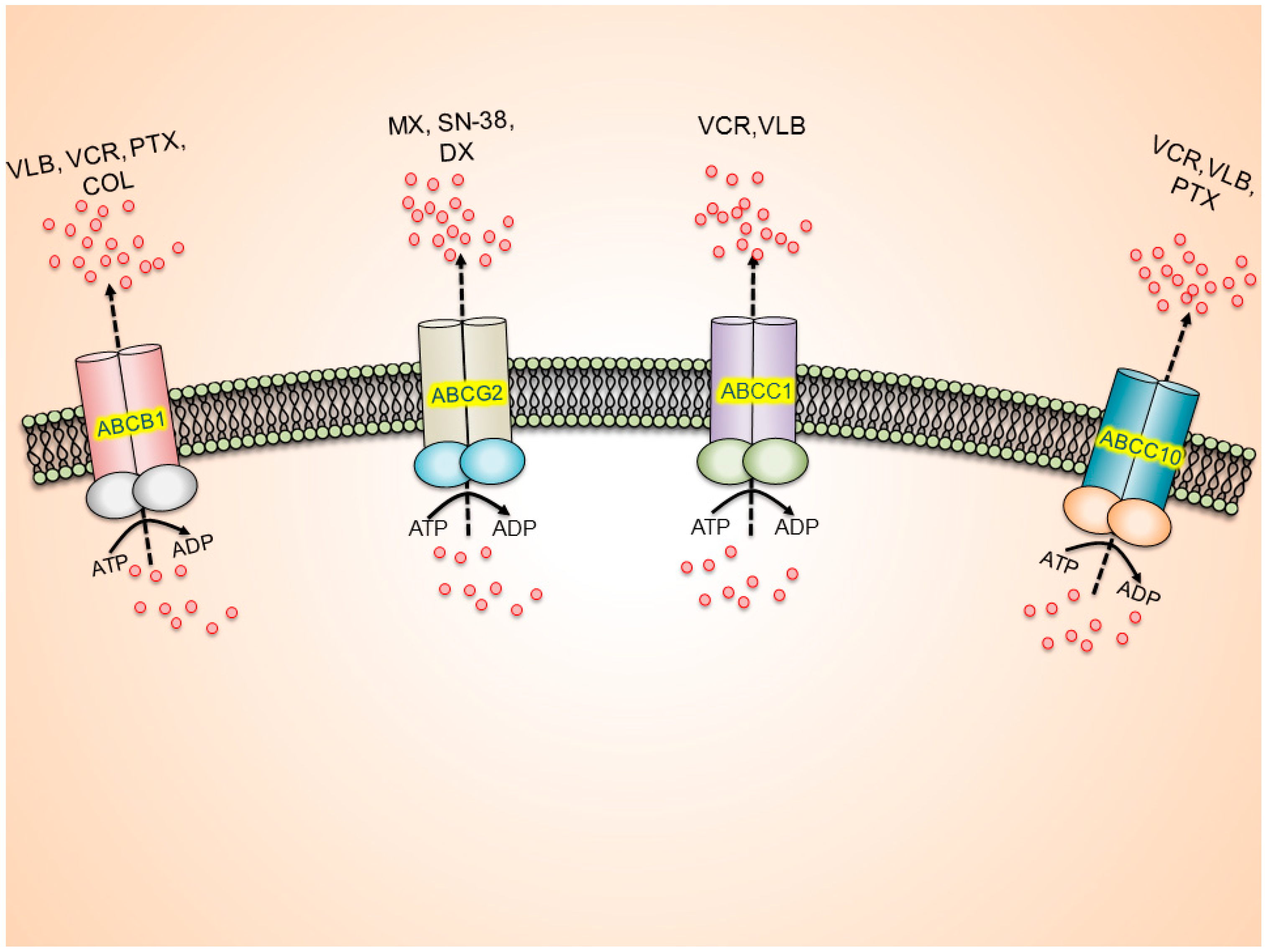
3.1. ATP-Binding Cassette (ABC) Transporters
3.1.1. ABCB1/P-glycoprotein (P-gp/MDR1)
3.1.2. ABCC/Multidrug Resistance Proteins (MRPs)
3.1.3. ABCG2/Breast Cancer Resistance Protein (BCRP)/Mitoxantrone Resistant Protein (MXR)
4. Role of ABC Drug Transporters in the Development of Resistance to TKIs
Impact of ABC Transporters on the Pharmacokinetics and Toxicity of TKIs
5. The ABC Transporter Mediated MDR Modulated by Tyrosine Kinase Inhibitors

5.1. BCR- ABL Tyrosine Kinase Inhibitor
5.1.1. Imatinib Mesylate (Gleevec®)

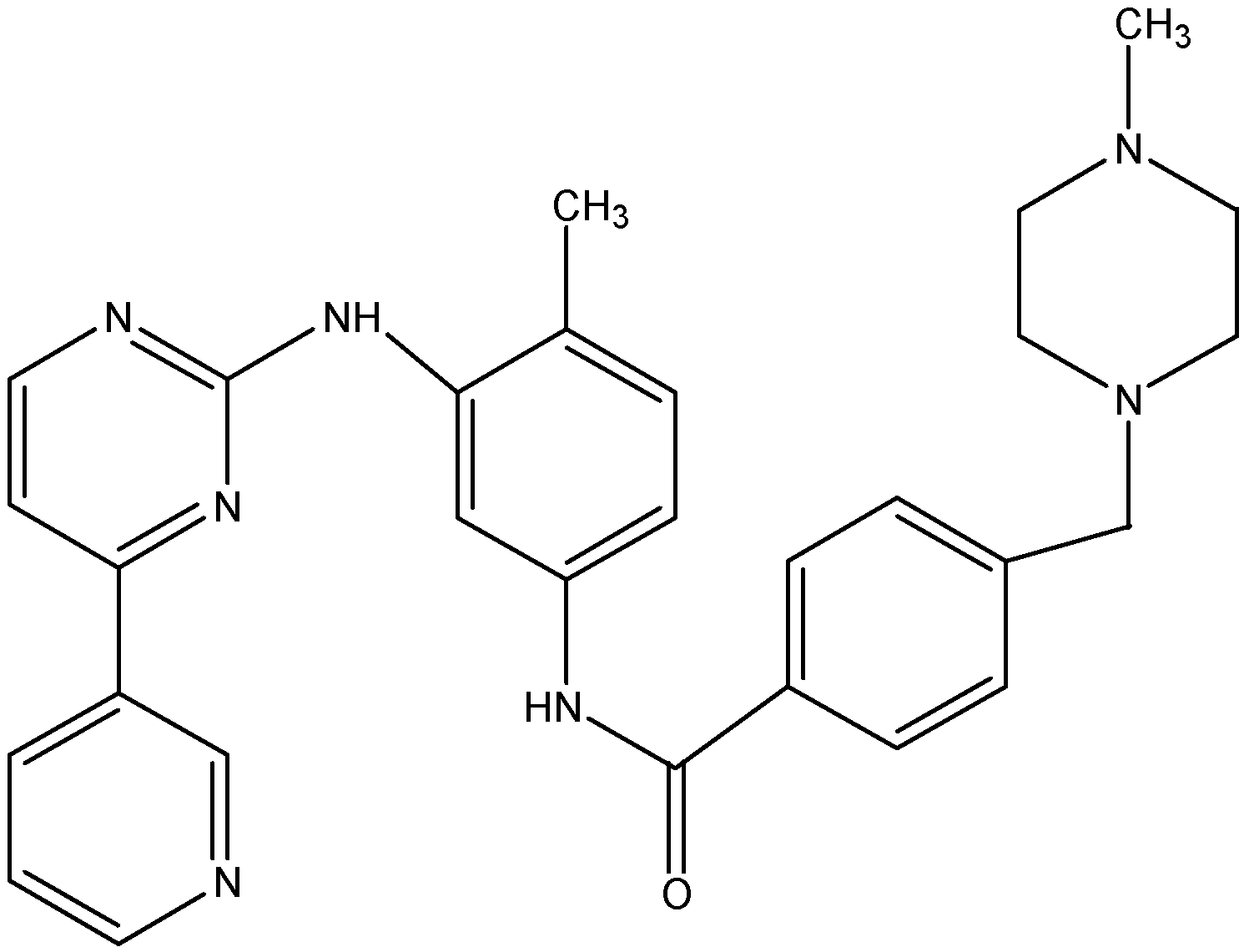
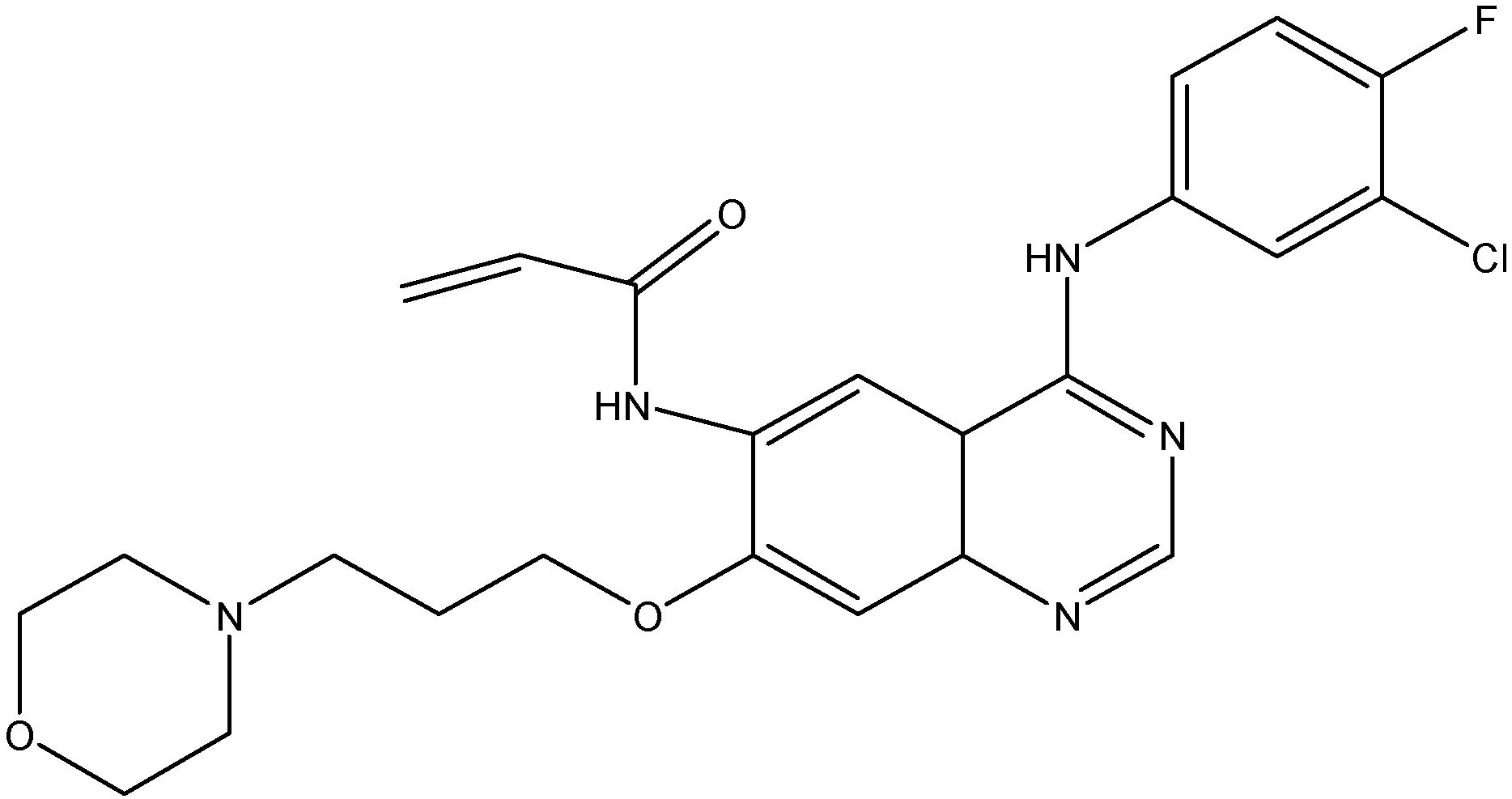
5.1.2. Nilotinib (AMN107)

5.1.3. Dasatinib (Sprycel®)

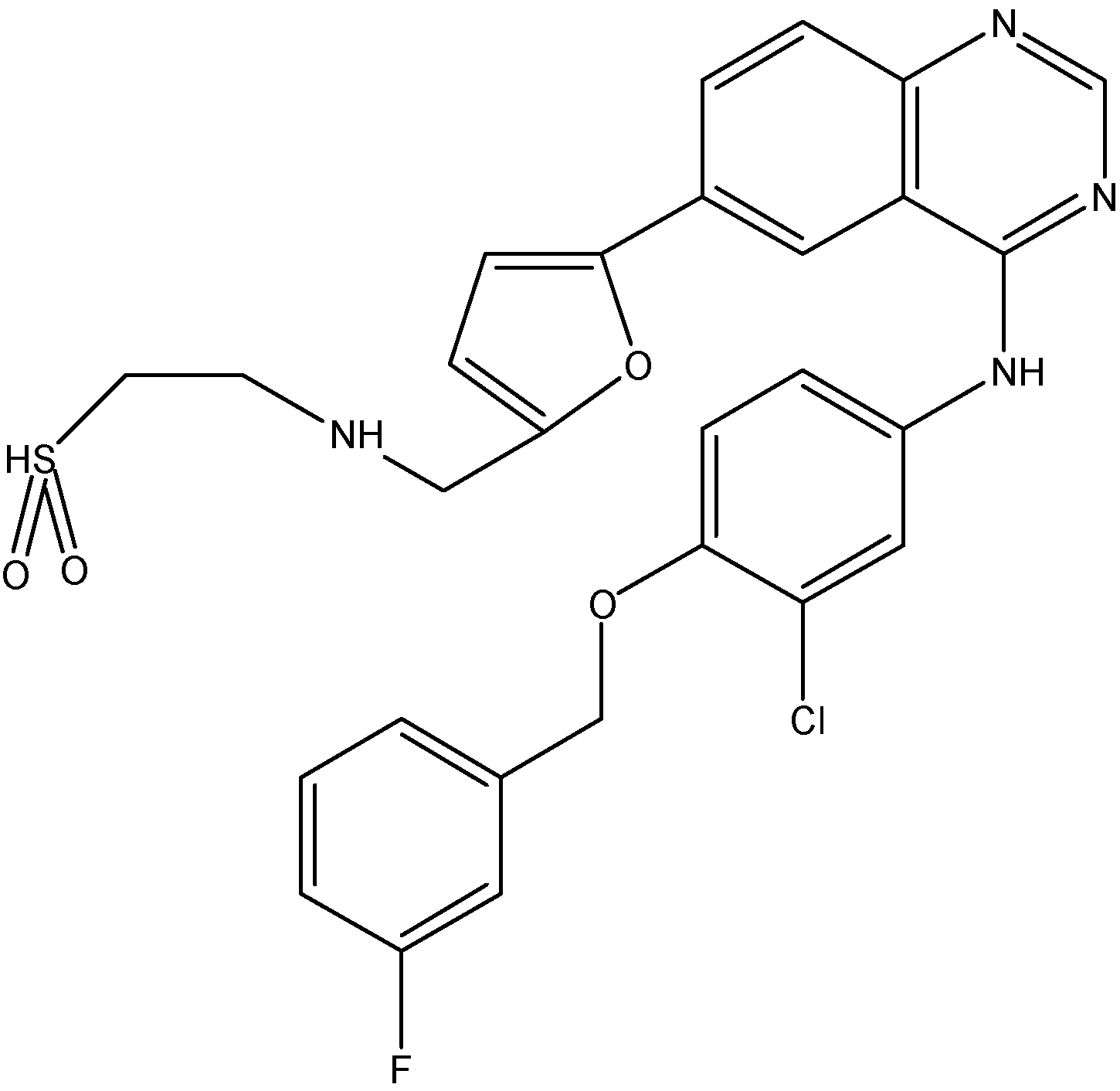
5.1.4. Ponatinib (Iclusig®)

5.2. Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors
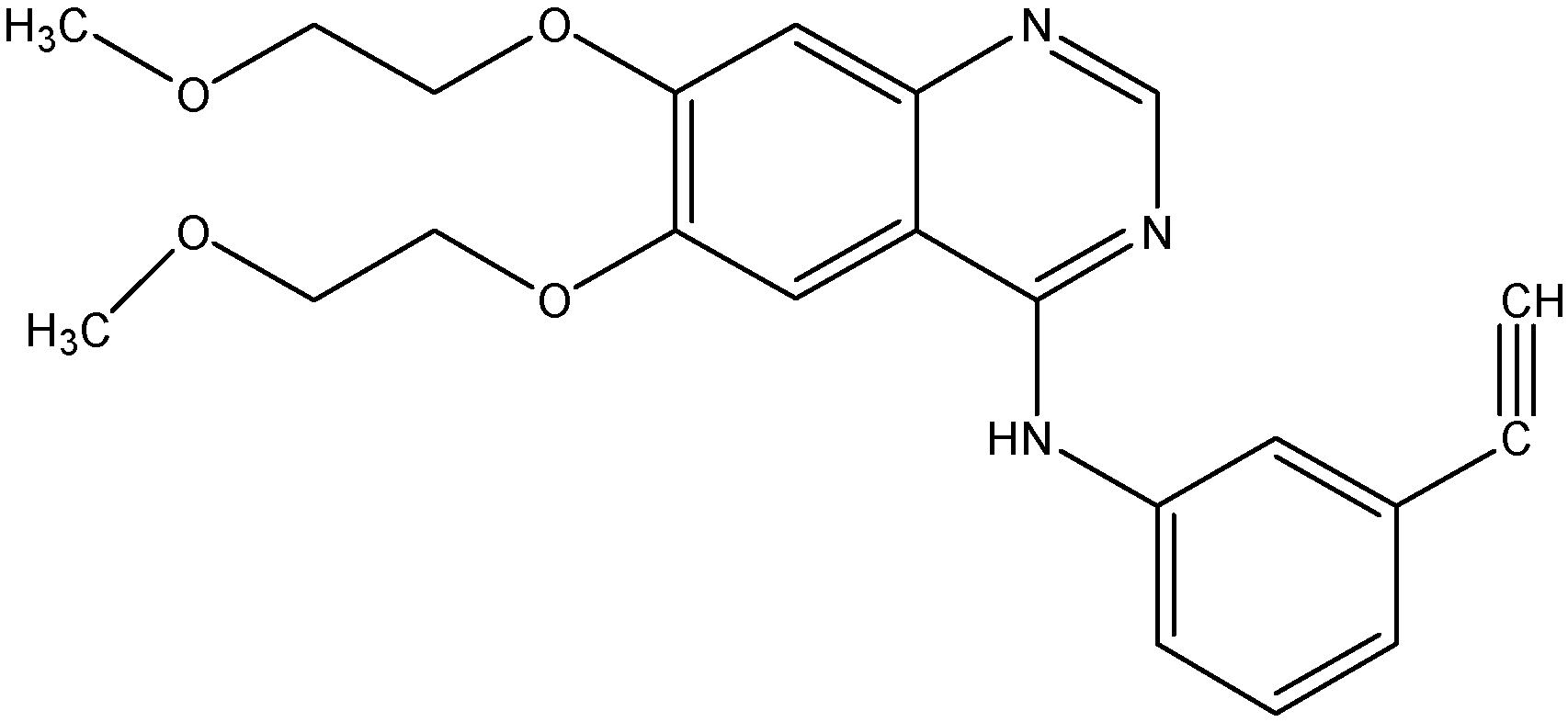
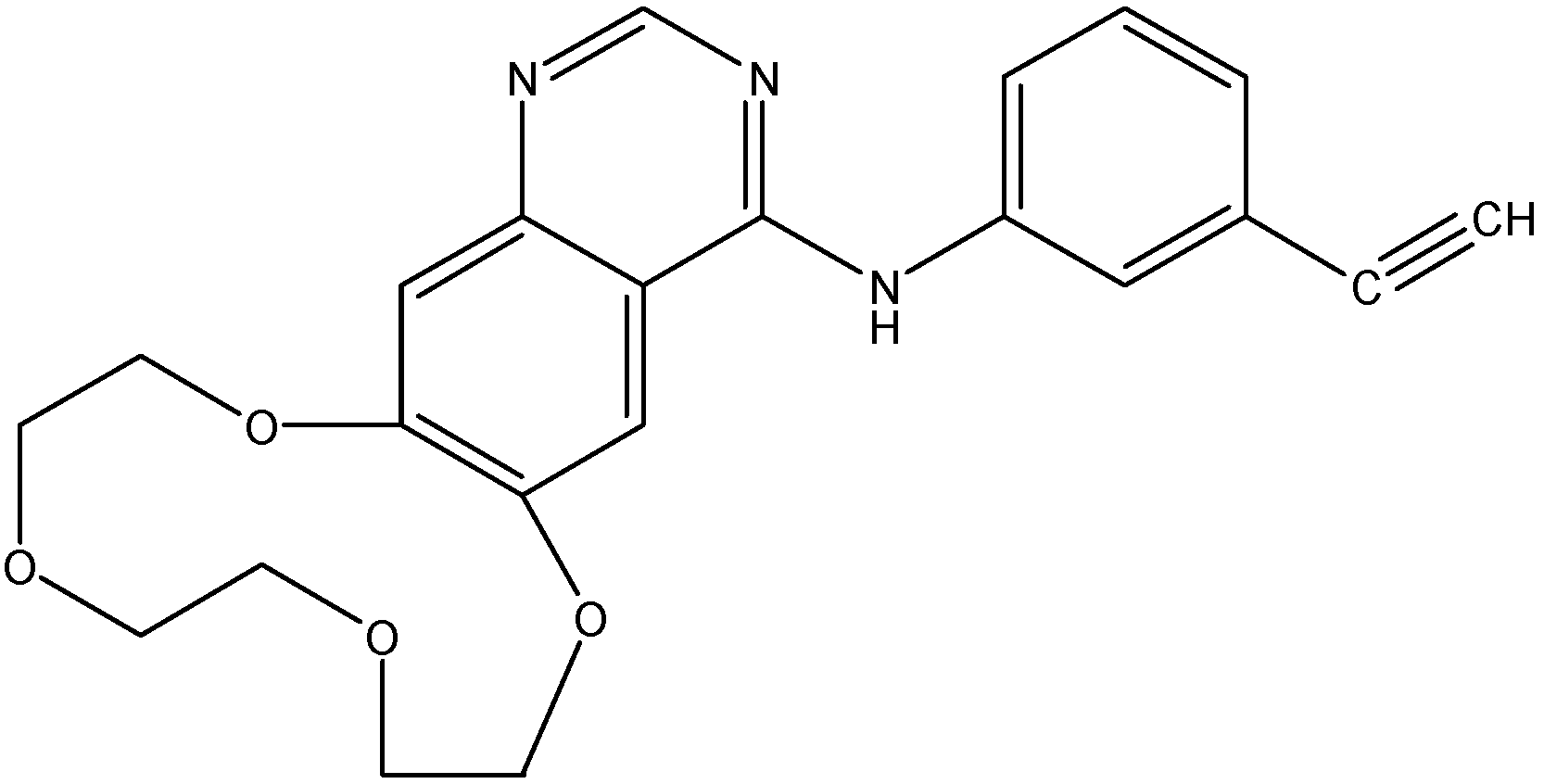
5.2.1. AST1306

5.2.2. Gefitinib (Iressa®)

5.2.3. Erlotinib (OSI-774; Tarceva®)

5.2.4. Lapatinib (GW-572016)
5.2.5. Canertinib (CI-1033)


5.2.6. Icotinib

5.3. Vascular Endothelial Growth Factor Tyrosine Kinase Inhibitors
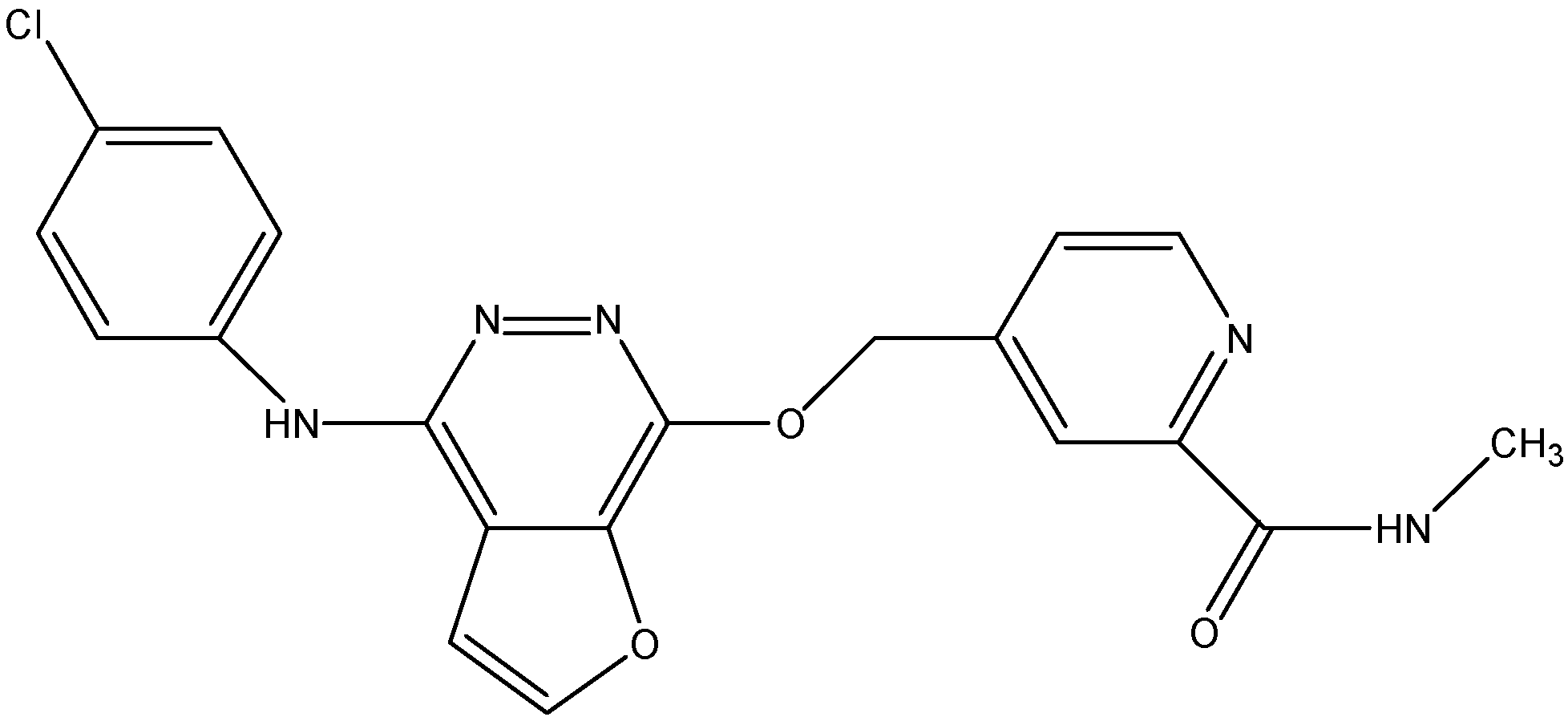
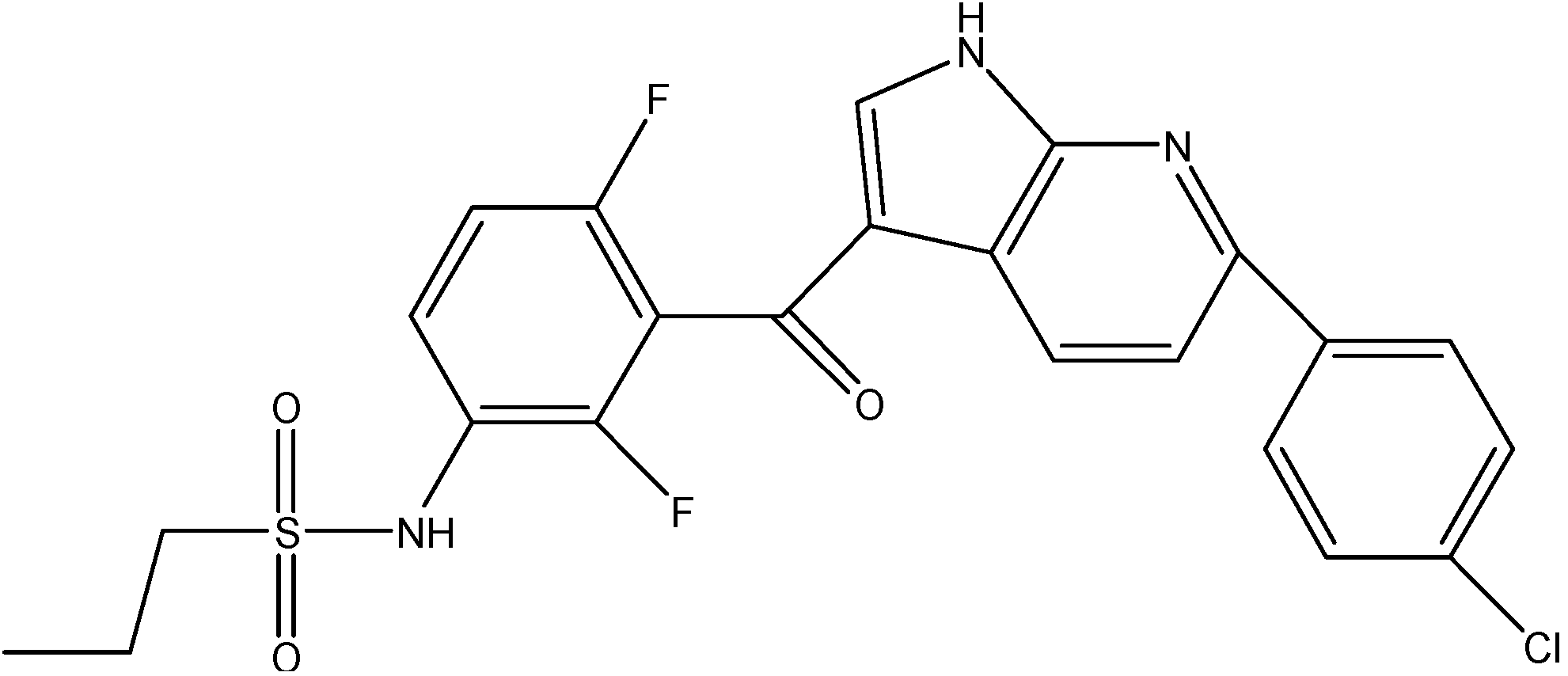
5.3.1. Telatinib

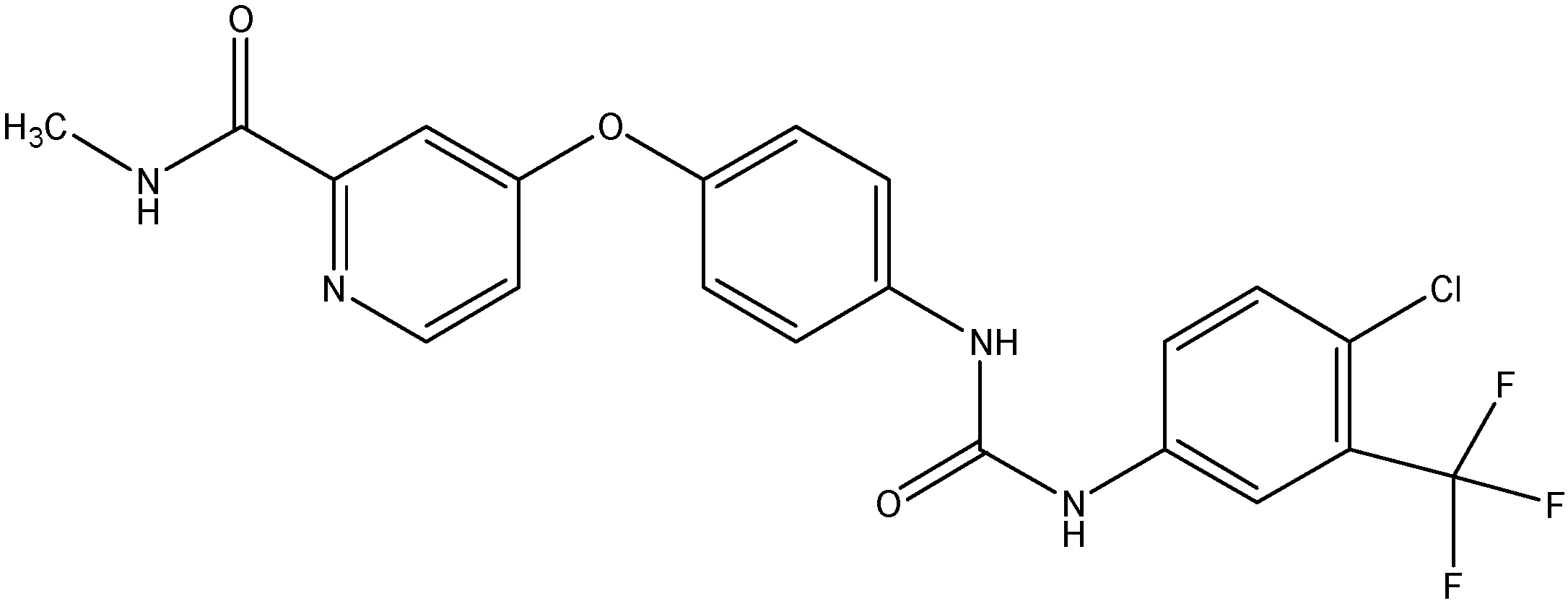
5.3.2. Sunitinib (SU11248)

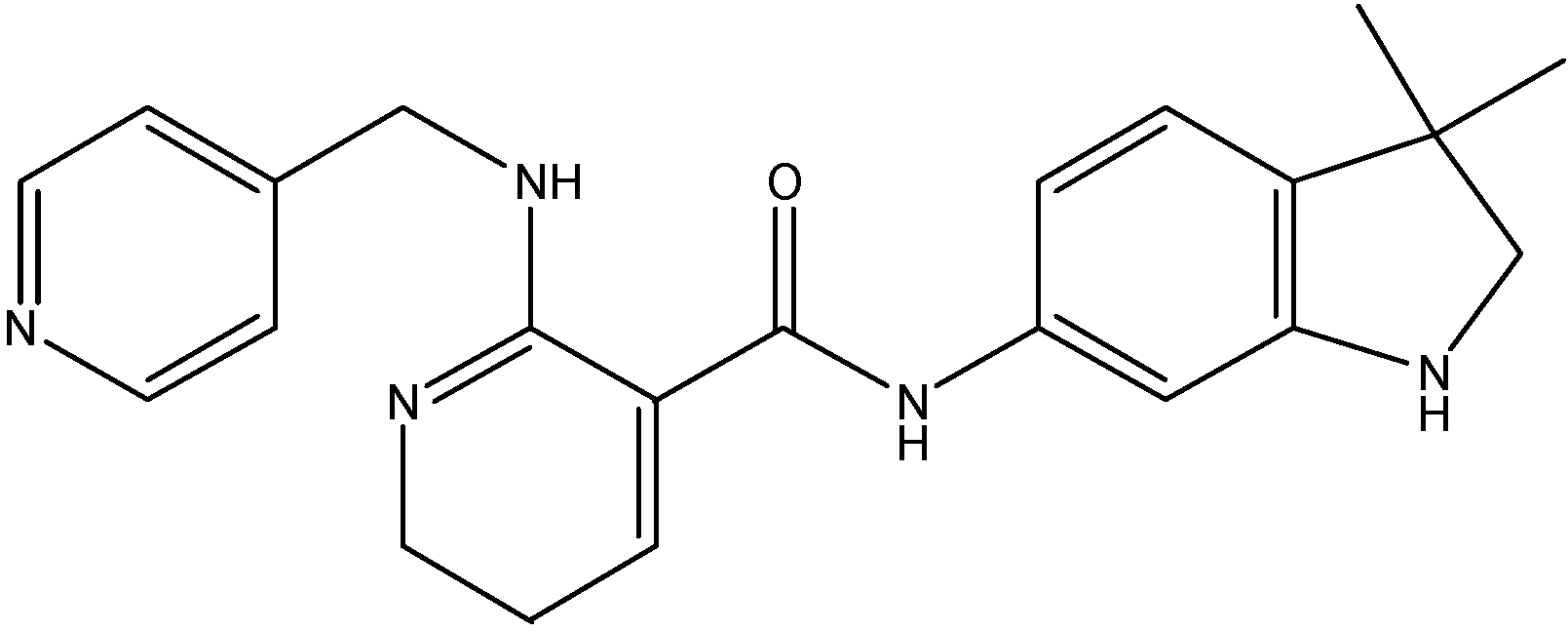
5.3.3. Sorafenib (BAY 43-9006)

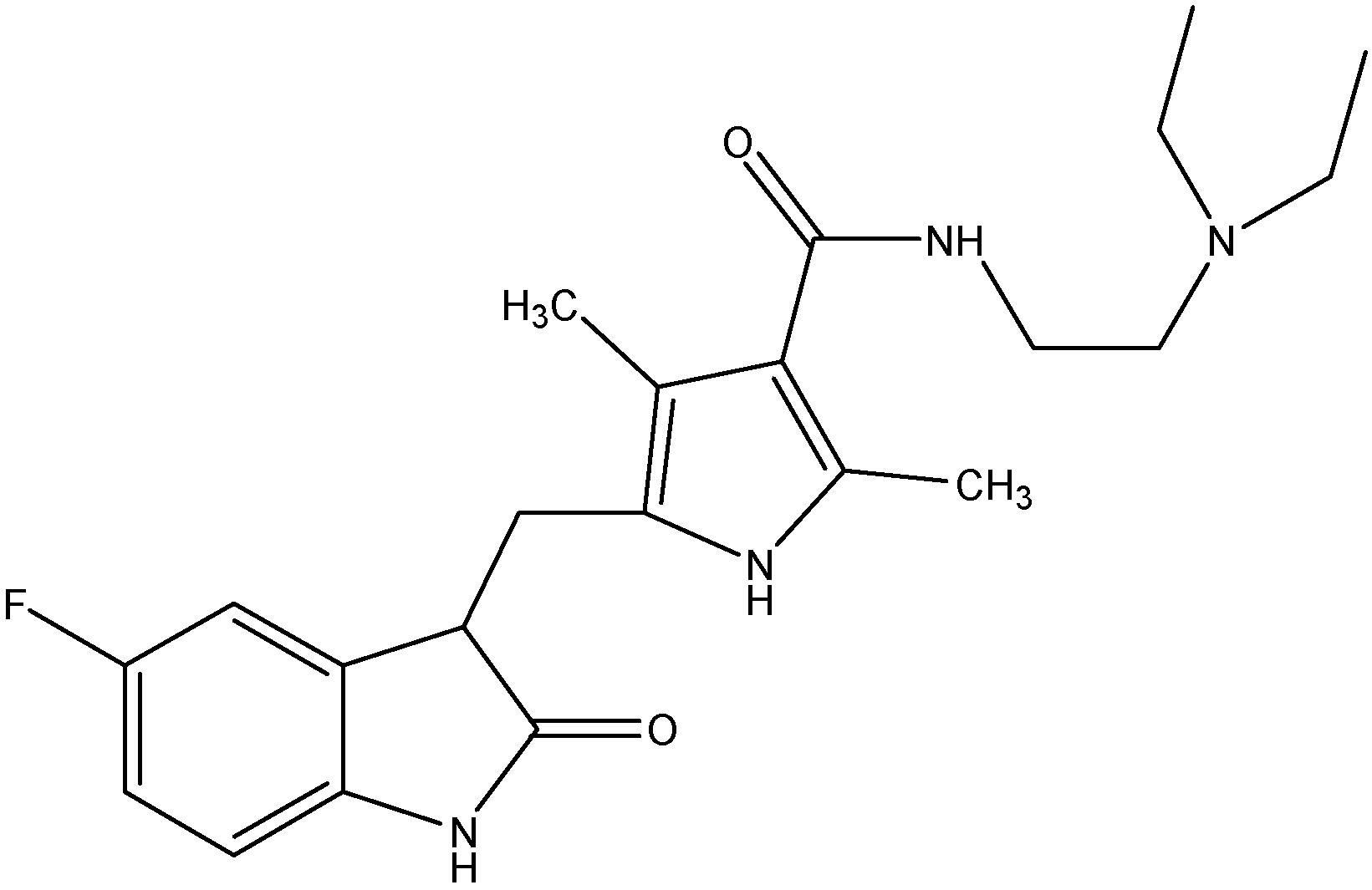
5.3.4. Motesanib

5.4. Platelet- Derived Growth Factor Inhibitors
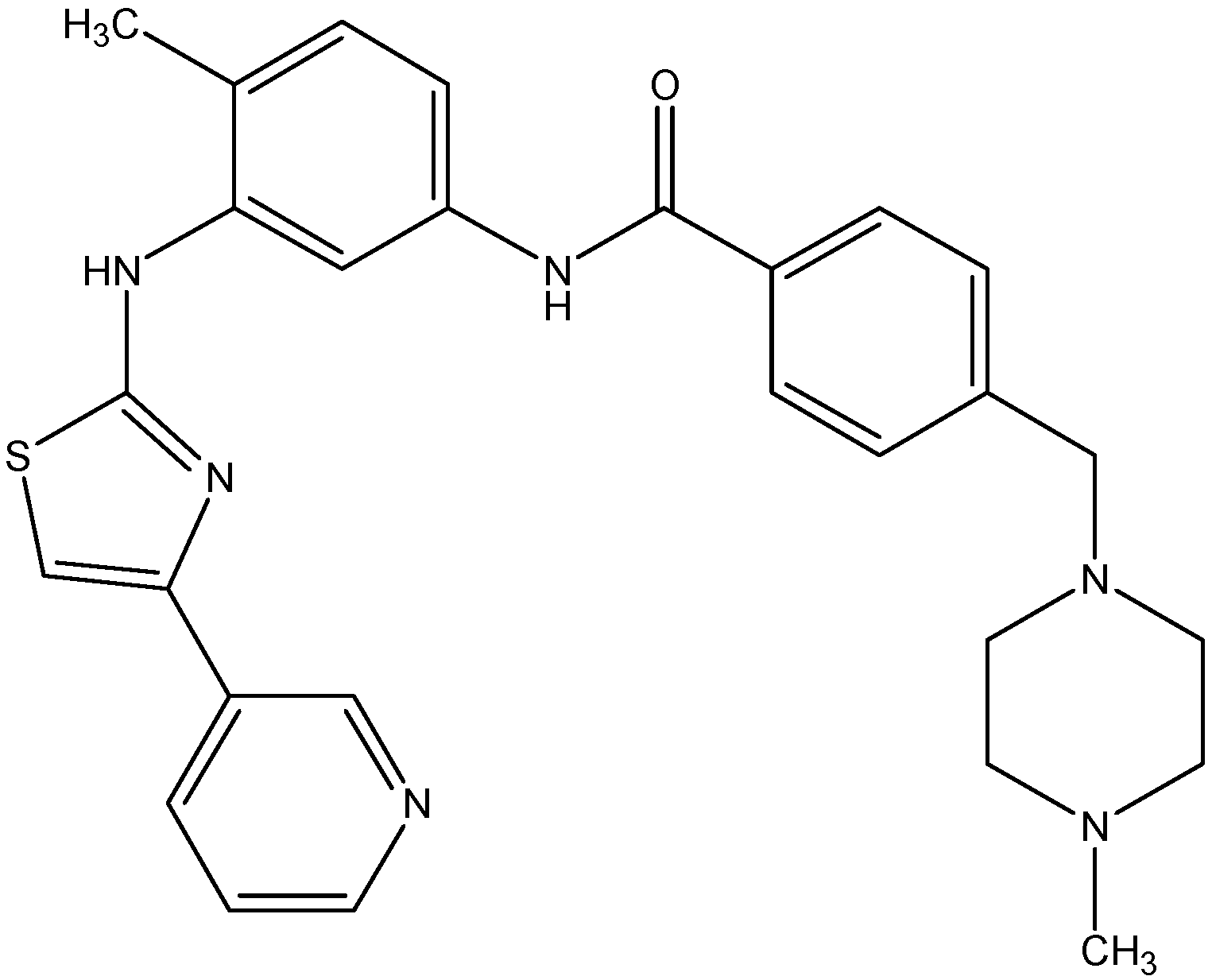
5.4.1. Masitinib

5.4.2. Linsitinib

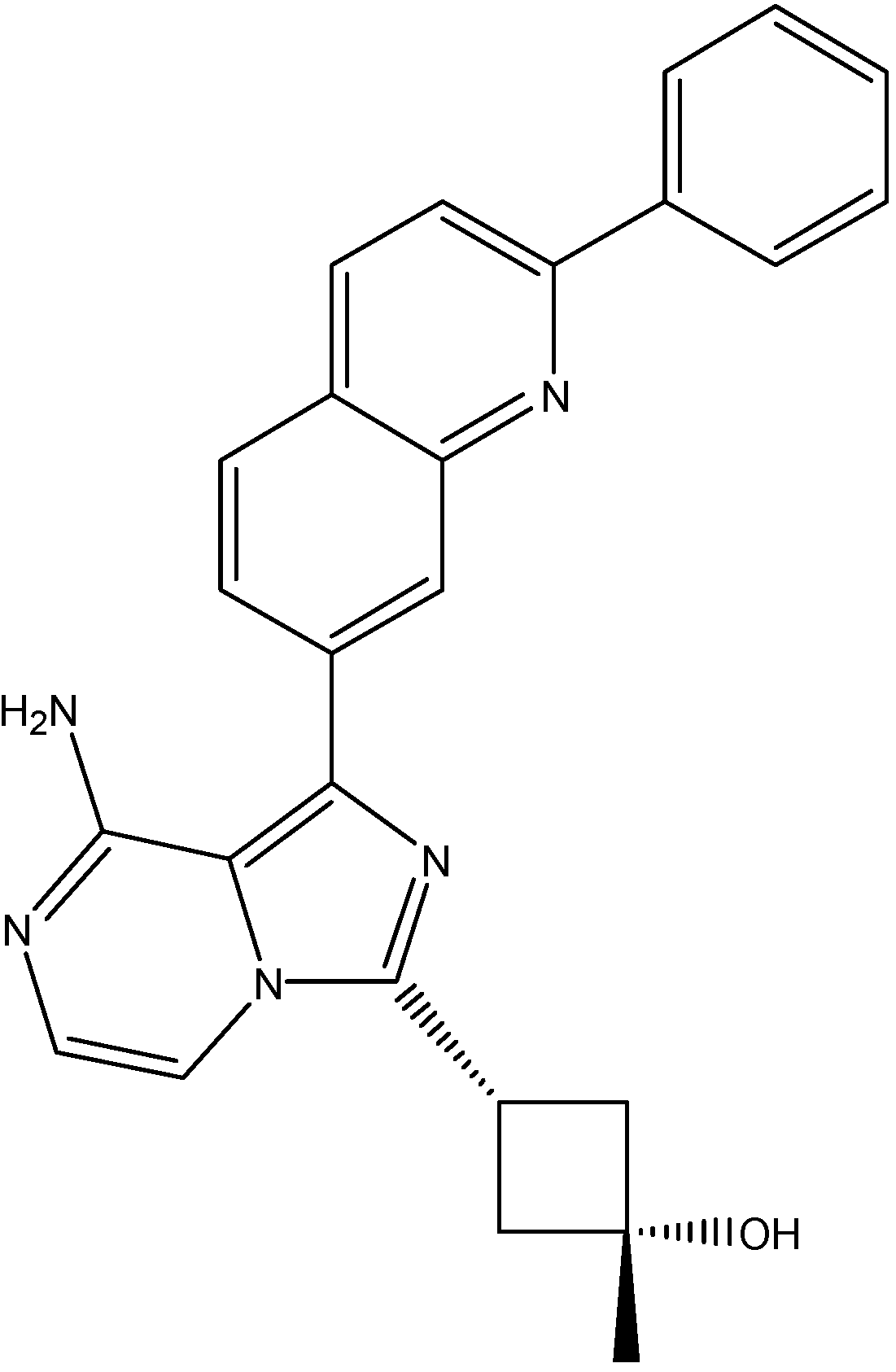
5.5. Fibroblast Growth Factor Receptor Inhibitors
PD173074

5.6. B-Raf/MEK/ERK Pathway Inhibitors
Vemurafenib (PLX 4032)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Tyrosine Kinase Target | Neoplasm(s) Targeted | Clinical Status | Association with ABC Transporters |
|---|---|---|---|---|
| Imatinib (Gleevec, Glivec) | BCR-ABL, c-KIT and PDGFR | CML and ALL, MDS/MPD, ASM HES, CEL, DFSP and GIST | Approved, 2001 | ABCB1 [122], ABCC1 [122], ABCG2 [121] and ABCC10 [123] |
| Nilotinib (Tasigna) | BCR-ABL | CML | Approved, 2007 | ABCB1 [93], ABCG2 [93] and ABCC10 [123] |
| Dasatinib (Sprycel) | BCR-ABL and Src | CML, ALL | Approved, 2006 | ABCB1 [117] and ABCG2 [128] |
| Gefitinib (Iressa) | EGFR | NSCLC | Approved, 2006 | ABCB1 [134] and ABCG2 [133] |
| Erlotinib (Tarceva) | EGFR andHER2 | NSCLC | Approved, 2004 | ABCB1, ABCG2 and ABCC10 [50,72] |
| Lapatinib (Tyverb) | EGFR, HER2 | HER2-positive breast cancer | Approved, 2007 | ABCB1, ABCG2 [137] and ABCC10 [72] |
| Canertinib (CI- 1033) | EGFR, HER2 and ErbB- 4 | Metastatic breast cancer. | Phase II | ABCB1 [108] and ABCG2 [139] |
| Icotinib | EGFR | NSCLC | Approved in china, 2011 | ABCG2 [141] |
| AST1306 | EGFRandHER2 | Solid tumors | Phase I | ABCG2 [131] |
| Sorafenib (BAY 43-9006) | VEGFR, EGFR | Renal and pancreatic cancer | Phase II | ABCC10 and ABCC11 [146] |
| Motesanib | PDGFR, VEGFR and c-KIT | NSCLC, GIST, breast cancer. | Phase III | ABCB1 and ABCG2 [147] |
| Sunitinib (Sutent) | FLT3, PDGFR, VEGFR and c-KIT | RCC, GIST | Approved, 2006 | ABCB1 and ABCG2 [96,143] |
| Telatinib | VEGFR-2, VEGFR-3 PDGFR-β and cKIT | Colorectal and gastric cancer | Phase II/III | ABCG2 [142] |
| Masitinib | c-Kit ,PDGFR α, β | Metastatic gastrointestinal stromal tumors | Phase II/III | ABCG2 and ABCC10 [148,149] |
| Linsitinib | IGF-1R/IR | Adrenocortical carcinoma | Phase III | ABCG2 and ABCC10 [150] |
| Ponatinib | FGFR1-4, FLT3 and PDGFR | CML and Ph+ ALL | Approved, 2012/suspended for vascular clots 2013 | ABCB1, ABCG2 [129] and ABCC10 [130] |
| PD173074 | FGFR | NSCLC | Phase I | ABCB1 [151] and ABCC10 [152] |
| Vemurafenib | B-Raf/MEK/ERK | Melanona | Approved, 2011 | ABCB1, ABCG2 and ABCC10 [154] |
6. Adverse Cutaneous Reactions of Tyrosine Kinase Inhibitors
7. Conclusions and Therapeutic Perspective
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Van der Geer, P.; Hunter, T.; Lindberg, R.A. Receptor protein-tyrosine kinases and their signal transduction pathways. Annu. Rev. Cell Biol. 1994, 10, 251–337. [Google Scholar]
- Krause, D.S.; van Etten, R.A. Tyrosine kinases as targets for cancer therapy. N. Engl. J. Med. 2005, 353, 172–187. [Google Scholar]
- Pytel, D.; Sliwinski, T.; Poplawski, T.; Ferriola, D.; Majsterek, I. Tyrosine kinase blockers: New hope for successful cancer therapy. Anti-Cancer Agents Med. Chem. 2009, 9, 66–76. [Google Scholar]
- Faivre, S.; Djelloul, S.; Raymond, E. New paradigms in anticancer therapy: Targeting multiple signaling pathways with kinase inhibitors. Semin. Oncol. 2006, 33, 407–420. [Google Scholar]
- Aman, P. Fusion oncogenes in tumor development. Semin. Cancer Biol. 2005, 15, 236–243. [Google Scholar] [CrossRef]
- Brozik, A.; Hegedus, C.; Erdei, Z.; Hegedus, T.; Ozvegy-Laczka, C.; Szakacs, G.; Sarkadi, B. Tyrosine kinase inhibitors as modulators of atp binding cassette multidrug transporters: Substrates, chemosensitizers or inducers of acquired multidrug resistance? Expert Opin. Drug Metab. Toxicol. 2011, 7, 623–642. [Google Scholar] [CrossRef]
- Lemos, C.; Jansen, G.; Peters, G.J. Drug transporters: Recent advances concerning BCRP and tyrosine kinase inhibitors. Br. J. Cancer 2008, 98, 857–862. [Google Scholar] [CrossRef]
- Wang, X.K.; Fu, L.W. Interaction of tyrosine kinase inhibitors with the MDR- related ABC transporter proteins. Curr. Drug Metab. 2010, 11, 618–628. [Google Scholar] [CrossRef]
- Quintieri, L.; Fantin, M.; Vizler, C. Identification of molecular determinants of tumor sensitivity and resistance to anticancer drugs. Adv. Exp. Med. Biol. 2007, 593, 95–104. [Google Scholar]
- Gottesman, M.M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Han, T.Y.; Giuliano, A.E.; Cabot, M.C. Ceramide glycosylation potentiates cellular multidrug resistance. FASEB J. 2001, 15, 719–730. [Google Scholar] [CrossRef]
- Lowe, S.W.; Ruley, H.E.; Jacks, T.; Housman, D.E. P53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell 1993, 74, 957–967. [Google Scholar] [CrossRef]
- Synold, T.W.; Dussault, I.; Forman, B.M. The orphan nuclear receptor sxr coordinately regulates drug metabolism and efflux. Nat. Med. 2001, 7, 584–590. [Google Scholar] [CrossRef]
- Deeley, R.G.; Westlake, C.; Cole, S.P. Transmembrane transport of endo- and xenobiotics by mammalian ATP-binding cassette multidrug resistance proteins. Physiol. Rev. 2006, 86, 849–899. [Google Scholar] [CrossRef]
- Bradbury, P.A.; Middleton, M.R. DNA repair pathways in drug resistance in melanoma. Anticancer Drugs 2004, 15, 421–426. [Google Scholar] [CrossRef]
- Dean, M.; Rzhetsky, A.; Allikmets, R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001, 11, 1156–1166. [Google Scholar] [CrossRef]
- Ambudkar, S.V.; Kim, I.W.; Xia, D.; Sauna, Z.E. The A-loop, a novel conserved aromatic acid subdomain upstream of the Walker A motif in ABC transporters, is critical for atp binding. FEBS Lett. 2006, 580, 1049–1055. [Google Scholar] [CrossRef]
- Wu, C.P.; Hsieh, C.H.; Wu, Y.S. The emergence of drug transporter-mediated multidrug resistance to cancer chemotherapy. Mol. Pharm. 2011, 8, 1996–2011. [Google Scholar] [CrossRef]
- Locher, K.P.; Borths, E. ABC transporter architecture and mechanism: Implications from the crystal structures of btucd and btuf. FEBS Lett. 2004, 564, 264–268. [Google Scholar] [CrossRef]
- Dano, K. Active outward transport of daunomycin in resistant ehrlich ascites tumor cells. Biochim. Biophys. Acta 1973, 323, 466–483. [Google Scholar] [CrossRef]
- Kristensen, G.B.; Vergote, I.; Stuart, G.; Del Campo, J.M.; Kaern, J.; Lopez, A.B.; Eisenhauer, E.; Aavall-Lundquist, E.; Ridderheim, M.; Havsteen, H.; et al. First-line treatment of ovarian cancer FIGO stages IIB-IV with paclitaxel/epirubicin/carboplatin versus paclitaxel/carboplatin. Int. J. Gynecol. Cancer 2003, 13 (Suppl. 2), 172–177. [Google Scholar]
- Soo, R.A.; Anderson, B.O.; Cho, B.C.; Yang, C.H.; Liao, M.; Lim, W.T.; Goldstraw, P.; Mok, T.S. First-line systemic treatment of advanced stage non-small-cell lung cancer in asia: Consensus statement from the asian oncology summit 2009. Lancet Oncol. 2009, 10, 1102–1110. [Google Scholar] [CrossRef]
- Oakman, C.; Pestrin, M.; Zafarana, E.; Cantisani, E.; Di Leo, A. Role of lapatinib in the first-line treatment of patients with metastatic breast cancer. Cancer Manag. Res. 2010, 2, 13–25. [Google Scholar]
- Pelzer, U.; Arnold, D.; Reitzig, P.; Herrenberger, J.; Korsten, F.W.; Kindler, M.; Stieler, J.; Dorken, B.; Riess, H.; Oettle, H. First-line treatment of pancreatic cancer patients with the combination of 5-fluorouracil/folinic acid plus gemcitabine: A multicenter phase ii trial by the CONKO-study group. Cancer Chemother. Pharmacol. 2011, 68, 1173–1178. [Google Scholar] [CrossRef]
- Pieters, R.; Hunger, S.P.; Boos, J.; Rizzari, C.; Silverman, L.; Baruchel, A.; Goekbuget, N.; Schrappe, M.; Pui, C.H. L-asparaginase treatment in acute lymphoblastic leukemia: A focus on erwinia asparaginase. Cancer 2011, 117, 238–249. [Google Scholar] [CrossRef]
- Schweighofer, C.D.; Wendtner, C.M. First-line treatment of chronic lymphocytic leukemia: Role of alemtuzumab. Onco. Targets Ther. 2010, 3, 53–67. [Google Scholar] [CrossRef]
- Borst, P.; Elferink, R.O. Mammalian abc transporters in health and disease. Annu. Rev. Biochem. 2002, 71, 537–592. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of atp-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Ludwig, J.; Xia, D.; Szakacs, G. Defeating drug resistance in cancer. Discov. Med. 2006, 6, 18–23. [Google Scholar]
- Glavinas, H.; Krajcsi, P.; Cserepes, J.; Sarkadi, B. The role of abc transporters in drug resistance, metabolism and toxicity. Curr. Drug Deliv. 2004, 1, 27–42. [Google Scholar] [CrossRef]
- Higgins, C.F. ABC transporters: From microorganisms to man. Annu. Rev. Cell Biol. 1992, 8, 67–113. [Google Scholar] [CrossRef]
- Shukla, S.; Wu, C.P.; Ambudkar, S.V. Development of inhibitors of ATP-binding cassette drug transporters: Present status and challenges. Expert Opin. Drug Metab. Toxicol. 2008, 4, 205–223. [Google Scholar] [CrossRef]
- Locher, K.P. Structure and mechanism of abc transporters. Curr. Opin. Struct. Biol. 2004, 14, 426–431. [Google Scholar] [CrossRef]
- Borths, E.L.; Locher, K.P.; Lee, A.T.; Rees, D.C. The structure of Escherichia coli BtuF and binding to its cognate ATP binding cassette transporter. Proc. Natl. Acad. Sci. USA 2002, 99, 16642–16647. [Google Scholar] [CrossRef]
- Gillet, J.P.; Efferth, T.; Remacle, J. Chemotherapy-induced resistance by ATP-binding cassette transporter genes. Biochim. Biophys. Acta 2007, 1775, 237–262. [Google Scholar]
- Wu, C.P.; Calcagno, A.M.; Ambudkar, S.V. Reversal of abc drug transporter-mediated multidrug resistance in cancer cells: Evaluation of current strategies. Curr. Mol. Pharmacol. 2008, 1, 93–105. [Google Scholar] [CrossRef]
- Liu, F.S. Mechanisms of chemotherapeutic drug resistance in cancer therapy—A quick review. Taiwan J. Obstet. Gynecol. 2009, 48, 239–244. [Google Scholar] [CrossRef]
- Tiwari, A.K.; Sodani, K.; Dai, C.L.; Ashby, C.R., Jr.; Chen, Z.S. Revisiting the abcs of multidrug resistance in cancer chemotherapy. Curr. Pharm. Biotechnol. 2011, 12, 570–594. [Google Scholar] [CrossRef]
- Dean, M.; Allikmets, R. Evolution of ATP-binding cassette transporter genes. Curr. Opin. Genet. Dev. 1995, 5, 779–785. [Google Scholar] [CrossRef]
- Hyde, S.C.; Emsley, P.; Hartshorn, M.J.; Mimmack, M.M.; Gileadi, U.; Pearce, S.R.; Gallagher, M.P.; Gill, D.R.; Hubbard, R.E.; Higgins, C.F. Structural model of ATP-binding proteins associated with cystic fibrosis, multidrug resistance and bacterial transport. Nature 1990, 346, 362–365. [Google Scholar] [CrossRef]
- Walker, J.E.; Saraste, M.; Runswick, M.J.; Gay, N.J. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982, 1, 945–951. [Google Scholar]
- Higgins, C.F.; Hiles, I.D.; Whalley, K.; Jamieson, D.J. Nucleotide binding by membrane components of bacterial periplasmic binding protein-dependent transport systems. EMBO J. 1985, 4, 1033–1039. [Google Scholar]
- Dean, M. ABC transporters, drug resistance, and cancer stem cells. J. Mammary Gland Biol. Neoplasia 2009, 14, 3–9. [Google Scholar] [CrossRef]
- Mao, Q.; Unadkat, J.D. Role of the breast cancer resistance protein (ABCG2) in drug transport. AAPS J. 2005, 7, E118–E133. [Google Scholar] [CrossRef]
- Sauna, Z.E.; Ambudkar, S.V. Characterization of the catalytic cycle of ATP hydrolysis by human P-glycoprotein. The two ATP hydrolysis events in a single catalytic cycle are kinetically similar but affect different functional outcomes. J. Biol. Chem. 2001, 276, 11653–11661. [Google Scholar]
- Gottesman, M.M.; Ambudkar, S.V. Overview: ABC transporters and human disease. J. Bioenerg. Biomembr. 2001, 33, 453–458. [Google Scholar] [CrossRef]
- Holland, I.B. ABC transporters, mechanisms and biology: An overview. Essays Biochem. 2011, 50, 1–17. [Google Scholar] [CrossRef]
- Goldstein, L.J.; Galski, H.; Fojo, A.; Willingham, M.; Lai, S.L.; Gazdar, A.; Pirker, R.; Green, A.; Crist, W.; Brodeur, G.M.; et al. Expression of a multidrug resistance gene in human cancers. J. Natl. Cancer Inst. 1989, 81, 116–124. [Google Scholar] [CrossRef]
- Dean, M.; Annilo, T. Evolution of the ATP-binding cassette (ABC) transporter superfamily in vertebrates. Annu. Rev. Genomics Hum. Genet. 2005, 6, 123–142. [Google Scholar] [CrossRef]
- Shi, Z.; Peng, X.X.; Kim, I.W.; Shukla, S.; Si, Q.S.; Robey, R.W.; Bates, S.E.; Shen, T.; Ashby, C.R., Jr.; Fu, L.W.; et al. Erlotinib (Tarceva, OSI-774) antagonizes ATP-binding cassette subfamily B member 1 and ATP-binding cassette subfamily G member 2-mediated drug resistance. Cancer Res. 2007, 67, 11012–11020. [Google Scholar] [CrossRef]
- Shi, Z.; Liang, Y.J.; Chen, Z.S.; Wang, X.H.; Ding, Y.; Chen, L.M.; Fu, L.W. Overexpression of Survivin and XIAP in MDR cancer cells unrelated to P-glycoprotein. Oncol. Rep. 2007, 17, 969–976. [Google Scholar]
- Sauna, Z.E.; Smith, M.M.; Muller, M.; Kerr, K.M.; Ambudkar, S.V. The mechanism of action of multidrug-resistance-linked P-glycoprotein. J. Bioenerg. Biomembr. 2001, 33, 481–491. [Google Scholar] [CrossRef]
- Sarkadi, B.; Homolya, L.; Szakacs, G.; Varadi, A. Human multidrug resistance ABCB and ABCG transporters: Participation in a chemoimmunity defense system. Physiol. Rev. 2006, 86, 1179–1236. [Google Scholar] [CrossRef]
- Figueroa-Gonzalez, G.; Jacobo-Herrera, N.; Zentella-Dehesa, A.; Pereda-Miranda, R. Reversal of multidrug resistance by morning glory resin glycosides in human breast cancer cells. J. Nat. Prod. 2011, 75, 93–97. [Google Scholar]
- Peng, X.X.; Tiwari, A.K.; Wu, H.C.; Chen, Z.S. Overexpression of P-glycoprotein induces acquired resistance to imatinib in chronic myeloid leukemia cells. Chin. J. Cancer 2011, 31, 110–118. [Google Scholar] [CrossRef]
- Mathias, A.A.; Hitti, J.; Unadkat, J.D. P-glycoprotein and breast cancer resistance protein expression in human placentae of various gestational ages. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R963–R969. [Google Scholar] [CrossRef]
- Zhou, S.F. Structure, function and regulation of P-glycoprotein and its clinical relevance in drug disposition. Xenobiotica 2008, 38, 802–832. [Google Scholar] [CrossRef]
- Krumpochova, P.; Sapthu, S.; Brouwers, J.F.; de Haas, M.; de Vos, R.; Borst, P.; van de Wetering, K. Transportomics: Screening for substrates of ABC transporters in body fluids using vesicular transport assays. FASEB J. 2011, 26, 738–747. [Google Scholar] [CrossRef]
- Leighton, J.C., Jr.; Goldstein, L.J. P-glycoprotein in adult solid tumors. Expression and prognostic significance. Hematol. Oncol. Clin. North Am. 1995, 9, 251–273. [Google Scholar]
- Perez-Sayans, M.; Somoza-Martin, J.M.; Barros-Angueira, F.; Diz, P.G.; Rey, J.M.; Garcia-Garcia, A. Multidrug resistance in oral squamous cell carcinoma: The role of vacuolar atpases. Cancer Lett. 2010, 295, 135–143. [Google Scholar]
- Marie, J.P. P-glycoprotein in adult hematologic malignancies. Hematol. Oncol. Clin. North Am. 1995, 9, 239–249. [Google Scholar]
- Verrelle, P.; Meissonnier, F.; Fonck, Y.; Feillel, V.; Dionet, C.; Kwiatkowski, F.; Plagne, R.; Chassagne, J. Clinical relevance of immunohistochemical detection of multidrug resistance p-glycoprotein in breast carcinoma. J. Natl. Cancer Inst. 1991, 83, 111–116. [Google Scholar] [CrossRef]
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in chinese hamster ovary cell mutants. Biochim. Biophys. Acta 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Schinkel, A.H.; Mol, C.A.; Wagenaar, E.; van Deemter, L.; Smit, J.J.; Borst, P. Multidrug resistance and the role of P-glycoprotein knockout mice. Eur. J. Cancer 1995, 31A, 1295–1298. [Google Scholar] [CrossRef]
- Kuo, M.T. Roles of multidrug resistance genes in breast cancer chemoresistance. Adv. Exp. Med. Biol. 2007, 608, 23–30. [Google Scholar]
- Leonard, G.D.; Fojo, T.; Bates, S.E. The role of abc transporters in clinical practice. Oncologist 2003, 8, 411–424. [Google Scholar] [CrossRef]
- Kruh, G.D.; Guo, Y.; Hopper-Borge, E.; Belinsky, M.G.; Chen, Z.S. ABCC10, ABCC11, and ABCC12. Pflügers Arch.-Eur. J. Physiol. 2007, 453, 675–684. [Google Scholar]
- Sodani, K.; Patel, A.; Kathawala, R.J.; Chen, Z.S. Multidrug resistance associated proteins in multidrug resistance. Chin. J. Cancer 2012, 31, 58–72. [Google Scholar] [CrossRef]
- Cole, S.P.; Bhardwaj, G.; Gerlach, J.H.; Mackie, J.E.; Grant, C.E.; Almquist, K.C.; Stewart, A.J.; Kurz, E.U.; Duncan, A.M.; Deeley, R.G. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science 1992, 258, 1650–1654. [Google Scholar] [CrossRef]
- Lovasz, N.; Ducza, E.; Gaspar, R.; Falkay, G. Ontogeny of sulfonylurea-binding regulatory subunits of katp channels in the pregnant rat myometrium. Reproduction 2011, 142, 175–181. [Google Scholar] [CrossRef]
- De Wet, H.; Fotinou, C.; Amad, N.; Dreger, M.; Ashcroft, F.M. The ATPase activities of sulfonylurea receptor 2A and sulfonylurea receptor 2B are influenced by the C-terminal 42 amino acids. FEBS J. 2010, 277, 2654–2662. [Google Scholar]
- Kuang, Y.H.; Shen, T.; Chen, X.; Sodani, K.; Hopper-Borge, E.; Tiwari, A.K.; Lee, J.W.; Fu, L.W.; Chen, Z.S. Lapatinib and erlotinib are potent reversal agents for MRP7 (ABCC10)-mediated multidrug resistance. Biochem. Pharmacol. 2010, 79, 154–161. [Google Scholar] [CrossRef]
- McGrath, T.; Latoud, C.; Arnold, S.T.; Safa, A.R.; Felsted, R.L.; Center, M.S. Mechanisms of multidrug resistance in HL60 cells. Analysis of resistance associated membrane proteins and levels of mdr gene expression. Biochem. Pharmacol. 1989, 38, 3611–3619. [Google Scholar]
- Borst, P.; Evers, R.; Kool, M.; Wijnholds, J. A family of drug transporters: The multidrug resistance-associated proteins. J. Natl. Cancer Inst. 2000, 92, 1295–1302. [Google Scholar] [CrossRef]
- Kruh, G.D.; Belinsky, M.G. The MRP family of drug efflux pumps. Oncogene 2003, 22, 7537–7552. [Google Scholar] [CrossRef]
- Chen, Z.S.; Hopper-Borge, E.; Belinsky, M.G.; Shchaveleva, I.; Kotova, E.; Kruh, G.D. Characterization of the transport properties of human multidrug resistance protein 7 (MRP7, ABCC10). Mol. Pharmacol. 2003, 63, 351–358. [Google Scholar] [CrossRef]
- Hopper-Borge, E.; Chen, Z.S.; Shchaveleva, I.; Belinsky, M.G.; Kruh, G.D. Analysis of the drug resistance profile of multidrug resistance protein 7 (ABCC10): Resistance to docetaxel. Cancer Res. 2004, 64, 4297–4930. [Google Scholar]
- Hopper-Borge, E.A.; Churchill, T.; Paulose, C.; Nicolas, E.; Jacobs, J.D.; Ngo, O.; Kuang, Y.; Grinberg, A.; Westphal, H.; Chen, Z.S.; et al. Contribution of ABCC10 (MRP7) to in vivo paclitaxel resistance as assessed in ABCC10(-/-) mice. Cancer Res. 2011, 71, 3649–3657. [Google Scholar]
- Hopper, E.; Belinsky, M.G.; Zeng, H.; Tosolini, A.; Testa, J.R.; Kruh, G.D. Analysis of the structure and expression pattern of MRP7 (ABCC10), a new member of the MRP subfamily. Cancer Lett. 2001, 162, 181–191. [Google Scholar] [CrossRef]
- Takayanagi, S.; Kataoka, T.; Ohara, O.; Oishi, M.; Kuo, M.T.; Ishikawa, T. Human ATP-binding cassette transporter ABCC10: Expression profile and p53-dependent upregulation. J. Exp. Ther. Oncol. 2004, 4, 239–246. [Google Scholar]
- Kathawala, R.J.; Wang, Y.J.; Ashby, C.R., Jr.; Chen, Z.S. Recent advances regarding the role of ABC subfamily C member 10 (ABCC10) in the efflux of antitumor drugs. Chin. J. Cancer 2014, 33, 223–230. [Google Scholar] [CrossRef]
- Matsuo, H.; Takada, T.; Ichida, K.; Nakamura, T.; Nakayama, A.; Takada, Y.; Okada, C.; Sakurai, Y.; Hosoya, T.; Kanai, Y.; et al. Identification of ABCG2 dysfunction as a major factor contributing to gout. Nucleosides Nucleotides Nucleic Acids 2011, 30, 1098–1104. [Google Scholar] [CrossRef]
- Rocchi, E.; Khodjakov, A.; Volk, E.L.; Yang, C.H.; Litman, T.; Bates, S.E.; Schneider, E. The product of the ABC half-transporter gene ABCG2 (BCRP/MXR/ABCP) is expressed in the plasma membrane. Biochem. Biophys. Res. Commun. 2000, 271, 42–46. [Google Scholar] [CrossRef]
- Litman, T.; Brangi, M.; Hudson, E.; Fetsch, P.; Abati, A.; Ross, D.D.; Miyake, K.; Resau, J.H.; Bates, S.E. The multidrug-resistant phenotype associated with overexpression of the new ABC half-transporter, MXR (ABCG2). J. Cell Sci. 2000, 113 Pt 11, 2011–2021. [Google Scholar]
- Ejendal, K.F.; Hrycyna, C.A. Multidrug resistance and cancer: The role of the human ABC transporter ABCG2. Curr. Protein Pept. Sci. 2002, 3, 503–511. [Google Scholar] [CrossRef]
- Robey, R.W.; Polgar, O.; Deeken, J.; To, K.W.; Bates, S.E. ABCG2: Determining its relevance in clinical drug resistance. Cancer Metastasis Rev. 2007, 26, 39–57. [Google Scholar] [CrossRef]
- Maliepaard, M.; Scheffer, G.L.; Faneyte, I.F.; van Gastelen, M.A.; Pijnenborg, A.C.; Schinkel, A.H.; van de Vijver, M.J.; Scheper, R.J.; Schellens, J.H. Subcellular localization and distribution of the breast cancer resistance protein transporter in normal human tissues. Cancer Res. 2001, 61, 3458–3464. [Google Scholar]
- Cooray, H.C.; Blackmore, C.G.; Maskell, L.; Barrand, M.A. Localisation of breast cancer resistance protein in microvessel endothelium of human brain. Neuroreport 2002, 13, 2059–2063. [Google Scholar] [CrossRef]
- Doyle, L.A.; Yang, W.; Abruzzo, L.V.; Krogmann, T.; Gao, Y.; Rishi, A.K.; Ross, D.D. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15665–15670. [Google Scholar] [CrossRef]
- Schinkel, A.H.; Jonker, J.W. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: An overview. Adv. Drug Deliv. Rev. 2003, 55, 3–29. [Google Scholar] [CrossRef]
- Dean, M.; Allikmets, R. Complete characterization of the human abc gene family. J. Bioenerg. Biomembr. 2001, 33, 475–479. [Google Scholar] [CrossRef]
- Miyake, K.; Mickley, L.; Litman, T.; Zhan, Z.; Robey, R.; Cristensen, B.; Brangi, M.; Greenberger, L.; Dean, M.; Fojo, T.; et al. Molecular cloning of cdnas which are highly overexpressed in mitoxantrone-resistant cells: Demonstration of homology to abc transport genes. Cancer Res. 1999, 59, 8–13. [Google Scholar]
- Tiwari, A.K.; Sodani, K.; Wang, S.R.; Kuang, Y.H.; Ashby, C.R., Jr.; Chen, X.; Chen, Z.S. Nilotinib (AMN107, Tasigna) reverses multidrug resistance by inhibiting the activity of the ABCB1/Pgp and ABCG2/BCRP/MXR transporters. Biochem. Pharmacol. 2009, 78, 153–161. [Google Scholar] [CrossRef]
- Chen, Z.S.; Robey, R.W.; Belinsky, M.G.; Shchaveleva, I.; Ren, X.Q.; Sugimoto, Y.; Ross, D.D.; Bates, S.E.; Kruh, G.D. Transport of methotrexate, methotrexate polyglutamates, and 17beta-estradiol 17-(beta-d-glucuronide) by ABCG2: Effects of acquired mutations at R482 on methotrexate transport. Cancer Res. 2003, 63, 4048–4054. [Google Scholar]
- Honjo, Y.; Hrycyna, C.A.; Yan, Q.W.; Medina-Perez, W.Y.; Robey, R.W.; van de Laar, A.; Litman, T.; Dean, M.; Bates, S.E. Acquired mutations in the MXR/BCRP/ABCP gene alter substrate specificity in MXR/BCRP/ABCP-overexpressing cells. Cancer Res. 2001, 61, 6635–6639. [Google Scholar]
- Dai, C.L.; Liang, Y.J.; Wang, Y.S.; Tiwari, A.K.; Yan, Y.Y.; Wang, F.; Chen, Z.S.; Tong, X.Z.; Fu, L.W. Sensitization of ABCG2-overexpressing cells to conventional chemotherapeutic agent by sunitinib was associated with inhibiting the function of ABCG2. Cancer Lett. 2009, 279, 74–83. [Google Scholar] [CrossRef]
- Pozza, A.; Perez-Victoria, J.M.; Sardo, A.; Ahmed-Belkacem, A.; Di Pietro, A. Purification of breast cancer resistance protein ABCG2 and role of arginine-482. Cell. Mol. Life Sci. 2006, 63, 1912–1922. [Google Scholar] [CrossRef]
- Ejendal, K.F.; Diop, N.K.; Schweiger, L.C.; Hrycyna, C.A. The nature of amino acid 482 of human ABCG2 affects substrate transport and ATP hydrolysis but not substrate binding. Protein Sci. 2006, 15, 1597–1607. [Google Scholar] [CrossRef]
- Bates, S.E.; Medina-Perez, W.Y.; Kohlhagen, G.; Antony, S.; Nadjem, T.; Robey, R.W.; Pommier, Y. ABCG2 mediates differential resistance to SN-38 (7-ethyl-10-hydroxycamptothecin) and homocamptothecins. J. Pharmacol. Exp. Ther. 2004, 310, 836–842. [Google Scholar] [CrossRef]
- Janne, P.A.; Gray, N.; Settleman, J. Factors underlying sensitivity of cancers to small-molecule kinase inhibitors. Nat. Rev. Drug Discov. 2009, 8, 709–723. [Google Scholar] [CrossRef]
- Assef, Y.; Rubio, F.; Colo, G.; del Monaco, S.; Costas, M.A.; Kotsias, B.A. Imatinib resistance in multidrug-resistant k562 human leukemic cells. Leuk. Res. 2009, 33, 710–716. [Google Scholar] [CrossRef]
- Burger, H.; van Tol, H.; Boersma, A.W.; Brok, M.; Wiemer, E.A.; Stoter, G.; Nooter, K. Imatinib mesylate (STI571) is a substrate for the breast cancer resistance protein (BCRP)/ABCG2 drug pump. Blood 2004, 104, 2940–2942. [Google Scholar] [CrossRef]
- Brendel, C.; Scharenberg, C.; Dohse, M.; Robey, R.W.; Bates, S.E.; Shukla, S.; Ambudkar, S.V.; Wang, Y.; Wennemuth, G.; Burchert, A.; et al. Imatinib mesylate and nilotinib (AMN107) exhibit high-affinity interaction with ABCG2 on primitive hematopoietic stem cells. Leukemia 2007, 21, 1267–1275. [Google Scholar] [CrossRef]
- Hegedus, C.; Ozvegy-Laczka, C.; Apati, A.; Magocsi, M.; Nemet, K.; Orfi, L.; Keri, G.; Katona, M.; Takats, Z.; Varadi, A.; et al. Interaction of nilotinib, dasatinib and bosutinib with ABCB1 and ABCG2: Implications for altered anti-cancer effects and pharmacological properties. Br. J. Pharmacol. 2009, 158, 1153–1164. [Google Scholar] [CrossRef]
- Hiwase, D.K.; Saunders, V.; Hewett, D.; Frede, A.; Zrim, S.; Dang, P.; Eadie, L.; To, L.B.; Melo, J.; Kumar, S.; et al. Dasatinib cellular uptake and efflux in chronic myeloid leukemia cells: Therapeutic implications. Clin. Cancer Res. 2008, 14, 3881–3888. [Google Scholar] [CrossRef]
- Huang, W.C.; Chen, Y.J.; Li, L.Y.; Wei, Y.L.; Hsu, S.C.; Tsai, S.L.; Chiu, P.C.; Huang, W.P.; Wang, Y.N.; Chen, C.H.; et al. Nuclear translocation of epidermal growth factor receptor by Akt-dependent phosphorylation enhances breast cancer-resistant protein expression in gefitinib-resistant cells. J. Biol. Chem. 2011, 286, 20558–20568. [Google Scholar] [CrossRef]
- Balabanov, S.; Gontarewicz, A.; Keller, G.; Raddrizzani, L.; Braig, M.; Bosotti, R.; Moll, J.; Jost, E.; Barett, C.; Rohe, I.; et al. ABCG2 overexpression represents a novel mechanism for acquired resistance to the multi-kinase inhibitor Danusertib in BCR-ABL-positive cells in vitro. PLoS One 2011, 6, e19164. [Google Scholar] [CrossRef]
- Erlichman, C.; Boerner, S.A.; Hallgren, C.G.; Spieker, R.; Wang, X.Y.; James, C.D.; Scheffer, G.L.; Maliepaard, M.; Ross, D.D.; Bible, K.C.; et al. The HER tyrosine kinase inhibitor CI1033 enhances cytotoxicity of 7-ethyl-10-hydroxycamptothecin and topotecan by inhibiting breast cancer resistance protein-mediated drug efflux. Cancer Res. 2001, 61, 739–748. [Google Scholar]
- Illmer, T.; Schaich, M.; Platzbecker, U.; Freiberg-Richter, J.; Oelschlagel, U.; von Bonin, M.; Pursche, S.; Bergemann, T.; Ehninger, G.; Schleyer, E. P-glycoprotein-mediated drug efflux is a resistance mechanism of chronic myelogenous leukemia cells to treatment with imatinib mesylate. Leukemia 2004, 18, 401–408. [Google Scholar] [CrossRef]
- Mahon, F.X.; Belloc, F.; Lagarde, V.; Chollet, C.; Moreau-Gaudry, F.; Reiffers, J.; Goldman, J.M.; Melo, J.V. MDR1 gene overexpression confers resistance to imatinib mesylate in leukemia cell line models. Blood 2003, 101, 2368–2373. [Google Scholar] [CrossRef]
- Mahon, F.X.; Hayette, S.; Lagarde, V.; Belloc, F.; Turcq, B.; Nicolini, F.; Belanger, C.; Manley, P.W.; Leroy, C.; Etienne, G.; et al. Evidence that resistance to nilotinib may be due to BCR-ABL, Pgp, or Src kinase overexpression. Cancer Res. 2008, 68, 9809–9816. [Google Scholar] [CrossRef]
- Czyzewski, K.; Styczynski, J. Imatinib is a substrate for various multidrug resistance proteins. Neoplasma 2009, 56, 202–207. [Google Scholar] [CrossRef]
- Shibayama, Y.; Nakano, K.; Maeda, H.; Taguchi, M.; Ikeda, R.; Sugawara, M.; Iseki, K.; Takeda, Y.; Yamada, K. Multidrug resistance protein 2 implicates anticancer drug-resistance to sorafenib. Biol. Pharm. Bull. 2011, 34, 433–435. [Google Scholar] [CrossRef]
- He, M.; Wei, M.J. Reversing multidrug resistance by tyrosine kinase inhibitors. Chin. J. Cancer 2012, 31, 126–133. [Google Scholar] [CrossRef]
- Marchetti, S.; de Vries, N.A.; Buckle, T.; Bolijn, M.J.; van Eijndhoven, M.A.; Beijnen, J.H.; Mazzanti, R.; van Tellingen, O.; Schellens, J.H. Effect of the ATP-binding cassette drug transporters ABCB1, ABCG2, and ABCC2 on erlotinib hydrochloride (Tarceva) disposition in in vitro and in vivo pharmacokinetic studies employing BCRP1-/-/MDR1a/1b-/- (triple-knockout) and wild-type mice. Mol. Cancer Ther. 2008, 7, 2280–2287. [Google Scholar] [CrossRef]
- Agarwal, S.; Sane, R.; Gallardo, J.L.; Ohlfest, J.R.; Elmquist, W.F. Distribution of gefitinib to the brain is limited by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2)-mediated active efflux. J. Pharmacol. Exp. Ther. 2010, 334, 147–155. [Google Scholar] [CrossRef]
- Chen, Y.; Agarwal, S.; Shaik, N.M.; Chen, C.; Yang, Z.; Elmquist, W.F. P-glycoprotein and breast cancer resistance protein influence brain distribution of dasatinib. J. Pharmacol. Exp. Ther. 2009, 330, 956–963. [Google Scholar] [CrossRef]
- Yang, J.J.; Milton, M.N.; Yu, S.; Liao, M.; Liu, N.; Wu, J.T.; Gan, L.; Balani, S.K.; Lee, F.W.; Prakash, S.; et al. P-glycoprotein and breast cancer resistance protein affect disposition of tandutinib, a tyrosine kinase inhibitor. Drug Metab. Lett. 2010, 4, 201–212. [Google Scholar]
- Ozvegy-Laczka, C.; Cserepes, J.; Elkind, N.B.; Sarkadi, B. Tyrosine kinase inhibitor resistance in cancer: Role of ABC multidrug transporters. Drug Resist. Updat. 2005, 8, 15–26. [Google Scholar]
- Hirota, S.; Isozaki, K.; Moriyama, Y.; Hashimoto, K.; Nishida, T.; Ishiguro, S.; Kawano, K.; Hanada, M.; Kurata, A.; Takeda, M.; et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998, 279, 577–580. [Google Scholar] [CrossRef]
- Houghton, P.J.; Germain, G.S.; Harwood, F.C.; Schuetz, J.D.; Stewart, C.F.; Buchdunger, E.; Traxler, P. Imatinib mesylate is a potent inhibitor of the ABCG2 (BCRP) transporter and reverses resistance to topotecan and SN-38 in vitro. Cancer Res. 2004, 64, 2333–2337. [Google Scholar] [CrossRef]
- Hegedus, T.; Orfi, L.; Seprodi, A.; Varadi, A.; Sarkadi, B.; Keri, G. Interaction of tyrosine kinase inhibitors with the human multidrug transporter proteins, MDR1 and MRP1. Biochim. Biophys. Acta 2002, 1587, 318–325. [Google Scholar] [CrossRef]
- Shen, T.; Kuang, Y.H.; Ashby, C.R.; Lei, Y.; Chen, A.; Zhou, Y.; Chen, X.; Tiwari, A.K.; Hopper-Borge, E.; Ouyang, J.; et al. Imatinib and nilotinib reverse multidrug resistance in cancer cells by inhibiting the efflux activity of the MRP7 (ABCC10). PLoS One 2009, 4, e7520. [Google Scholar] [CrossRef]
- Shukla, S.; Sauna, Z.E.; Ambudkar, S.V. Evidence for the interaction of imatinib at the transport-substrate site(s) of the multidrug-resistance-linked ABC drug transporters ABCB1 (P-glycoprotein) and ABCG2. Leukemia 2008, 22, 445–447. [Google Scholar] [CrossRef]
- Shukla, S.; Skoumbourdis, A.P.; Walsh, M.J.; Hartz, A.M.; Fung, K.L.; Wu, C.P.; Gottesman, M.M.; Bauer, B.; Thomas, C.J.; Ambudkar, S.V. Synthesis and characterization of a BODIPY conjugate of the BCR-ABL kinase inhibitor Tasigna (nilotinib): Evidence for transport of Tasigna and its fluorescent derivative by ABC drug transporters. Mol. Pharm. 2011, 8, 1292–1302. [Google Scholar] [CrossRef]
- Hiwase, D.K.; White, D.; Zrim, S.; Saunders, V.; Melo, J.V.; Hughes, T.P. Nilotinib-mediated inhibition of ABCB1 increases intracellular concentration of dasatinib in CML cells: Implications for combination TKI therapy. Leukemia 2010, 24, 658–660. [Google Scholar] [CrossRef]
- Tiwari, A.K.; Sodani, K.; Dai, C.L.; Abuznait, A.H.; Singh, S.; Xiao, Z.J.; Patel, A.; Talele, T.T.; Fu, L.; Kaddoumi, A.; et al. Nilotinib potentiates anticancer drug sensitivity in murine ABCB1-, ABCG2-, and ABCC10-multidrug resistance xenograft models. Cancer Lett. 2013, 328, 307–317. [Google Scholar] [CrossRef]
- Giannoudis, A.; Davies, A.; Lucas, C.M.; Harris, R.J.; Pirmohamed, M.; Clark, R.E. Effective dasatinib uptake may occur without human organic cation transporter 1 (hOCT1): Implications for the treatment of imatinib-resistant chronic myeloid leukemia. Blood 2008, 112, 3348–3354. [Google Scholar] [CrossRef]
- Sen, R.; Natarajan, K.; Bhullar, J.; Shukla, S.; Fang, H.B.; Cai, L.; Chen, Z.S.; Ambudkar, S.V.; Baer, M.R. The novel BCR-ABL and FLT3 inhibitor ponatinib is a potent inhibitor of the MDR-associated ATP-binding cassette transporter ABCG2. Mol. Cancer Ther. 2012, 11, 2033–2044. [Google Scholar] [CrossRef]
- Sun, Y.L.; Kumar, P.; Sodani, K.; Patel, A.; Pan, Y.; Baer, M.R.; Chen, Z.S.; Jiang, W.Q. Ponatinib enhances anticancer drug sensitivity in MRP7-overexpressing cells. Oncol. Rep. 2014, 31, 1605–1612. [Google Scholar]
- Zhang, H.; Wang, Y.J.; Zhang, Y.K.; Wang, D.S.; Kathawala, R.J.; Patel, A.; Talele, T.T.; Chen, Z.S.; Fu, L.W. AST1306, a potent EGFR inhibitor, antagonizes ATP-binding cassette subfamily G member 2-mediated multidrug resistance. Cancer Lett. 2014, 350, 61–68. [Google Scholar] [CrossRef]
- Arteaga, C.L.; Johnson, D.H. Tyrosine kinase inhibitors-zd1839 (iressa). Curr. Opin. Oncol. 2001, 13, 491–498. [Google Scholar] [CrossRef]
- Ozvegy-Laczka, C.; Hegedus, T.; Varady, G.; Ujhelly, O.; Schuetz, J.D.; Varadi, A.; Keri, G.; Orfi, L.; Nemet, K.; Sarkadi, B. High-affinity interaction of tyrosine kinase inhibitors with the ABCG2 multidrug transporter. Mol. Pharmacol. 2004, 65, 1485–1495. [Google Scholar]
- Kitazaki, T.; Oka, M.; Nakamura, Y.; Tsurutani, J.; Doi, S.; Yasunaga, M.; Takemura, M.; Yabuuchi, H.; Soda, H.; Kohno, S. Gefitinib, an EGFR tyrosine kinase inhibitor, directly inhibits the function of P-glycoprotein in multidrug resistant cancer cells. Lung Cancer 2005, 49, 337–343. [Google Scholar] [CrossRef]
- Moyer, J.D.; Barbacci, E.G.; Iwata, K.K.; Arnold, L.; Boman, B.; Cunningham, A.; DiOrio, C.; Doty, J.; Morin, M.J.; Moyer, M.P.; et al. Induction of apoptosis and cell cycle arrest by CP-358,774, an inhibitor of epidermal growth factor receptor tyrosine kinase. Cancer Res. 1997, 57, 4838–4848. [Google Scholar]
- Herbst, R.S. Erlotinib (Tarceva): An update on the clinical trial program. Semin. Oncol. 2003, 30, 34–46. [Google Scholar] [CrossRef]
- Dai, C.L.; Tiwari, A.K.; Wu, C.P.; Su, X.D.; Wang, S.R.; Liu, D.G.; Ashby, C.R., Jr.; Huang, Y.; Robey, R.W.; Liang, Y.J.; et al. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res. 2008, 68, 7905–7914. [Google Scholar] [CrossRef]
- Thomas, S.M.; Grandis, J.R. Pharmacokinetic and pharmacodynamic properties of EGFR inhibitors under clinical investigation. Cancer Treat. Rev. 2004, 30, 255–268. [Google Scholar] [CrossRef]
- Minocha, M.; Khurana, V.; Qin, B.; Pal, D.; Mitra, A.K. Enhanced brain accumulation of pazopanib by modulating P-gp and BCRP1 mediated efflux with canertinib or erlotinib. Int. J. Pharm. 2012, 436, 127–134. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, L.; Liu, X.; Zhou, C.; Zhang, S.; Wang, D.; Li, Q.; Qin, S.; Hu, C.; Zhang, Y.; et al. Icotinib versus gefitinib in previously treated advanced non-small-cell lung cancer (ICOGEN): A randomised, double-blind phase 3 non-inferiority trial. Lancet Oncol. 2013, 14, 953–961. [Google Scholar] [CrossRef]
- Wang, D.S.; Patel, A.; Shukla, S.; Zhang, Y.K.; Wang, Y.J.; Kathawala, R.J.; Robey, R.W.; Zhang, L.; Yang, D.H.; Talele, T.T.; et al. Icotinib antagonizes ABCG2-mediated multidrug resistance, but not the pemetrexed resistance mediated by thymidylate synthase and ABCG2. Oncotarget 2014, 5, 4529–4542. [Google Scholar]
- Sodani, K.; Patel, A.; Anreddy, N.; Singh, S.; Yang, D.H.; Kathawala, R.J.; Kumar, P.; Talele, T.T.; Chen, Z.S. Telatinib reverses chemotherapeutic multidrug resistance mediated by ABCG2 efflux transporter in vitro and in vivo. Biochem. Pharmacol. 2014, 89, 52–61. [Google Scholar] [CrossRef]
- Shukla, S.; Robey, R.W.; Bates, S.E.; Ambudkar, S.V. Sunitinib (Sutent, SU11248), a small-molecule receptor tyrosine kinase inhibitor, blocks function of the ATP-binding cassette (ABC) transporters p-glycoprotein (ABCB1) and ABCG2. Drug Metab. Dispos. 2009, 37, 359–365. [Google Scholar] [CrossRef]
- Eskens, F.A. Angiogenesis inhibitors in clinical development; where are we now and where are we going? Br. J. Cancer 2004, 90, 1–7. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef]
- Hu, S.; Chen, Z.; Franke, R.; Orwick, S.; Zhao, M.; Rudek, M.A.; Sparreboom, A.; Baker, S.D. Interaction of the multikinase inhibitors sorafenib and sunitinib with solute carriers and ATP-binding cassette transporters. Clin. Cancer Res. 2009, 15, 6062–6069. [Google Scholar] [CrossRef]
- Wang, Y.J.; Kathawala, R.J.; Zhang, Y.K.; Patel, A.; Kumar, P.; Shukla, S.; Fung, K.L.; Ambudkar, S.V.; Talele, T.T.; Chen, Z.S. Motesanib (AMG706), a potent multikinase inhibitor, antagonizes multidrug resistance by inhibiting the efflux activity of the ABCB1. Biochem. Pharmacol. 2014, 90, 367–378. [Google Scholar] [CrossRef]
- Kathawala, R.J.; Sodani, K.; Chen, K.; Patel, A.; Abuznait, A.H.; Anreddy, N.; Sun, Y.L.; Kaddoumi, A.; Ashby, C.R., Jr.; Chen, Z.S. Masitinib antagonizes ATP-binding cassette subfamily c member 10-mediated paclitaxel resistance: A preclinical study. Mol. Cancer Ther. 2014, 13, 714–723. [Google Scholar] [CrossRef]
- Kathawala, R.J.; Chen, J.J.; Zhang, Y.K.; Wang, Y.J.; Patel, A.; Wang, D.S.; Talele, T.T.; Ashby, C.R., Jr.; Chen, Z.S. Masitinib antagonizes ATP-binding cassette subfamily g member 2-mediated multidrug resistance. Int. J. Oncol. 2014, 44, 1634–1642. [Google Scholar]
- Zhang, H.; Kathawala, R.J.; Wang, Y.J.; Zhang, Y.K.; Patel, A.; Shukla, S.; Robey, R.W.; Talele, T.T.; Ashby, C.R., Jr.; Ambudkar, S.V.; et al. Linsitinib (OSI-906) antagonizes ATP-binding cassette subfamily G member 2 and subfamily C member 10-mediated drug resistance. Int. J. Biochem. Cell Biol. 2014, 51, 111–119. [Google Scholar] [CrossRef]
- Patel, A.; Tiwari, A.K.; Chufan, E.E.; Sodani, K.; Anreddy, N.; Singh, S.; Ambudkar, S.V.; Stephani, R.; Chen, Z.S. PD173074, a selective FGFR inhibitor, reverses ABCB1-mediated drug resistance in cancer cells. Cancer Chemother. Pharmacol. 2013, 72, 189–199. [Google Scholar] [CrossRef]
- Anreddy, N.; Patel, A.; Sodani, K.; Kathawala, R.J.; Chen, E.P.; Wurpel, J.N.; Chen, Z.S. PD173074,a selective FGFR inhibitor, reverses MRP7 (ABCC10)-mediated MDR. Acta Pharm. Sin. B 2014, 4, 202–207. [Google Scholar] [CrossRef]
- Luke, J.J.; Hodi, F.S. Vemurafenib and braf inhibition: A new class of treatment for metastatic melanoma. Clin. Cancer Res. 2012, 18, 9–148. [Google Scholar] [CrossRef]
- Vispute, S.G.; Chen, J.J.; Sun, Y.L.; Sodani, K.S.; Singh, S.; Pan, Y.; Talele, T.; Ashby, C.R., Jr.; Chen, Z.S. Vemurafenib (PLX4032, Zelboraf®), a BRAF inhibitor, modulates ABCB1-, ABCG2-, and ABCC10-mediated multidrug resistance. J. Cancer Res. Updates 2013, 2, 306–317. [Google Scholar]
- Brazzelli, V.; Grasso, V.; Borroni, G. Imatinib, dasatinib and nilotinib: A review of adverse cutaneous reactions with emphasis on our clinical experience. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 1471–1480. [Google Scholar] [CrossRef]
- Heuchel, R.; Berg, A.; Tallquist, M.; Ahlen, K.; Reed, R.K.; Rubin, K.; Claesson-Welsh, L.; Heldin, C.H.; Soriano, P. Platelet-derived growth factor beta receptor regulates interstitial fluid homeostasis through phosphatidylinositol-3' kinase signaling. Proc. Natl. Acad. Sci. USA 1999, 96, 11410–11415. [Google Scholar] [CrossRef]
- Tamura, A.; An, R.; Hagiya, Y.; Hoshijima, K.; Yoshida, T.; Mikuriya, K.; Ishikawa, T. Drug-induced phototoxicity evoked by inhibition of human ABC transporter ABCG2: Development of in vitro high-speed screening systems. Expert Opin. Drug Metab. Toxicol. 2008, 4, 255–272. [Google Scholar] [CrossRef]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Anreddy, N.; Gupta, P.; Kathawala, R.J.; Patel, A.; Wurpel, J.N.D.; Chen, Z.-S. Tyrosine Kinase Inhibitors as Reversal Agents for ABC Transporter Mediated Drug Resistance. Molecules 2014, 19, 13848-13877. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules190913848
Anreddy N, Gupta P, Kathawala RJ, Patel A, Wurpel JND, Chen Z-S. Tyrosine Kinase Inhibitors as Reversal Agents for ABC Transporter Mediated Drug Resistance. Molecules. 2014; 19(9):13848-13877. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules190913848
Chicago/Turabian StyleAnreddy, Nagaraju, Pranav Gupta, Rishil J. Kathawala, Atish Patel, John N. D. Wurpel, and Zhe-Sheng Chen. 2014. "Tyrosine Kinase Inhibitors as Reversal Agents for ABC Transporter Mediated Drug Resistance" Molecules 19, no. 9: 13848-13877. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules190913848