
New Diethyl Ammonium Salt of Thiobarbituric Acid Derivative: Synthesis, Molecular Structure Investigations and Docking Studies

 ,
,  , ,
, , 
Abstract
:
1. Introduction
2. Results
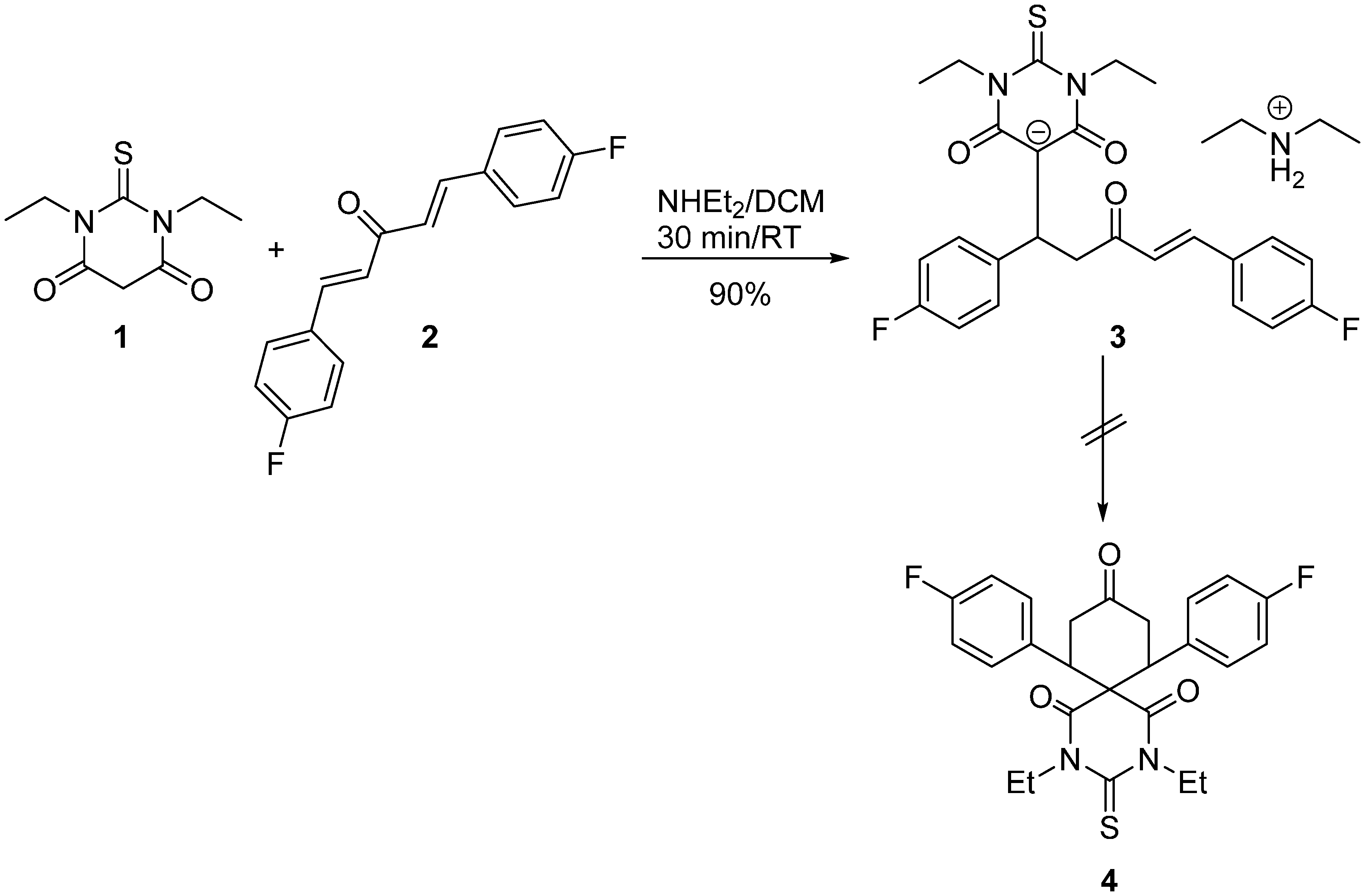
Synthesis

3. Discussion
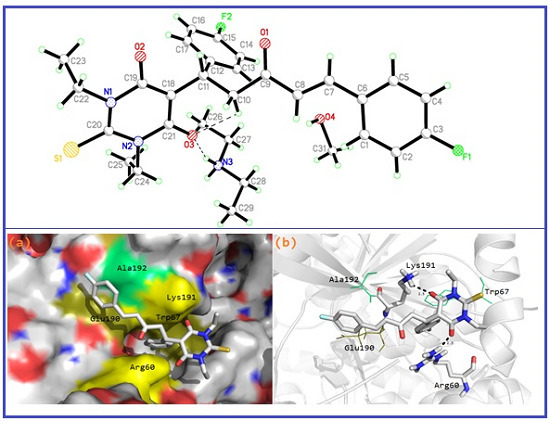
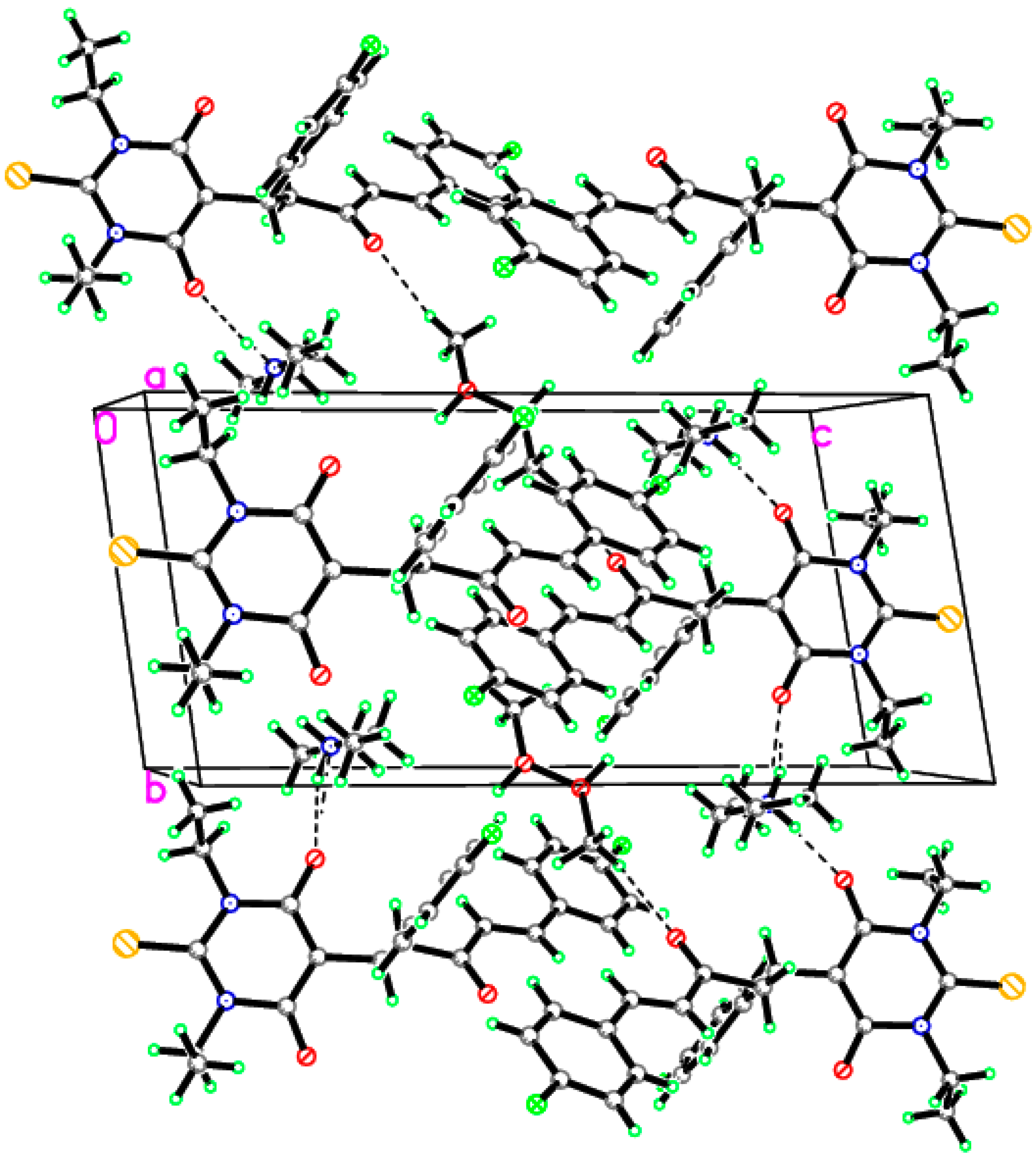
3.1. Single–Crystal X-ray Diffraction Study

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal Data | |
|---|---|
| Chemical formula | (C25H23F2N2O3S)·(C4H12N)·CH3OH |
| Mr | 375.12 |
| Crystal system, space group | Triclinic, P-1 |
| Temperature (K) | 150 |
| a, b, c (Å) | 8.9182(4), 9.2271(5), 18.2932(9) |
| α, β, γ (°) | 81.772(2), 80.176(2), 80.538(2) |
| V (Å3) | 1452.79(13) |
| Z | 2 |
| Radiation type | Mo Kα |
| µ (mm−1) | 0.16 |
| Crystal size (mm) | 0.31 × 0.26 × 0.20 |
| Data Collection | |
| Diffractometer | D8 Venture area detector |
| Absorption correction | multi-scanSADABS V2014/3 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 72,300, 7527, 5725 |
| Rint | 0.055 |
| Refinement | |
| R[F2 > 2σ(F2)], wR(F2), S | 0.050, 0.149, 1.05 |
| No. of reflections | 7527 |
| No. of parameters | 371 |
| No. of restraints | 0 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e·Å−3) | 0.66, −0.54 |
| S1–C20 | 1.6829(17) | N1–C20 | 1.366(2) |
|---|---|---|---|
| F1–C3 | 1.356(2) | N1–C22 | 1.474(2) |
| F2–C15 | 1.370(2) | N2–C20 | 1.369(2) |
| O1–C9 | 1.216(2) | N2–C21 | 1.415(2) |
| O2–C19 | 1.254(2) | N2–C24 | 1.475(2) |
| O3–C21 | 1.256(2) | N3–C28 | 1.488(2) |
| N1–C19 | 1.421(2) | N3–C27 | 1.487(2) |
| C19–N1–C20 | 124.08(13) | N1–C19–C18 | 117.36(14) |
| C19–N1–C22 | 116.13(12) | O2–C19–N1 | 116.20(14) |
| C20–N1–C22 | 119.57(13) | O2–C19–C18 | 126.42(15) |
| C20–N2–C21 | 123.59(13) | S1–C20–N2 | 121.62(12) |
| C20–N2–C24 | 119.25(13) | N1–C20–N2 | 116.11(14) |
| C21–N2–C24 | 117.04(13) | S1–C20–N1 | 122.26(12) |
| C27–N3–C28 | 113.94(13) | N2–C21–C18 | 117.87(14) |
| F1–C3–C4 | 118.14(17) | O3–C21–N2 | 117.81(14) |
| F1–C3–C2 | 118.57(17) | O3–C21–C18 | 124.32(15) |
| O1–C9–C10 | 122.01(17) | N1–C22–C23 | 111.24(14) |
| O1–C9–C8 | 122.96(17) | N2–C24–C25 | 112.24(14) |
| F2–C15–C14 | 118.54(17) | N3–C27–C26 | 110.43(15) |
| F2–C15–C16 | 118.58(16) | N3–C28–C29 | 110.50(15) |


| D–H···A | D–H | H···A | D···A | D–H···A |
|---|---|---|---|---|
| N3–H1N3···O2 i | 0.89(2) | 1.83(2) | 2.7123(19) | 174.3(19) |
| N3–H3O···O3 | 0.84(3) | 1.89(3) | 2.6919(19) | 159(2) |
| C1–H1A···F2 ii | 0.9300 | 2.5100 | 3.428(2) | 171.00 |
| C10–H10B···O3 | 0.9700 | 2.4900 | 3.060(2) | 117.00 |
| Symmetry codes: (i) x, y − 1, z; (ii) x − 1, y, z. | ||||
3.2. Molecular Modeling Study


| Parameter a | Calc. | Exp | Parameter a | Calc. | Exp | ||||
|---|---|---|---|---|---|---|---|---|---|
| a | b | c | a | b | c | ||||
| R(1-41) | 1.697 | 1.696 | 1.696 | 1.683 | A(4-23-24) | 122.8 | 122.5 | 122.8 | 122.0 |
| R(2-13) | 1.347 | 1.347 | 1.755 | 1.356 | A(5-40-7) | 118.6 | 118.6 | 118.7 | 116.2 |
| R(3-34) | 1.355 | 1.354 | 1.766 | 1.370 | A(5-40-39) | 124.8 | 124.8 | 124.8 | 126.4 |
| R(4-23) | 1.224 | 1.224 | 1.223 | 1.216 | A(6-42-8) | 116.7 | 116.7 | 116.8 | 117.8 |
| R(5-40) | 1.237 | 1.236 | 1.237 | 1.254 | A(6-42-39) | 124.8 | 124.6 | 124.7 | 124.3 |
| R(6-42) | 1.286 | 1.289 | 1.285 | 1.256 | A(40-7-41) | 124.5 | 124.5 | 124.5 | 124.1 |
| R(7-40) | 1.433 | 1.432 | 1.432 | 1.421 | A(40-7-43) | 115.0 | 115.0 | 115.0 | 116.1 |
| R(7-41) | 1.373 | 1.373 | 1.373 | 1.366 | A(7-40-39) | 116.6 | 116.6 | 116.5 | 117.4 |
| R(7-43) | 1.479 | 1.480 | 1.480 | 1.474 | A(41-7-43) | 120.5 | 120.5 | 120.5 | 119.6 |
| R(8-41) | 1.384 | 1.385 | 1.385 | 1.369 | A(7-41-8) | 116.1 | 116.1 | 116.2 | 116.1 |
| R(8-42) | 1.425 | 1.424 | 1.425 | 1.415 | A(7-43-46) | 112.2 | 112.2 | 112.3 | 111.2 |
| R(8-50) | 1.478 | 1.478 | 1.478 | 1.475 | A(41-8-42) | 123.5 | 123.4 | 123.5 | 123.6 |
| R(9-11) | 1.389 | 1.389 | 1.389 | 1.388 | A(41-8-50) | 119.5 | 119.5 | 119.5 | 119.2 |
| R(9-18) | 1.410 | 1.410 | 1.409 | 1.403 | A(42-8-50) | 116.8 | 116.9 | 116.8 | 117.0 |
| R(11-13) | 1.393 | 1.393 | 1.397 | 1.368 | A(8-42-39) | 118.5 | 118.7 | 118.4 | 117.9 |
| R(13-14) | 1.390 | 1.390 | 1.394 | 1.368 | A(8-50-53) | 114.1 | 114.1 | 114.0 | 112.2 |
| R(14-16) | 1.392 | 1.392 | 1.392 | 1.387 | A(11-9-18) | 121.2 | 121.2 | 121.3 | 121.3 |
| R(16-18) | 1.408 | 1.408 | 1.407 | 1.394 | A(9-11-13) | 118.8 | 118.8 | 119.3 | 117.7 |
| R(18-19) | 1.462 | 1.461 | 1.462 | 1.462 | A(9-18-16) | 117.9 | 118.0 | 117.8 | 118.4 |
| R(19-21) | 1.348 | 1.347 | 1.347 | 1.334 | A(9-18-19) | 123.3 | 123.3 | 123.4 | 122.7 |
| R(21-23) | 1.491 | 1.488 | 1.492 | 1.472 | A(11-13-14) | 121.9 | 122.0 | 121.0 | 123.3 |
| R(23-24) | 1.521 | 1.523 | 1.521 | 1.513 | A(13-14-16) | 118.5 | 118.5 | 119.0 | 118.7 |
| R(24-27) | 1.537 | 1.537 | 1.537 | 1.525 | A(14-16-18) | 121.6 | 121.6 | 121.6 | 120.6 |
| R(27-29) | 1.531 | 1.531 | 1.529 | 1.527 | A(16-18-19) | 118.8 | 118.7 | 118.8 | 118.9 |
| R(27-39) | 1.525 | 1.524 | 1.524 | 1.513 | A(18-19-21) | 128.4 | 128.3 | 128.2 | 126.0 |
| R(29-30) | 1.399 | 1.400 | 1.399 | 1.389 | A(19-21-23) | 120.7 | 120.7 | 120.7 | 122.7 |
| R(29-37) | 1.407 | 1.407 | 1.406 | 1.393 | A(21-23-24) | 115.5 | 115.6 | 115.5 | 115.0 |
| R(30-32) | 1.400 | 1.401 | 1.399 | 1.396 | A(23-24-27) | 115.5 | 115.2 | 115.5 | 114.8 |
| R(32-34) | 1.387 | 1.388 | 1.391 | 1.368 | A(24-27-29) | 114.9 | 115.0 | 114.9 | 113.9 |
| R(34-35) | 1.392 | 1.392 | 1.396 | 1.368 | A(24-27-39) | 111.0 | 111.2 | 111.0 | 112.0 |
| R(35-37) | 1.394 | 1.394 | 1.394 | 1.385 | A(29-27-39) | 113.6 | 113.7 | 113.4 | 111.1 |
| R(39-40) | 1.430 | 1.431 | 1.430 | 1.391 | A(27-29-30) | 124.0 | 124.2 | 124.1 | 124.2 |
| R(39-42) | 1.387 | 1.386 | 1.387 | 1.397 | A(27-29-37) | 118.3 | 118.1 | 118.3 | 117.9 |
| R(43-46) | 1.528 | 1.528 | 1.528 | 1.514 | A(27-39-40) | 117.1 | 117.2 | 117.2 | 119.5 |
| R(50-53) | 1.528 | 1.529 | 1.528 | 1.513 | A(27-39-42) | 122.5 | 122.5 | 122.4 | 119.9 |
| R(57-62) | 1.501 | 1.501 | 1.501 | 1.487 | A(30-29-37) | 117.7 | 117.6 | 117.6 | 117.9 |
| R(57-65) | 1.501 | 1.498 | 1.501 | 1.488 | A(29-30-32) | 121.7 | 121.6 | 121.7 | 121.1 |
| R(58-62) | 1.523 | 1.523 | 1.523 | 1.507 | A(29-37-35) | 121.6 | 121.7 | 121.7 | 121.7 |
| R(65-68) | 1.523 | 1.524 | 1.523 | 1.508 | A(30-32-34) | 118.7 | 118.8 | 119.1 | 118.2 |
| A(1-41-7) | 122.3 | 122.3 | 122.3 | 122.3 | A(32-34-35) | 121.5 | 121.5 | 120.7 | 122.9 |
| A(1-41-8) | 121.5 | 121.5 | 121.5 | 121.6 | A(34-35-37) | 118.8 | 118.8 | 119.2 | 118.1 |
| A(2-13-11) | 118.9 | 118.9 | 119.4 | 118.6 | A(40-39-42) | 120.4 | 120.3 | 120.5 | 120.6 |
| A(2-13-14) | 119.2 | 119.2 | 119.6 | 118.1 | A(62-57-65) | 114.3 | 114.4 | 114.3 | 113.9 |
| A(3-34-32) | 119.3 | 119.3 | 119.6 | 118.5 | A(57-62-58) | 111.9 | 111.8 | 112.0 | 110.4 |
| A(3-34-35) | 119.2 | 119.3 | 119.7 | 118.6 | A(57-65-68) | 111.3 | 110.9 | 111.3 | 110.5 |
| A(4-23-21) | 121.7 | 122.0 | 121.7 | 123.0 | |||||
| Atom | 3a | 3b | 3c | Atom | 3a | 3b | 3c |
|---|---|---|---|---|---|---|---|
| S1 | −0.2768 | −0.2719 | −0.3166 | C41 | 0.2791 | 0.2778 | 0.3354 |
| F (Cl)2 | −0.3284 | −0.3287 | (−0.0121) | C42 | 0.6153 | 0.6144 | 0.5547 |
| F (Cl)3 | −0.3414 | −0.3389 | (−0.0473) | C43 | −0.2630 | −0.2632 | −0.1403 |
| O4 | −0.5583 | −0.5598 | −0.4727 | H44 | 0.2599 | 0.2605 | 0.1816 |
| O5 | −0.6582 | −0.6546 | −0.5683 | H45 | 0.2577 | 0.2580 | 0.1864 |
| O6 | −0.7931 | −0.7953 | −0.6889 | C46 | −0.6861 | −0.6863 | −0.4477 |
| N7 | −0.4497 | −0.4495 | −0.5040 | H47 | 0.2332 | 0.2341 | 0.1434 |
| N8 | −0.4511 | −0.4511 | −0.4954 | H48 | 0.2361 | 0.2361 | 0.1645 |
| C9 | −0.1856 | −0.1845 | −0.1721 | H49 | 0.2332 | 0.2332 | 0.1564 |
| H10 | 0.2391 | 0.2384 | 0.1436 | C50 | −0.2637 | −0.2643 | −0.1389 |
| C11 | −0.2998 | −0.2998 | −0.1306 | H51 | 0.2621 | 0.2628 | 0.1924 |
| H12 | 0.2538 | 0.2533 | 0.1601 | H52 | 0.2514 | 0.2512 | 0.1659 |
| C13 | 0.4323 | 0.4327 | −0.0649 | C53 | −0.6750 | −0.6752 | −0.4451 |
| C14 | −0.3021 | −0.3019 | −0.1318 | H54 | 0.2332 | 0.2342 | 0.1444 |
| H15 | 0.2549 | 0.2548 | 0.1619 | H55 | 0.1875 | 0.1858 | 0.0970 |
| C16 | −0.1854 | −0.1842 | −0.1868 | H56 | 0.2550 | 0.2562 | 0.1955 |
| H17 | 0.2443 | 0.2443 | 0.1519 | N57 | −0.6390 | −0.6411 | −0.6438 |
| C18 | −0.1037 | −0.1032 | 0.1717 | C58 | −0.7043 | −0.7042 | −0.4778 |
| C19 | −0.1471 | −0.1425 | −0.1564 | H59 | 0.2594 | 0.2603 | 0.1823 |
| H20 | 0.2503 | 0.2498 | 0.1704 | H60 | 0.2575 | 0.2573 | 0.2000 |
| C21 | −0.3233 | −0.3204 | −0.2074 | H61 | 0.2321 | 0.2329 | 0.1597 |
| H22 | 0.2216 | 0.2203 | 0.1306 | C62 | −0.2595 | −0.2599 | −0.1682 |
| C23 | 0.5456 | 0.5440 | 0.4455 | H63 | 0.2334 | 0.2345 | 0.1685 |
| C24 | −0.5416 | −0.5400 | −0.4057 | H64 | 0.2671 | 0.2661 | 0.2257 |
| H25 | 0.2705 | 0.2700 | 0.1740 | C65 | −0.2604 | −0.2585 | −0.1658 |
| H26 | 0.2575 | 0.2553 | 0.1759 | H66 | 0.2725 | 0.2578 | 0.2305 |
| C27 | −0.2774 | −0.2793 | −0.2173 | H67 | 0.2309 | 0.2365 | 0.1638 |
| H28 | 0.3017 | 0.3043 | 0.1997 | C68 | −0.7069 | −0.7080 | −0.4834 |
| C29 | −0.0184 | −0.0165 | 0.1929 | H69 | 0.2591 | 0.2544 | 0.1814 |
| C30 | −0.2356 | −0.2376 | −0.2040 | H70 | 0.2269 | 0.2318 | 0.1490 |
| H31 | 0.2364 | 0.2361 | 0.1371 | H71 | 0.2615 | 0.2653 | 0.2055 |
| C32 | −0.3189 | −0.3196 | −0.1538 | H72 | 0.4312 | 0.4317 | 0.3686 |
| H33 | 0.2480 | 0.2473 | 0.1414 | H73 | 0.4901 | 0.4902 | 0.4600 |
| C34 | 0.3958 | 0.3977 | −0.0780 | C74 | −0.3071 | −0.2206 | |
| C35 | −0.3077 | −0.3082 | −0.1371 | H75 | 0.1922 | 0.1420 | |
| H36 | 0.2497 | 0.2511 | 0.1531 | H76 | 0.1981 | 0.1510 | |
| C37 | −0.2096 | −0.2103 | −0.1905 | H77 | 0.2179 | 0.1726 | |
| H38 | 0.2606 | 0.2606 | 0.1761 | O78 | −0.7670 | −0.6131 | |
| C39 | −0.2761 | −0.2740 | −0.0583 | H79 | 0.4766 | 0.3999 | |
| C40 | 0.6489 | 0.6496 | 0.5807 |
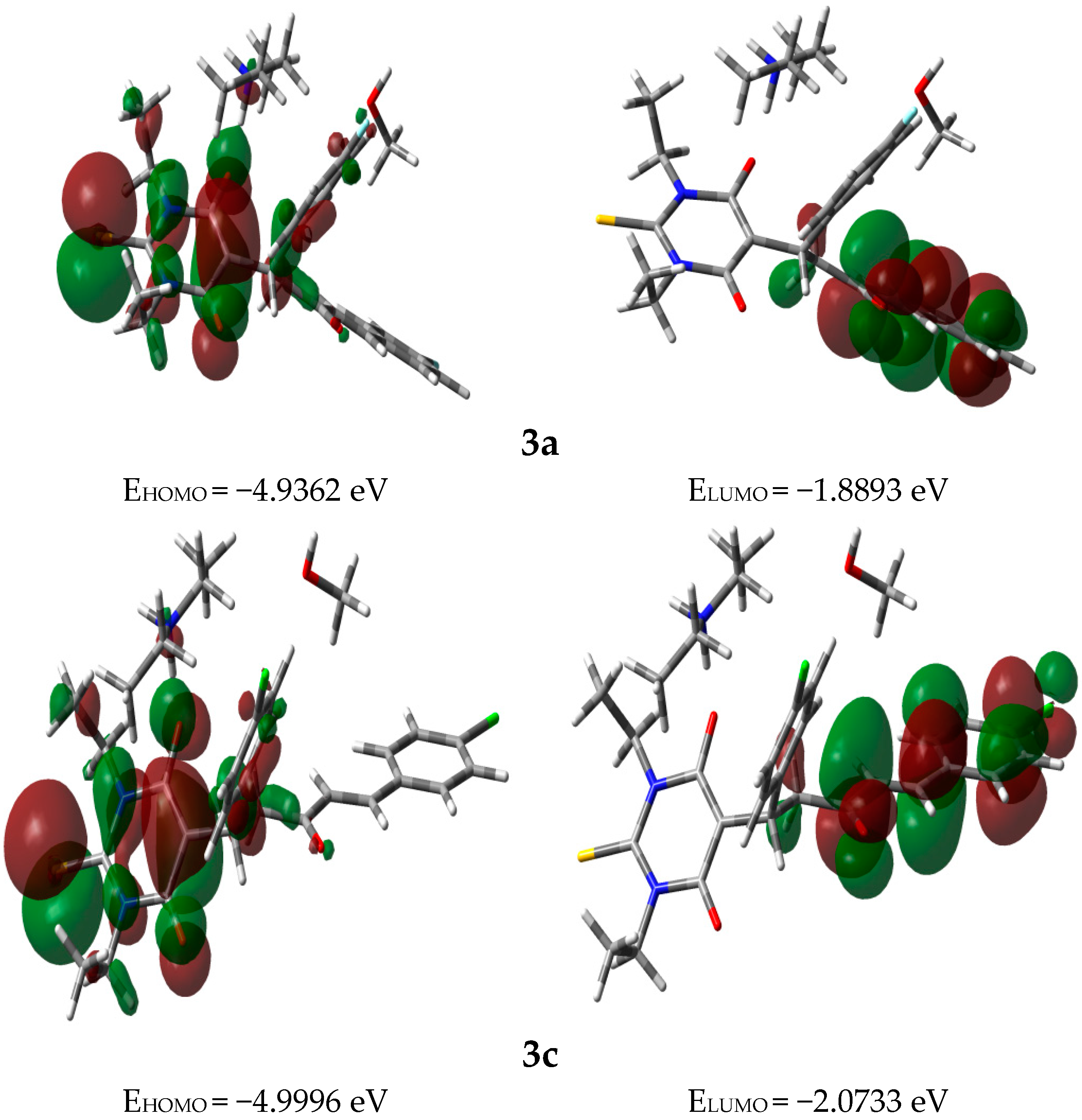
3.3. Frontier Molecular Orbitals (FMOs)

| Compound | Η | S | |||
|---|---|---|---|---|---|
| 3a | −3.4127 | 3.4127 | 1.5234 | 0.3282 | 3.8225 |
| 3c | −3.5364 | 3.5364 | 1.4632 | 0.3417 | 4.2737 |
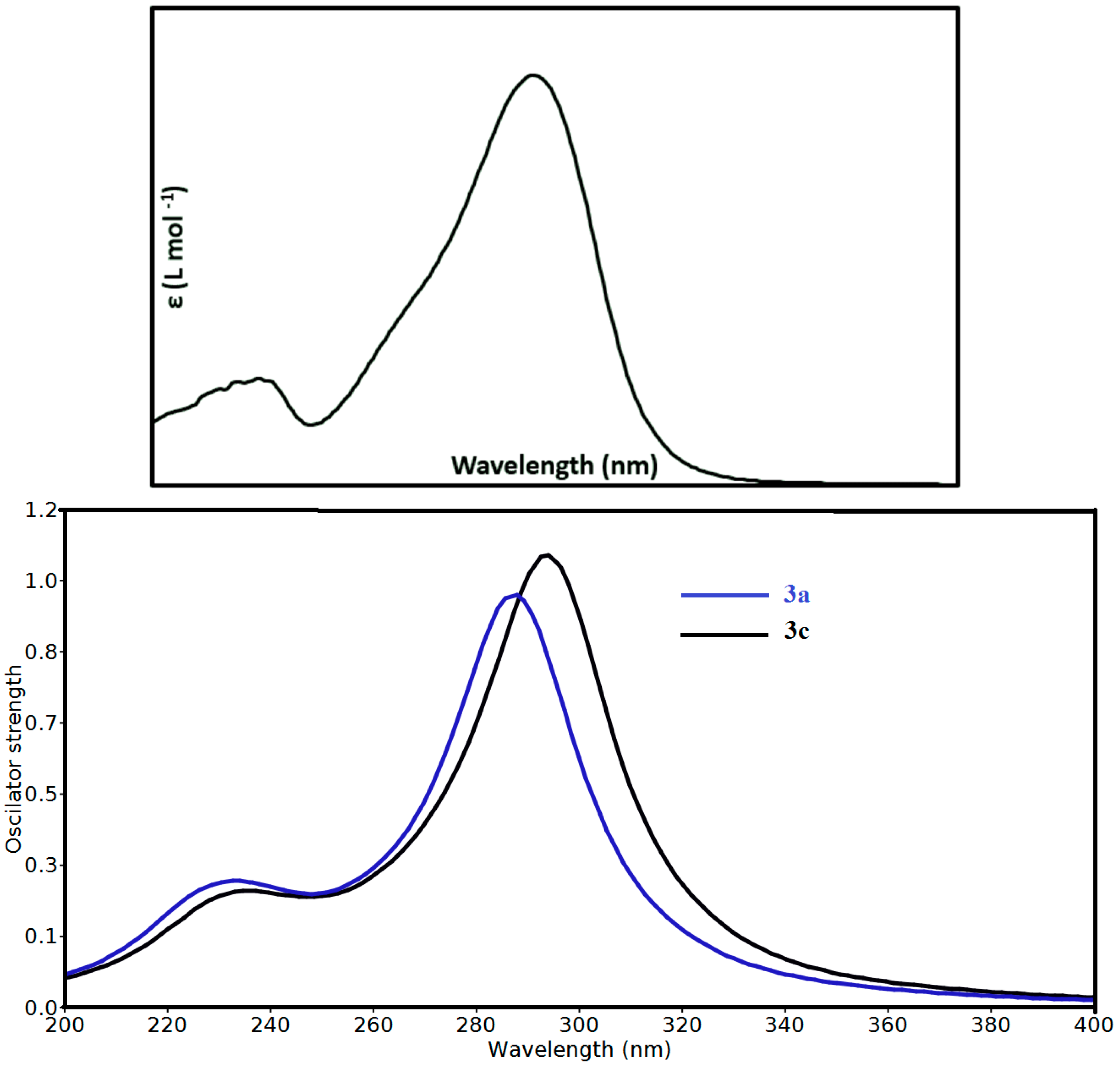
3.4. Electronic Spectra and TD-DFT Calculations

3.5. Charge Decomposition Analysis (CDA)
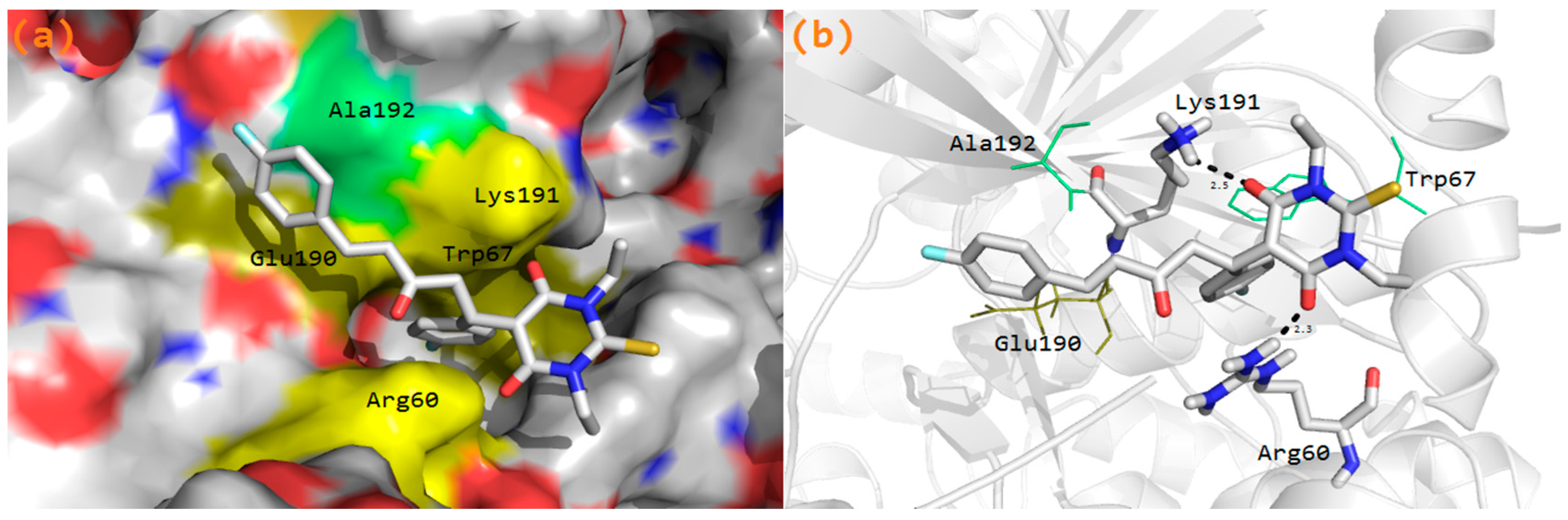
3.6. Molecular Docking Study


4. Materials and Methods
4.1. General
4.2. Synthesis of Diethylammonium (E)-5-(1,5-bis(4-Fluorophenyl)-3-oxopent-4-en-1-yl)-1,3-diethyl-4,6-dioxo-2-thioxohexahydropyrimidin-5-ide 3
4.3. Single-Crystal X-Ray Diffraction Studies
4.4. Computational Methods
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Archana, S.; Srivastava, V.K.; Kumar, A. Synthesis of newer indolyl/phenothiazinyl substituted 2-oxo/thiobarbituric acid derivatives as potent anticonvulsant agents. Arzneim. Forsch. Drug Res. 2002, 52, 787–791. [Google Scholar] [CrossRef]
- Goodman and Gilman’s. In The Pharmacological Basis of Therapeutics, 9th ed.; McGraw-Hill: New York, NY, USA, 1996; p. 471.
- Goel, B.; Sharma, S.; Bajaj, K.; Bansal, D.; Singh, T.; Malik, N.; Lata, S.; Tyagi, C.; Panwar, H.; Agarwal, A.; et al. Synthesis and CNS depressant of newer spirobarbiturates. Indian J. Pharm. Sci. 2005, 67, 194–199. [Google Scholar]
- Osman, A.N.; Kandeel, M.M.; Ahmed, M. Synthesis and anticonvulsant activity of some spiro compounds derived from barbituric and thiobarbituric acids: Part I. Indian J. Chem. 1996, 35B, 1073–1078. [Google Scholar]
- Sarma, G.V.S.R.; Rao, J.V.; Suresh, B. Synthesis and pharmacological evaluation of 1-[2,6,8-trisubstituted (3H)-oxoquinazolin-3-yl]-3-(4-substituted phenyl) thiobarbiturates. Indian J. Pharm. Sci. 1999, 61, 105–109. [Google Scholar]
- Gupta, K.P.; Gupta, R.C.; Bhargava, K.P.; Ali, B. Inhibition of brain respiration by new anticonvulsant benzylidenethiobarbiturates. Eur. J. Med. Chem. 1982, 17, 448–452. [Google Scholar]
- Padmavati, V.; Reddy, B.J.M.; Venketa Subbaiah, D.R.C.; Padmaja, A. Michael adducts-synthons for a new class of 1,4-dispirocyclohexane derivatives. Indian J. Chem. 2006, 45B, 808–812. [Google Scholar] [CrossRef]
- Levina, R.Y.; Velichko, F.K. Advances in the chemistry of barbituric acids. Russ. Chem. Rev. 1960, 29, 437–438. [Google Scholar] [CrossRef]
- Barakat, A.; Al-Majid, A.M.; Lotfy, G.; Arshad, F.; Yousuf, S.; Choudhary, M.I.; Ashraf, S.; Ul-Haq, Z. Synthesis and dynamics studies of barbituric acid derivatives as urease inhibitors. Chem. Cent. J. 2015. [Google Scholar] [CrossRef]
- Central Nervous System Drugs, Sedatives and Hypnotics, Barbiturates. In Facts and Comparisons Drug Information; Olin, B.R. (Ed.) Facts and Comparisons: St. Louis, MO, USA, 1993; pp. 1398–1413.
- Naguib, F.N.M.; Levesque, D.L.; Wang, E.C.; Panzica, R.P.; El Kouni, M.H. 5-Benzylbarbituric acid derivatives, potent and specific inhibitors of uridine phosphorylase. Biochem. Pharmacol. 1993, 30, 1273–1283. [Google Scholar] [CrossRef]
- Skiles, W.J.; Gonnella, N.C.; Jeng, A.Y. The design, structure, and clinical update of small molecular weight matrix metalloproteinase inhibitors. Curr. Med. Chem. 2004, 11, 2911–2977. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sanchez-Rosello, M.; Acena, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in pharmaceutical industry: Fluorine-containing drugs introduced to the market in the last decade. Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef] [PubMed]
- Begue, J.P.; Bonnet-Delpon, D. Recent advances (1995–2005) in fluorinated pharmaceuticals based on natural products. J. Fluorine Chem. 2006, 127, 992–1012. [Google Scholar] [CrossRef]
- Muller, K.; Faeh, C.; Diederich, F. Fluorine in pharmaceuticals: Looking beyond intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- Hagmann, W.K. The many roles for fluorine in medicinal chemistry. J. Med. Chem. 2008, 51, 4359–4369. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A.; Al-Majid, A.M.; Al-Najjar, H.J.; Mabkhot, Y.N.; Ghabbour, H.A.; Fun, H.-K. An efficient and green procedure for synthesis of rhodanine derivatives by Aldol-thia-Michael protocol using aqueous diethylamine medium. RSC Adv. 2014, 4, 4909–4916. [Google Scholar] [CrossRef]
- Al-Majid, A.M.; Barakat, A.; Al-Najjar, H.J.; Mabkhot, Y.N.; Ghabbour, H.A.; Fun, H.-K. Tandem Aldol-Michael reactions in aqueous diethylamine medium: A greener and efficient approach to bis-pyrimidine derivatives. Int. J. Mol. Sci. 2013, 14, 23762–23773. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A.; Al-Majid, A.M.; Al-Najjar, H.J.; Mabkhot, Y.N.; Javaid, S.; Yousuf, S.; Choudhary, M.I. Zwitterionic pyrimidinium adducts as antioxidants with therapeutic potential as nitric oxide scavenger. Eur. J. Med. Chem. 2014, 84, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A.; Islam, M.S.; Al Majid, A.M.A.; Al-Othman, Z.A. Highly enantioselective Friedel–Crafts alkylation of indoles with α,β-unsaturated ketones with simple Cu(II)–oxazoline–imidazoline catalysts. Tetrahedron 2013, 69, 5185–5192. [Google Scholar] [CrossRef]
- Barakat, A.; Islam, M.S.; Al-Majid, A.M.; Soliman, S.M.; Mabkhot, Y.N.; Al-Othman, Z.A.; Ghabbour, H.A.; Fun, H.-K. Synthesis of novel 5-monoalkylbarbiturate derivatives: New access to 1,2-oxazepines. Tetrahedron Lett. 2015. [Google Scholar] [CrossRef]
- Barakat, A.; Islam, M.S.; Al Majid, A.M.A.; Ghabbour, H.A.; Fun, H.-K.; Javed, K.; Imad, R.; Yousuf, S.; Choudhary, M.I.; Wadood, A. Synthesis, in vitro biological activities and in silico study of dihydropyrimidines derivatives. Bioorg. Med. Chem. 2015, 23, 6740–6748. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.S.; Al-Majid, A.M.; Barakat, A.; Soliman, S.M.; Ghabbour, H.A.; Quah, C.K.; Fun, H.K. Synthesis, molecular structure and spectroscopic investigations of novel fluorinated spiro heterocycles. Molecules 2015, 20, 8223–8241. [Google Scholar] [CrossRef] [PubMed]
- Sidir, I.; Sidir, Y.G.; Kumalar, M.; Tasal, E. Ab initio Hartree–Fock and density functional theory investigations on the conformational stability, molecular structure and vibrational spectra of 7-acetoxy-6-(2,3-dibromopropyl)-4,8-dimethylcoumarin molecule. J. Mol. Struct. 2010, 964, 134–151. [Google Scholar] [CrossRef]
- Sebastian, S.; Sundaraganesan, N. The spectroscopic (FT-IR, FT-IR gas phase, FT-Raman and UV) and NBO analysis of 4-Hydroxypiperidine by density functional method. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2010, 753, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Joe, I.H.; Kostova, I.; Ravikumar, C.; Amalanathan, M.; Cîntă Pînzaru, S. Theoretical and vibrational spectral investigation of sodium salt of acenocoumarol. J. Raman Spectrosc. 2009, 40, 1033–1038. [Google Scholar] [CrossRef]
- Fukui, K.; Yonezawa, T.; Shingu, H.J. A molecular–orbital theory of reactivity in aromatic hydrocarbons. J. Chem. Phys. 1952, 20, 722–725. [Google Scholar] [CrossRef]
- Padmaja, L.; Ravikumar, C.; Sajan, D.; Joe, I.H.; Jayakumar, V.S.; Pettit, G.R.; Nielsen, O.F. Density functional study on the structural conformations and intramolecular charge transfer from the vibrational spectra of the anticancer drug combretastatin-A2. J. Raman Spectrosc. 2009, 40, 419–428. [Google Scholar] [CrossRef]
- Ravikumar, C.; Joe, I.H.; Jayakumar, V.S. Charge transfer interactions and nonlinear optical properties of push–pull chromophore benzaldehyde phenylhydrazone: A vibrational approach. Chem. Phys. Lett. 2008, 460, 552–558. [Google Scholar] [CrossRef]
- Pearson, R.G. Absolute electronegativity and hardness: Applications to organic chemistry. J. Org. Chem. 1989, 54, 1430–1432. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Geerlings, P.; de Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1873. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Szentpaly, L.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Chattaraj, K.; Giri, S. Stability, reactivity, and aromaticity of compounds of a multivalent super atom. J. Phys. Chem. A 2007, 111, 11116–11121. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Singh, R.N.; Kumar, A.; Tiwari, R.K.; Rawat, P.; Gupta, V.P. A combined experimental and quantum chemical (DFT and AIM) study on molecular structure, spectroscopic properties, NBO and multiple interaction analysis in a novel ethyl 4-[2-(carbamoyl)hydrazinylidene]-3,5-dimethyl-1H-pyrrole-2-carboxylate and its dimer. J. Mol. Struct. 2013, 1035, 427–440. [Google Scholar] [CrossRef]
- Dapprich, S.; Frenking, G. Investigation of donor-acceptor interactions: A charge decomposition analysis using fragment molecular orbitals. J. Phys. Chem. 1995, 99, 9352–9362. [Google Scholar] [CrossRef]
- Lagunin, A.; Stepanchikova, A.; Filimonov, D.; Prorikov, V. PASS: Prediction of activity spectra for biologically active substances. Bioinformatics 2000, 16, 747–748. [Google Scholar] [CrossRef] [PubMed]
- Treadway, J.L.; Mendys, P.; Hoover, D.J. Glycogen phosphorylase inhibitors for treatment of type 2 diabetes mellitus. Exp. Opin. Investig. Drugs 2001, 10, 439–454. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE). Available online: http://www.chemcomp.com (accessed on 15 March 2015).
- Oikonomakos, N.G.; Skamnaki, V.T.; Tsitsanou, K.E.; Gavalas, N.G.; Johnson, L.N. A new allosteric site in glycogen phosphorylase b as a target for drug interactions. Structure 2000, 15, 575–584. [Google Scholar] [CrossRef]
- Paul, M.K.; Mukhopadhyay, A.K. Tyrosine kinase—role and significance in cancer. Int. J. Med. Sci. 2004, 1, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Structure validation in chemical crystallography. Acta Cryst. 2009, D65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- CCDC 1039271 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: [email protected]).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian–03, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Dennington, R.D., II; Keith, T.A.; Millam, J. Gauss View, Version 4.1; Semichem Inc.: Shawnee Mission, KS, USA, 2007. [Google Scholar]
- Zhurko, G.A.; Zhurko, D.A. Chemcraft. Lite Version Build 08. Available online: http://www.chemcraftprog.com/ (accessed on 1 April 2005).
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO, Version 3.1; University of Wisconsin: Madison, WI, USA, 1998. [Google Scholar]
- Sample Availability: Sample of the compound 3 is available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barakat, A.; Al-Majid, A.M.; Soliman, S.M.; Lotfy, G.; Ghabbour, H.A.; Fun, H.-K.; Wadood, A.; Warad, I.; Sloop, J.C. New Diethyl Ammonium Salt of Thiobarbituric Acid Derivative: Synthesis, Molecular Structure Investigations and Docking Studies. Molecules 2015, 20, 20642-20658. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules201119710
Barakat A, Al-Majid AM, Soliman SM, Lotfy G, Ghabbour HA, Fun H-K, Wadood A, Warad I, Sloop JC. New Diethyl Ammonium Salt of Thiobarbituric Acid Derivative: Synthesis, Molecular Structure Investigations and Docking Studies. Molecules. 2015; 20(11):20642-20658. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules201119710
Chicago/Turabian StyleBarakat, Assem, Abdullah Mohammed Al-Majid, Saied M. Soliman, Gehad Lotfy, Hazem A. Ghabbour, Hoong-Kun Fun, Abdul Wadood, Ismail Warad, and Joseph C. Sloop. 2015. "New Diethyl Ammonium Salt of Thiobarbituric Acid Derivative: Synthesis, Molecular Structure Investigations and Docking Studies" Molecules 20, no. 11: 20642-20658. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules201119710