
Recent Advances in Developing Inhibitors for Hypoxia-Inducible Factor Prolyl Hydroxylases and Their Therapeutic Implications

Abstract
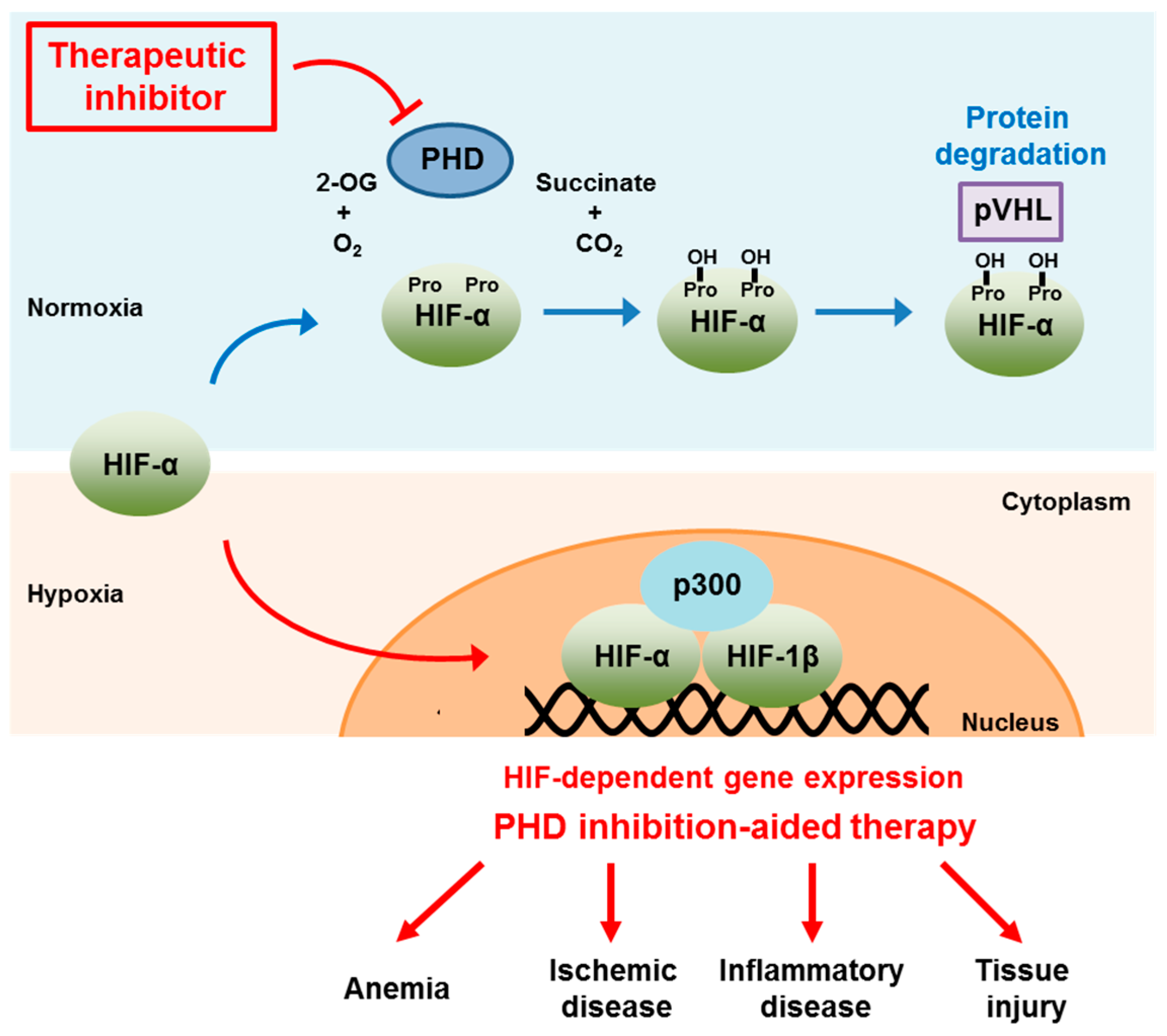
:1. Introduction

2. PHD2 Structure
3. Assays for Measuring PHD Activity

{kind=link}
| Method | Description | Advantage | Disadvantage | Ref. |
|---|---|---|---|---|
| Radioactive isotope-based | Detection of reaction products using [14C]-labeled 2-OG Detection of interactions between VHL and HIF-1α, either of which is labeled with [35S]-methionine | Highly sensitive | Use of radioactive materials Can be non-specific | [4,31,32,33] |
| Fluorescence-based | Detection of fluorescent derivatives of 2-OG Detection of increased FP of HIF-1α peptides upon binding to VHL Detection of TR-FRET by interactions between VHL and HIF-1α Detection of electrochemi-luminescence by interaction between VHL and HIF-1α | Suitable for HTS Easy to perform | Can be non-specific Cannot be used for fluorescent inhibitors | [34,35,36,37] |
| MS-based | Detection of reaction products using [14C]-labeled 2-OG Detection of interactions between HIF and PHD using [35S]-methionine labeled proteins Detection of inhibitors after dissociation from PHDing | Highly sensitive | Difficult for HTS Can be time-consuming | [38,39,40] |
| Cell-based | Detection of ODD domain-mediated luciferase activity | Physiological environment | May not be appropriate for bulky molecules Can be non-specific, and hard to interpret | [41] |
3.1. Radioactive Isotope-Based Assays
3.2. Fluorescence-Based Assays
3.3. MS-Based Assays
3.4. Cell-Based Assays
4. Development of PHD Inhibitors
4.1. SBDD for PHD Inhibitors
| Compound Number | Compound Structure | IC50 | Producer |
|---|---|---|---|
| 1 |  | 2.4 μM | Proctor & Gamble |
| 2 |  | 0.017 μM | Proctor & Gamble |
| 3 (TM6008) |  | N/A | Tokai University |
| 4 (TM6089) |  | N/A | Tokai University |
| 5 (JNJ-42041935) |  | 0.1 μM | Janssen |
| 6 |  | 0.05 μM | Janssen |
| 7 |  | <25 μM | Crystal-Genomics |
| 8 |  | 2.1 μM | Crystal-Genomics |
| 9 |  | 0.003 μM | Amgen |
4.2. PHD Inhibitors Developed by HTS Methods
| Compound Number | Compound Structure | IC50 | Producer |
|---|---|---|---|
| 10 |  | EC50 for EPO 1–20 nM | GSK |
| 11 |  | N/A | Amgen |
| 12 |  | 0.7 nM | Merck |
| 13 |  | 70 nM | Bayer |
| 14 |  | 2 μM | Cornell University |
| 15 |  | 10 μM | Cornell University |
| 16 |  | 4 nM | Merck |
| 17 |  | 0.2 nM | Merck |
5. Therapeutic Implications
5.1. Anemia
| Drug | Patient Population (Clinical Trial Phase) | Purpose of Study | Clinical Trials. Gov. Number (Status) |
|---|---|---|---|
| FG-4592 | Subjects with anemia associated with chronic kidney disease without dialysis (phase III) | Evaluate the efficacy for treatment of anemia correction and hemoglobin correction as well as their maintenance | NCT01750190 (recruiting) |
| AKB-6548 | Subjects with end stage renal disease undergoing chronic hemodialysis (phase II) | Evaluate the hemoglobin response (efficacy), safety, and tolerability | NCT02260193 (activated, but not recruiting) |
| GSK-1278863 | Hemodialysis dependent subjects with anemia associated with chronic kidney diseases, chronically hyporesponsive to recombinant human EPO (phase II) | Evaluate the safety and efficacy after switching from recombinant human EPO | NCT02075463 (recruiting) |
| BAY-85-3934 | Subjects with anemia associated with chronic kidney disease on dialysis, in USA and Japan (phase II) | Evaluate the efficacy and safety of oral BAY-85-3934 and active comparator in the long term treatment | NCT02064426 (recruiting) |
5.2. Ischemic Disease
5.3. Inflammatory Disease
5.4. Tissue Injury
6. Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell Biol. 1992, 12, 5447–5454. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.H.; Zhao, X.Y.; Han, Y.H.; Zhang, J.; Wang, L.S.; Xia, L.; Zhao, K.W.; Zheng, Y.; Guo, M.; Chen, G.Q. Proteomics-based identification of two novel direct targets of hypoxia-inducible factor-1 and their potential roles in migration/invasion of cancer cells. Proteomics 2009, 9, 3901–3912. [Google Scholar] [CrossRef] [PubMed]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF-1α and HIF-2α: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2011, 12, 9–22. [Google Scholar] [PubMed]
- Epstein, A.C.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. C. Elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef]
- Huang, L.E.; Arany, Z.; Livingston, D.M.; Bunn, H.F. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its α subunit. J. Biol. Chem. 1996, 271, 32253–32259. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [PubMed]
- Kamura, T.; Sato, S.; Iwai, K.; Czyzyk-Krzeska, M.; Conaway, R.C.; Conaway, J.W. Activation of HIF-1α ubiquitination by a reconstituted von Hippel-Lindau (VHL) tumor suppressor complex. Proc. Natl. Acad. Sci. USA 2000, 97, 10430–10435. [Google Scholar] [CrossRef] [PubMed]
- Paltoglou, S.; Roberts, B.J. HIF-1α and EPAS ubiquitination mediated by the VHL tumour suppressor involves flexibility in the ubiquitination mechanism, similar to other ring E3 ligases. Oncogene 2007, 26, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Hirsila, M.; Koivunen, P.; Gunzler, V.; Kivirikko, K.I.; Myllyharju, J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J. Biol. Chem. 2003, 278, 30772–30780. [Google Scholar] [CrossRef] [PubMed]
- Oliver, K.; Taylor, C.; Cummins, E. Hypoxia regulation of NFκB signalling during inflammation: The role of hydroxylases. Arthritis Res. Therapy 2009, 11, 215. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Gu, J.; Li, L.; Liu, J.; Luo, B.; Cheung, H.; Boehm, J.S.; Ni, M.; Geisen, C.; Root, D.E.; et al. Control of cyclin D1 and breast tumorigenesis by the EglN2 prolyl hydroxylase. Cancer Cell 2009, 16, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Place, T.L.; Domann, F.E. Prolyl-hydroxylase 3: Evolving roles for an ancient signaling protein. Hypoxia 2013, 2013, 13–17. [Google Scholar] [PubMed]
- Koditz, J.; Nesper, J.; Wottawa, M.; Stiehl, D.P.; Camenisch, G.; Franke, C.; Myllyharju, J.; Wenger, R.H.; Katschinski, D.M. Oxygen-dependent atf-4 stability is mediated by the PHD3 oxygen sensor. Blood 2007, 110, 3610–3617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Pi, X.; Mishra, A.; Fong, G.; Peng, J.; Patterson, C. PHD3-dependent hydroxylation of HCLK2 promotes the DNA damage response. J. Clin. Investig. 2012, 122, 2827–2836. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Lin, B.; Wang, Y.; Zhong, J.; O’Meally, R.; Cole, R.N.; Pandey, A.; Levchenko, A.; Semenza, G.L. PHD3-mediated prolyl hydroxylation of nonmuscle actin impairs polymerization and cell motility. Mol. Biol. Cell 2014, 25, 2788–2796. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Garvalov, B.K.; Foss, F.; Henze, A.T.; Bethani, I.; Graf-Hochst, S.; Singh, D.; Filatova, A.; Dopeso, H.; Seidel, S.; Damm, M.; et al. PHD3 regulates EGFR internalization and signalling in tumours. Nat. Commun. 2014, 5, 5577. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.; Wottawa, M.; Farhat, K.; Zieseniss, A.; Schnelle, M.; Le-Huu, S.; von Ahlen, M.; Malz, C.; Camenisch, G.; Katschinski, D.M. Prolyl hydroxylase domain (PHD) 2 affects cell migration and F-actin formation via RhoA/rho-associated kinase-dependent cofilin phosphorylation. J. Biol. Chem. 2010, 285, 33756–33763. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Sohn, H.; Park, Z.; Oh, S.; Kang, Y.; Lee, K.; Kang, M.; Jang, Y.; Yang, S.; Hong, Y.; et al. A lactate-induced response to hypoxia. Cell 2015, 161, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Ho, V.C.; Takeda, H.; Duan, L.; Nagy, A.; Fong, G. Placental but not heart defects are associated with elevated hypoxia-inducible factor α levels in mice lacking prolyl hydroxylase domain protein 2. Mol. Cell. Biol. 2006, 26, 8336–8346. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Cowan, A.; Fong, G. Essential role for prolyl hydroxylase domain protein 2 in oxygen homeostasis of the adult vascular system. Circulation 2007, 116, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Katschinski, D.M. In vivo functions of the prolyl-4-hydroxylase domain oxygen sensors: Direct route to the treatment of anaemia and the protection of ischaemic tissues. Acta Physiol. 2009, 195, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- McMullin, M.F. HIF pathway mutations and erythrocytosis. Expert Rev. Hematol. 2010, 3, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010, 29, 625–634. [Google Scholar] [CrossRef] [PubMed]
- McDonough, M.A.; Li, V.; Flashman, E.; Chowdhury, R.; Mohr, C.; Lienard, B.M.R.; Zondlo, J.; Oldham, N.J.; Clifton, I.J.; Lewis, J.; et al. Cellular oxygen sensing: Crystal structure of hypoxia-inducible factor prolyl hydroxylase (PHD2). Proc. Natl. Acad. Sci. USA 2006, 103, 9814–9819. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; McDonough, M.A.; Mecinovic, J.; Loenarz, C.; Flashman, E.; Hewitson, K.S.; Domene, C.; Schofield, C.J. Structural basis for binding of hypoxia-inducible factor to the oxygen-sensing prolyl hydroxylases. Structure 2009, 17, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Ehrismann, D.; Flashman, E.; Genn, D.N.; Mathioudakis, N.; Hewitson, K.S.; Ratcliffe, P.J.; Schofield, C.J. Studies on the activity of the hypoxia-inducible-factor hydroxylases using an oxygen consumption assay. Biochem. J. 2007, 401, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Bex, C.; Knauth, K.; Dambacher, S.; Buchberger, A. A yeast two-hybrid system reconstituting substrate recognition of the von Hippel-Lindau tumor suppressor Protein. Nucleic Acids Res. 2007, 35, e142. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Yum, S.; Ha, N.; Jung, Y. Von Hippel-Lindau β-domain-luciferase fusion protein as a bioluminescent hydroxyproline sensor for a hypoxia-inducible factor prolyl hydroxylase assay. Anal. Biochem. 2010, 407, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Kivirikko, I.; Myllyla, R. Posttranslational enzymes in the biosynthesis of collagen: Intracellular enzymes. Methods Enzymol. 1982, 82 (Pt A), 245–304. [Google Scholar] [PubMed]
- Bruick, R.K.; McKnight, S.L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 2001, 294, 1337–1340. [Google Scholar] [CrossRef] [PubMed]
- Thirstrup, K.; Christensen, S.; Moller, H.A.; Ritzen, A.; Bergstrom, A.L.; Sager, T.N.; Jensen, H.S. Endogenous 2-oxoglutarate levels impact potencies of competitive HIF prolyl hydroxylase inhibitors. Pharm. Res. 2011, 64, 268–273. [Google Scholar] [CrossRef] [PubMed]
- McNeill, L.A.; Bethge, L.; Hewitson, K.S.; Schofield, C.J. A fluorescence-based assay for 2-oxoglutarate-dependent oxygenases. Anal. Biochem. 2005, 336, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Ahn, D.R.; Kim, S.Y.; Lee, M.J.; Yang, E.G. HIF-1α Peptide Derivatives with modifications at the hydroxyproline residue as activators of HIF-1α. Bioorg. Med. Chem. Lett. 2009, 19, 4403–4405. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Choi, Y.K.; Kim, J.W.; Park, Y.K.; Yang, E.G.; Ahn, D.R. Inhibition of a prolyl hydroxylase domain (PHD) by substrate analog peptides. Bioorg. Med. Chem. Lett. 2011, 21, 4325–4328. [Google Scholar] [CrossRef] [PubMed]
- Dao, J.H.; Kurzeja, R.J.; Morachis, J.M.; Veith, H.; Lewis, J.; Yu, V.; Tegley, C.M.; Tagari, P. Kinetic characterization and identification of a novel inhibitor of hypoxia-inducible factor prolyl hydroxylase 2 using a time-resolved fluorescence resonance energy transfer-based assay technology. Anal. Biochem. 2009, 384, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, C.J.; Loenarz, C.; Mecinoviċ, J.; Yeoh, K.K.; Hindley, N.; Liėnard, B.M.; Sobott, F.; Schofield, C.J.; Flashman, E. Application of a proteolysis/mass spectrometry method for investigating the effects of inhibitors on hydroxylase structure. J. Med. Chem. 2009, 52, 2799–2805. [Google Scholar] [CrossRef] [PubMed]
- Flagg, S.C.; Martin, C.B.; Taabazuing, C.Y.; Holmes, B.E.; Knapp, M.J. Screening chelating inhibitors of HIF-prolyl hydroxylase domain 2 (PHD2) and factor inhibiting HIF (FIH). J. Inorg. Biochem. 2012, 113, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Vachal, P.; Miao, S.; Pierce, J.M.; Guiadeen, D.; Colandrea, V.J.; Wyvratt, M.J.; Salowe, S.P.; Sonatore, L.M.; Milligan, J.A.; Hajdu, R.; et al. 1,3,8-triazaspiro[4.5]decane-2,4-diones as efficacious pan-inhibitors of hypoxia-inducible factor prolyl hydroxylase 1–3 (HIF PHD1–3) for the treatment of anemia. J. Med. Chem. 2012, 55, 2945–2959. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, N.A.; Rakhman, I.; Moroz, N.; Basso, M.; Payappilly, J.; Kazakov, S.; Hernandez-Guzman, F.; Gaisina, I.N.; Kozikowski, A.P.; Ratan, R.R.; et al. Utilization of an in vivo reporter for high throughput identification of branched small molecule regulators of hypoxic adaptation. Chem. Biol. 2010, 17, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Kaule, G.; Guenzler, V. Assay for 2-oxoglutarate decarboxylating enzymes based on the determination of [1–14C] succinate: Application to prolyl 4-hydroxylase. Anal. Biochem. 1990, 184, 291–297. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Leung, I.K.H.; Demetriades, M.; Hardy, A.P.; Lejeune, C.; Smart, T.J.; Szöllössi, A.; Kawamura, A.; Schofield, C.J.; Claridge, T.D.W. Reporter ligand NMR screening method for 2-oxoglutarate oxygenase inhibitors. J. Med. Chem. 2013, 56, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Wang, X.; Cao, J.; Geng, Z.; Wang, Z. Effect of proline analogues on activity of human prolyl hydroxylase and the regulation of HIF signal transduction pathway. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Park, H.; Yang, E.G. A fluorescence polarization-based interaction assay for hypoxia-inducible factor prolyl hydroxylases. Biochem. Biophys. Res. Commun. 2005, 337, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Lee, H.Y.; Ahn, D.R.; Kim, S.Y.; Kim, S.; Lee, K.B.; Lee, Y.M.; Park, H.; Yang, E.G. Baicalein induces functional hypoxia-inducible factor-1α and angiogenesis. Mol. Pharmacol. 2008, 74, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.M.; Choi, K.O.; Lee, N.; Oh, M.; Park, H. The zinc chelator, N,N,N′,N′-tetrakis (2-pyridylmethyl) ethylenediamine, increases the level of nonfunctional HIF-1α protein in normoxic cells. Biochem. Biophys. Res. Commun. 2006, 343, 1002–1008. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.M.; Choi, K.O.; Park, Y.K.; Cho, H.; Yang, E.G.; Park, H. Clioquinol, a Cu(II)/Zn(II) chelator, inhibits both ubiquitination and asparagine hydroxylation of hypoxia-inducible factor-1α, leading to expression of vascular endothelial growth factor and erythropoietin in normoxic cells. J. Biol. Chem. 2006, 281, 34056–34063. [Google Scholar] [CrossRef] [PubMed]
- Handbook of Assay Developmenet in Drug Discovery; Minor, L.K. (Ed.) CRC Press: Cleveland, OH, USA, 2006; pp. 25–38.
- Warshakoon, N.C.; Wu, S.; Boyer, A.; Kawamoto, R.; Sheville, J.; Bhatt, R.T.; Renock, S.; Xu, K.; Pokross, M.; Zhou, S.; et al. Design and synthesis of substituted pyridine derivatives as HIF-1α prolyl hydroxylase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 5616–5620. [Google Scholar] [CrossRef] [PubMed]
- Warshakoon, N.C.; Wu, S.; Boyer, A.; Kawamoto, R.; Sheville, J.; Renock, S.; Xu, K.; Pokross, M.; Zhou, S.; Winter, C.; et al. Structure-based design, synthesis, and sar evaluation of a new series of 8-hydroxyquinolines as HIF-1α prolyl hydroxylase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 5517–5522. [Google Scholar] [CrossRef] [PubMed]
- Warshakoon, N.C.; Wu, S.; Boyer, A.; Kawamoto, R.; Renock, S.; Xu, K.; Pokross, M.; Evdokimov, A.G.; Zhou, S.; Winter, C.; et al. Design and synthesis of a series of novel pyrazolylpyridines as HIF-1α prolyl hydroxylase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 5687–5690. [Google Scholar] [CrossRef] [PubMed]
- Warshakoon, N.C.; Wu, S.; Boyer, A.; Kawamoto, R.; Sheville, J.; Renock, S.; Xu, K.; Pokross, M.; Evdokimov, A.G.; Walter, R.; et al. A novel series of imidazo[1,2-a]pyridine derivatives as HIF-1α prolyl hydroxylase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 5598–5601. [Google Scholar] [CrossRef] [PubMed]
- Demetriades, M.; Leung, I.K.H.; Chowdhury, R.; Chan, M.C.; McDonough, M.A.; Yeoh, K.K.; Tian, Y.; Claridge, T.D.W.; Ratcliffe, P.J.; Woon, E.C.Y.; et al. Dynamic combinatorial chemistry employing boronic acids/boronate esters leads to potent oxygenase inhibitors. Angew. Chem. Int. Ed. 2012, 51, 6672–6675. [Google Scholar] [CrossRef] [PubMed]
- Nangaku, M.; Izuhara, Y.; Takizawa, S.; Yamashita, T.; Fujii-Kuriyama, Y.; Ohneda, O.; Yamamoto, M.; van Ypersele de Strihou, C.; Hirayama, N.; Miyata, T. A novel class of prolyl hydroxylase inhibitors induces angiogenesis and exerts organ protection against ischemia. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2548–2554. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Haberberger, T.; Gervasi, D.C.; Michelson, K.S.; Gunzler, V.; Kondo, K.; Yang, H.; Sorokina, I.; Conaway, R.C.; Conaway, J.W.; et al. Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc. Natl. Acad. Sci. USA 2002, 99, 13459–13464. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Ichiki, T.; Narabayashi, E. Inhibition of prolyl hydroxylase domain-containing protein suppressed lipopolysaccharide-induced TNF-α expression. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 2132–2137. [Google Scholar] [CrossRef] [PubMed]
- Kontani, S.; Nagata, E.; Uesugi, T.; Moriya, Y.; Fujii, N.; Miyata, T.; Takizawa, S. A novel prolyl hydroxylase inhibitor protects against cell death after hypoxia. Neurochem. Res. 2013, 38, 2588–2594. [Google Scholar] [CrossRef] [PubMed]
- Hocutt, F.M.; Leonard, B.E.; Peltier, H.M.; Phuong, V.K.; Rabinowitz, M.H.; Rosen, M.D.; Tarantino, K.T.; Venkatesan, H.; Zhao, L.X. Benzimidazoles as Prolyl Hydroxylase Inhibitors. Patent WO2009134750 A1, 5 November 2009. [Google Scholar]
- Barrett, T.D.; Palomino, H.L.; Brondstetter, T.I.; Kanelakis, K.C.; Wu, X.; Haug, P.V.; Yan, W.; Young, A.; Hua, H.; Hart, J.C.; et al. Pharmacological characterization of 1-(5-chloro-6-(trifluoromethoxy)-1H-benzoimidazol-2-yl)-1H-pyrazole-4-carboxylic acid (JNJ-42041935), a potent and selective hypoxia-inducible factor prolyl hydroxylase inhibitor. Mol. Pharmacol. 2011, 79, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Rosen, M.D.; Venkatesan, H.; Peltier, H.M.; Bembenek, S.D.; Kanelakis, K.C.; Zhao, L.X.; Leonard, B.E.; Hocutt, F.M.; Wu, X.; Palomino, H.L.; et al. Benzimidazole-2-pyrazole HIF prolyl 4-hydroxylase inhibitors as oral erythropoietin secretagogues. ACS Med. Chem. Lett. 2010, 1, 526–529. [Google Scholar] [CrossRef] [PubMed]
- Minamishima, Y.A.; Kaelin, W.G., Jr. Reactivation of hepatic EPO synthesis in mice after PHD loss. Science 2010, 329, 407. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, M.H.; Rosen, M.D.; Tarantino, K.T.; Venkatesan, H. 1-(4-Aminoquinazolin-2-yl)-1H-pyrazole-4-carboxylic Acid Derivatives as Prolyl Hydroxylase Inhibitors and Their Preparation and Use for the Treatment of Diseases. U.S. Patent WO2012021830 A1, 16 February 2012. [Google Scholar]
- Kim, S.N.; Hong, Y.R.; Shin, D.; Ro, S.; Cho, J.M.; Lee, S.; Chang, H.J. Preparation of Pyridine Derivatives for Treating HIF-Related Disorders. Patent WO2009037570 A2, 26 March 2009. [Google Scholar]
- Hong, Y.R.; Kim, H.T.; Ro, S.; Cho, J.M.; Lee, S.H.; Kim, I.S.; Jung, Y.H. Discovery of novel 2-[2-(3-hydroxy-pyridin-2-yl)-thiazol-4-yl]-acetamide derivatives as HIF prolyl 4-hydroxylase inhibitors; SAR, synthesis and modeling evaluation. Bioorg. Med. Chem. Lett. 2014, 24, 3142–3145. [Google Scholar] [CrossRef] [PubMed]
- Frohn, M.; Viswanadhan, V.; Pickrell, A.J.; Golden, J.E.; Muller, K.M.; Buerli, R.W.; Biddlecome, G.; Yoder, S.C.; Rogers, N.; Dao, J.H.; et al. Structure-guided design of substituted aza-benzimidazoles as potent hypoxia inducible factor-1α prolyl hydroxylase-2 inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 5023–5026. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, M.H. Inhibition of hypoxia-inducible factor prolyl hydroxylase domain oxygen sensors: tricking the body into mounting orchestrated survival and repair responses. J. Med. Chem. 2013, 56, 9369–9402. [Google Scholar] [CrossRef] [PubMed]
- Chai, D.; Colon, M.; Duffy, K.J.; Fitch, D.M.; Tedesco, R.; Zimmerman, M.N. Preparation of N-[(4-Hydroxy-2-oxo-1,2-dihydro-3-quinolinyl)carbonyl]glycine Derivatives as Prolyl Hydroxylase Inhibitors. U.S. Patent WO2007038571 A2, 5 April 2007. [Google Scholar]
- Brackley, J.A., III; Shaw, A.N.; Tedesco, R.; Wang, Y.; Wiggall, K.J.; Yu, H. N-Glycinyl Pyridinedicarboxamide Derivatives as HIF Prolyl hydroxylase Inhibitors and Their Preparation, Pharmaceutical Compositions and Use in the Treatment of Anemia. U.S. Patent WO2009158315 A1, 30 December 2009. [Google Scholar]
- Schulz, M.J.; Wang, Y.; Ghergurovich, J.M. Prolyl Hydroxylase Inhibitors. U.S. Patent WO2010059555 A1, 27 May 2010. [Google Scholar]
- Allen, J.R.; Burli, R.; Bryan, M.C.; Cao, G.; Neira, S.C.; Reed, A.B. Preparation of Quinolinones and Azaquinolinones as Prolyl Hydroxylase Inhibitors. U.S. Patent WO2008130600 A2, 30 October 2008. [Google Scholar]
- Clements, M.J.; Debenham, J.S.; Hale, J.J.; Madsen-Duggan, C.B.; Walsh, T.F. Preparation of Substituted 4-Hydroxypyrimidine-5-carboxamides as HIF Prolyl Hydroxylase Inhibitors. U.S. Patent WO2009117269 A1, 24 September 2009. [Google Scholar]
- Clements, M.J.; Debenham, J.S.; Hale, J.J.; Madsen-Duggan, C. Substituted 4-Hydroxypyrimidine-5-carboxamides and Use as Treatment for Anemia and Like Conditions. U.S. Patent WO2011002624 A1, 6 January 2011. [Google Scholar]
- Zhou, C.; Zou, W.; Hua, Y.; Dang, Q. Pyrimidine Derivatives as HIF Prolyl Hydroxylase Inhibitors and Their Preparation and Use for the Treatment of Anemia. U.S. Patent WO2011130908 A1, 27 October 2011. [Google Scholar]
- Flamme, I.; Ergueden, J.; Oehme, F.; Thede, K.; Karig, G.; Kuhl, A.; Wild, H.; Schuhmacher, J.; Kolkhof, P.; Baerfacker, L.; et al. Preparation of 4-Pyridin-3-yl-1,2-dihydro-3H-pyrazol-3-ones as Cardiovascular Agents. U.S. Patent WO2006114213 A1, 2 November 2006. [Google Scholar]
- Jeske, M.; Flamme, I.; Stoll, F.; Beck, H.; Akbaba, M. Preparation of Dihydropyrazolones as HIF Prolyl Hydroxylase Inhibitors. U.S. Patent WO2009129945 A1, 29 October 2009. [Google Scholar]
- Theriault, J.R.; Felts, A.S.; Bates, B.S.; Perez, J.R.; Palmer, M.; Gilbert, S.R.; Dawson, E.S.; Engers, J.L.; Lindsley, C.W.; Emmitte, K.A. Discovery of a new molecular probe ML228: An activator of the hypoxia inducible factor (HIF) pathway. Bioorg. Med. Chem. Lett. 2012, 22, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Colandrea, V.J.; Hale, J.J. Prolyl hydroxylase domain-containing protein inhibitors as stabilizers of hypoxia-inducible factor: Small molecule-based therapeutics for anemia. Expert Opin. Ther. Pat. 2010, 20, 1219–1245. [Google Scholar] [CrossRef] [PubMed]
- Muchnik, E. Kaplan, HIF prolyl hydroxylase inhibitors for aneima. J. Expert Opin. Investig. Drugs 2011, 20, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Eckle, T.; Kohler, D.; Lehmann, R.; El Kasmi, K.; Eltzschig, H.K. Hypoxia-inducible factor-1 is central to cardioprotection: A new paradigm for ischemic preconditioning. Circulation 2008, 118, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Eckle, T.; Hartmann, K.; Bonney, S.; Reithel, S.; Mittelbronn, M.; Walker, L.A.; Lowes, B.D.; Han, J.; Borchers, C.H.; Buttrick, P.M.; et al. Adora2b-elicited Per2 stabilization promotes a HIF-dependent metabolic switch crucial for myocardial adaptation to ischemia. Nat. Med. 2012, 18, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Luo, W.; Zhan, H.; Semenza, G.L. Hypoxia-inducible factor 1 is required for remote ischemic preconditioning of the heart. Proc. Natl. Acad. Sci. USA 2013, 110, 17462–17467. [Google Scholar] [CrossRef] [PubMed]
- Karuppagounder, S.S.; Ratan, R.R. Hypoxia-inducible factor prolyl hydroxylase inhibition: Robust new target or another big bust for stroke therapeutics? J. Cereb. Blood Flow Metab. 2012, 32, 1347–1361. [Google Scholar] [CrossRef] [PubMed]
- Siddiq, A.; Ayoub, I.A.; Chavez, J.C.; Aminova, L.; Shah, S.; LaManna, J.C.; Patton, S.M.; Connor, J.R.; Cherny, R.A.; Volitakis, I.; et al. Hypoxia-inducible factor prolyl 4-hydroxylase inhibition. A target for neuroprotection in the central nervous system. J. Biol. Chem. 2005, 280, 41732–41743. [Google Scholar] [CrossRef] [PubMed]
- Baranova, O.; Miranda, L.F.; Pichiule, P.; Dragatsis, I.; Johnson, R.S.; Chavez, J.C. Neuron-specific inactivation of the hypoxia inducible factor 1α increases brain injury in a mouse model of transient focal cerebral ischemia. J. Neurosci. 2007, 27, 6320–6332. [Google Scholar] [CrossRef] [PubMed]
- Nagel, S.; Papadakis, M.; Chen, R.; Hoyte, L.C.; Brooks, K.J.; Gallichan, D.; Sibson, N.R.; Pugh, C.; Buchan, A.M. Neuroprotection by dimethyloxalylglycine following permanent and transient focal cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 2011, 31, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Fraisl, P.; Aragones, J.; Carmeliet, P. Inhibition of oxygen sensors as a therapeutic strategy for ischaemic and inflammatory disease. Nat. Rev. Drug Discov. 2009, 8, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Bratton, D.L.; Colgan, S.P. Targeting hypoxia signalling for the treatment of ischaemic and inflammatory diseases. Nat. Rev. Drug Discov. 2014, 13, 852–869. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [PubMed]
- Bartels, K.; Grenz, A.; Eltzschig, H.K. Hypoxia and inflammation are two sides of the same coin. Proc. Natl. Acad. Sci. USA 2013, 110, 18351–18352. [Google Scholar] [CrossRef] [PubMed]
- Scholz, C.C.; Cavadas, M.A.; Tambuwala, M.M.; Hams, E.; Rodriguez, J.; von Kriegsheim, A.; Cotter, P.; Bruning, U.; Fallon, P.G.; Cheong, A.; et al. Regulation of IL-1β-induced NF-κB by hydroxylases links key hypoxic and inflammatory signaling pathways. Proc. Natl. Acad. Sci. USA 2013, 110, 18490–18495. [Google Scholar] [CrossRef] [PubMed]
- Cummins, E.P.; Seeballuck, F.; Keely, S.J.; Mangan, N.E.; Callanan, J.J.; Fallon, P.G.; Taylor, C.T. The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology 2008, 134, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.; Keely, S.; Karhausen, J.; Gerich, M.E.; Furuta, G.T.; Colgan, S.P. Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology 2008, 134, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Botusan, I.R.; Sunkari, V.G.; Savu, O.; Catrina, A.I.; Gruenler, J.; Lindberg, S.; Pereira, T.; Ylae-Herttuala, S.; Poellinger, L.; Brismar, K.; et al. Stabilization of HIF-1α is critical to improve wound healing in diabetic mice. Proc. Natl. Acad. Sci. USA 2008, 105, 19426–19431. [Google Scholar] [CrossRef] [PubMed]
- Kalucka, J.; Ettinger, A.; Franke, K.; Mamlouk, S.; Singh, R.P.; Farhat, K.; Muschter, A.; Olbrich, S.; Breier, G.; Katschinski, D.M.; et al. Loss of epithelial hypoxia-inducible factor prolyl hydroxylase 2 accelerates skin wound healing in mice. Mol. Cell Biol. 2013, 33, 3426–3438. [Google Scholar] [CrossRef] [PubMed]
- Thangarajah, H.; Vial, I.N.; Grogan, R.H.; Yao, D.; Shi, Y.; Januszyk, M.; Galiano, R.D.; Chang, E.I.; Galvez, M.G.; Glotzbach, J.P.; et al. HIF-1α dysfunction in diabetes. Cell Cycle 2010, 9, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yan, X.; Cheng, L.; Dai, J.; Wang, C.; Han, P.; Chai, Y. Wound healing improvement with PHD-2 silenced fibroblasts in diabetic mice. PLoS ONE 2013, 8, e84548. [Google Scholar] [CrossRef] [PubMed]
- Myllyharju, J. Prolyl 4-hydroxylases, master regulators of the hypoxia response. Acta Physiol. 2013, 208, 148–165. [Google Scholar] [CrossRef] [PubMed]
- Ruthenborg, R.J.; Ban, J.J.; Wazir, A.; Takeda, N.; Kim, J.W. Regulation of wound healing and fibrosis by hypoxia and hypoxia-inducible factor-1. Mol. Cells 2014, 37, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Seeley, T.A.; Liu, D.Y.; Klauss, S.J. HIF Prolyl Hydroxylase Modifiers and Methods for Treatment of Cancer. U.S. Patent WO2006138511 A2, 28 December 2006. [Google Scholar]
- Mazzone, M.; Dettori, D.; Leite de Oliveira, R.; Loges, S.; Schmidt, T.; Jonckx, B.; Tian, Y.; Lanahan, A.A.; Pollard, P.; Ruiz de Almodovar, C.; et al. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell 2009, 136, 839–851. [Google Scholar] [CrossRef] [PubMed]
- Eckle, T.; Brodsky, K.; Bonney, M.; Packard, T.; Han, J.; Borchers, C.H.; Mariani, T.J.; Kominsky, D.J.; Mittelbronn, M.; Eltzschig, H.K. HIF-1α reduces acute lung injury by optimizing carbohydrate metabolism in the alveolar epithelium. PLoS Biol. 2013, 11, e1001665. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.Y.; Yang, E.G. Recent Advances in Developing Inhibitors for Hypoxia-Inducible Factor Prolyl Hydroxylases and Their Therapeutic Implications. Molecules 2015, 20, 20551-20568. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules201119717
Kim SY, Yang EG. Recent Advances in Developing Inhibitors for Hypoxia-Inducible Factor Prolyl Hydroxylases and Their Therapeutic Implications. Molecules. 2015; 20(11):20551-20568. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules201119717
Chicago/Turabian StyleKim, So Yeon, and Eun Gyeong Yang. 2015. "Recent Advances in Developing Inhibitors for Hypoxia-Inducible Factor Prolyl Hydroxylases and Their Therapeutic Implications" Molecules 20, no. 11: 20551-20568. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules201119717