
Synthesis of Tertiary and Quaternary Amine Derivatives from Wood Resin as Chiral NMR Solvating Agents

Abstract
:
1. Introduction


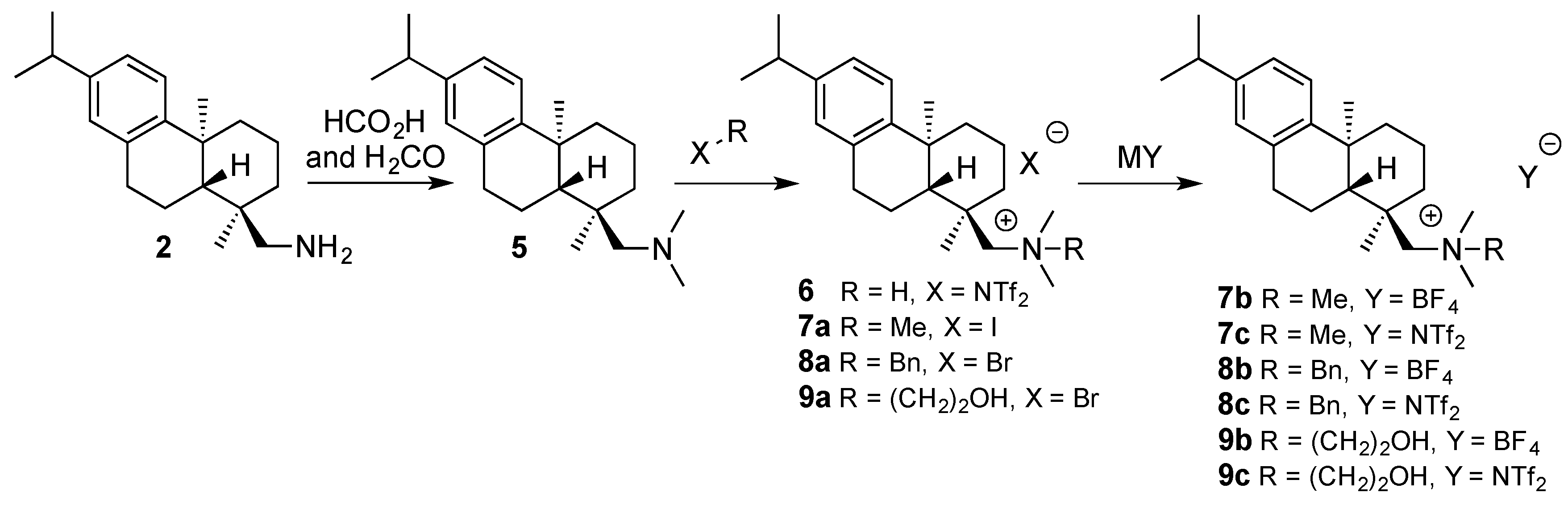
2. Results and Discussion


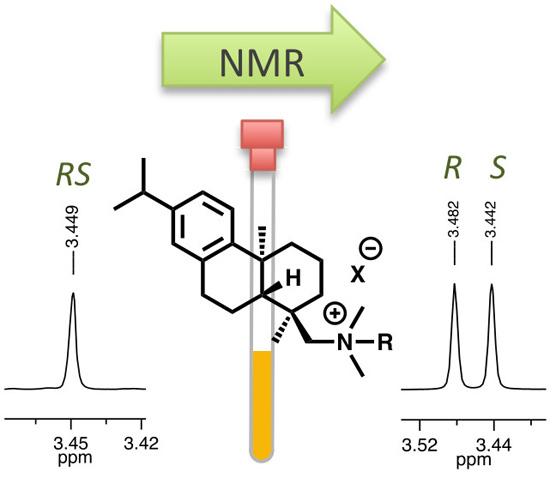{kind=link}
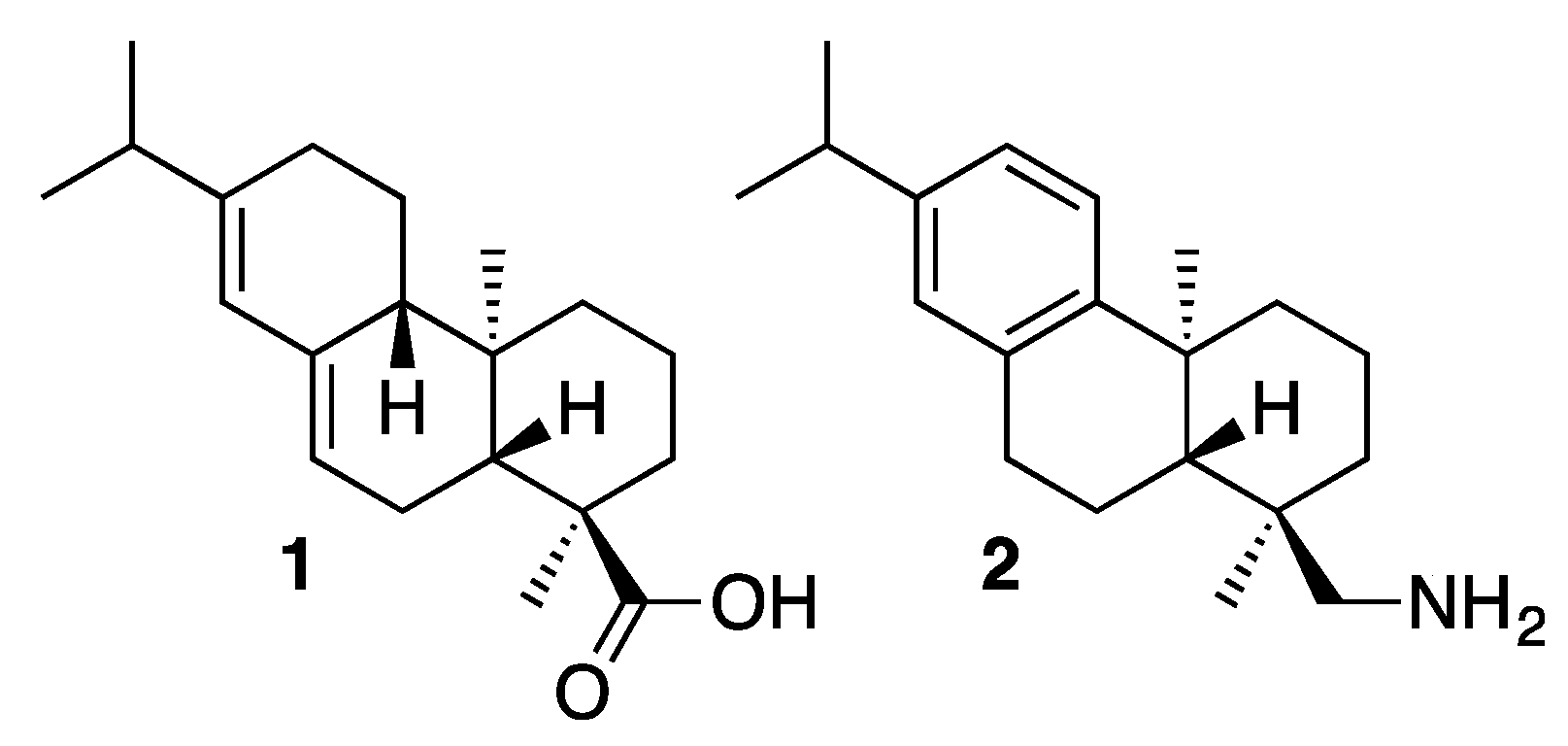{kind=link}
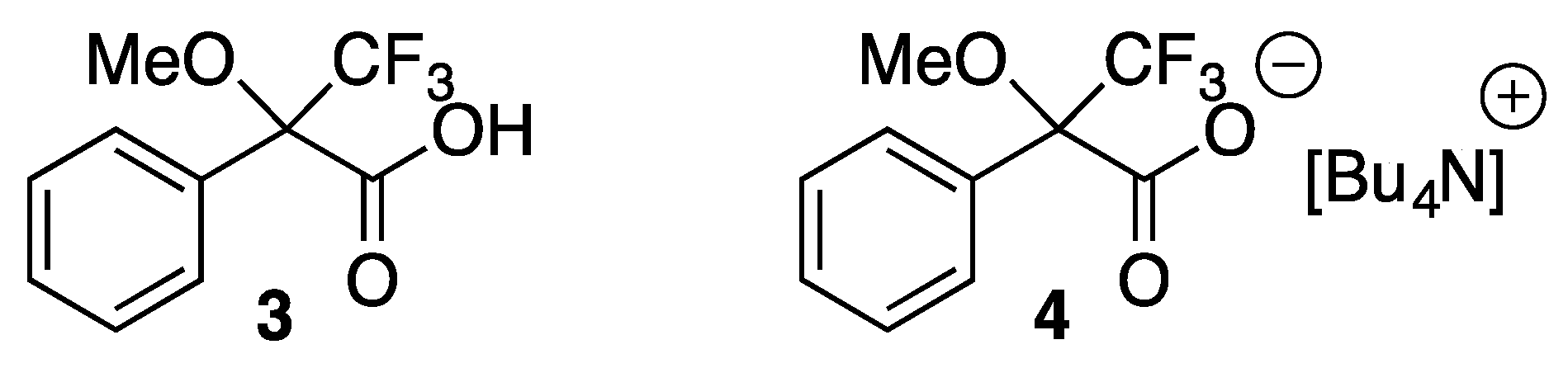{kind=link}
{kind=link}
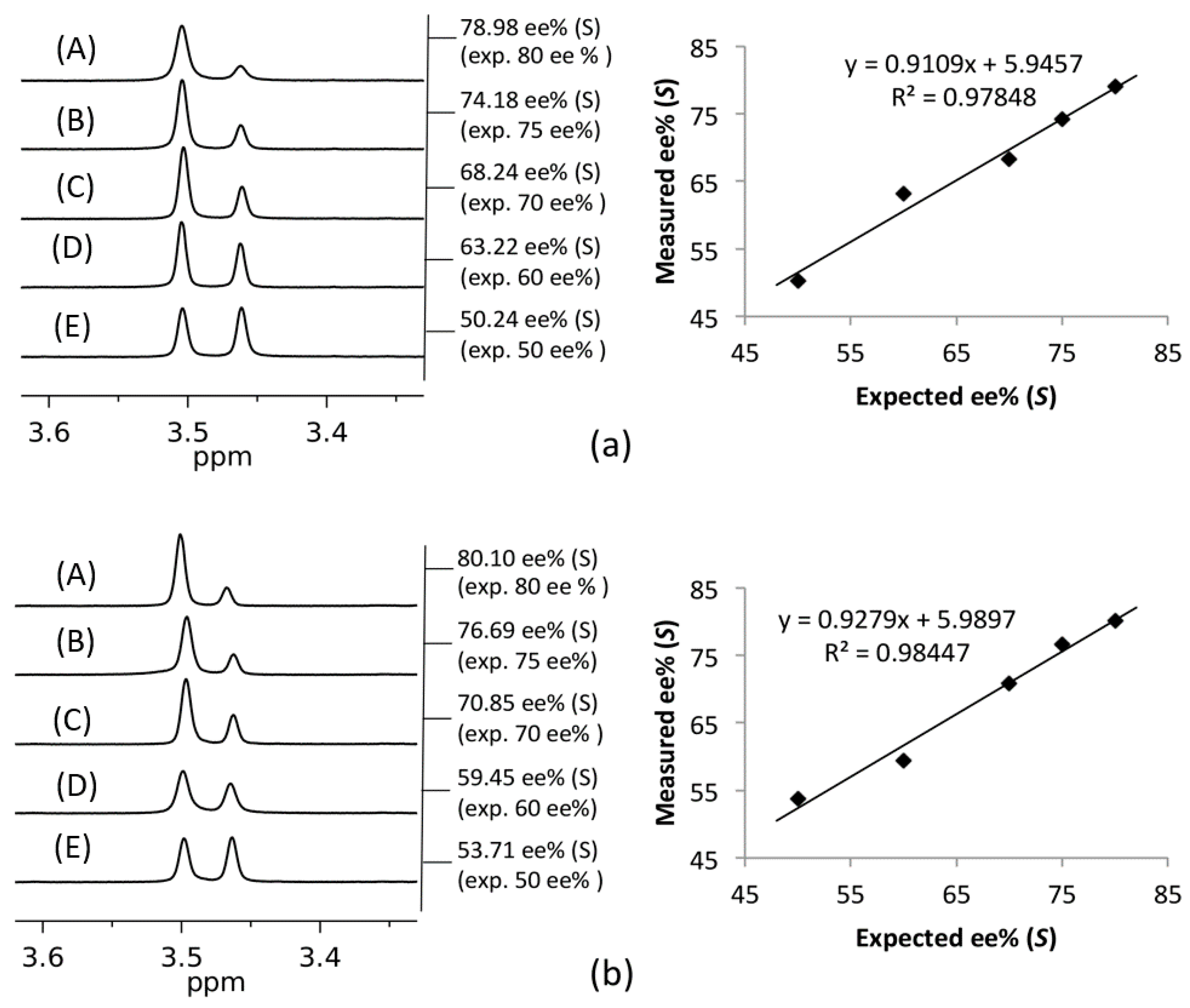{kind=link}
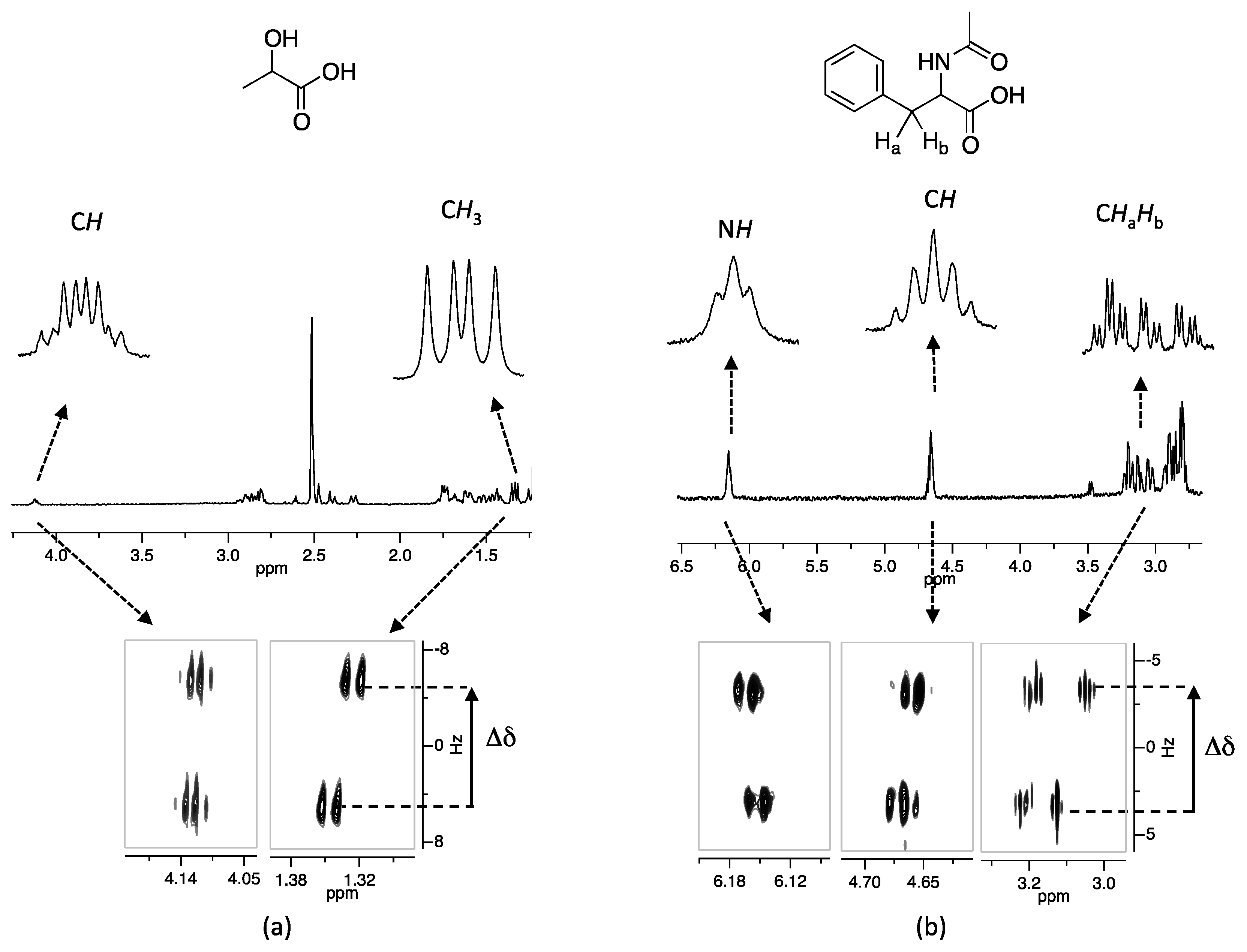{kind=link}
{kind=link}
| CSA | R | X | mp (°C) | (a) | (b) |
|---|---|---|---|---|---|
| 5 | - | - | liquid at rt. | +52.05 (c) | +161.71 |
| 6 | H | NTf2 | 114.2 | +7.45 | +44.29 |
| 7a | Me | I | 250.3 | +20.91 | +95.19 |
| 7b | Me | BF4 | 246.8 | +19.93 (d) | +82.77 |
| 7c | Me | NTf2 | 46.6 | +7.79 | +47.41 |
| 8a | Bn | Br | 188.6 | +7.19 | +34.83 |
| 8b | Bn | BF4 | 179.9 | +2.28 | +11.18 |
| 8c | Bn | NTf2 | 48.8 | +1.70 | +11.67 |
| 9a | (CH2)2OH | Br | 213.5 | +16.45 (e) | +72.12 |
| 9b | (CH2)2OH | BF4 | 188.5 | +18.73 | +83.43 |
| 9c | (CH2)2OH | NTf2 | viscous liquid at rt. | +6.56 | +41.90 |
| CSA | Host:Guest | 3 (ppm, (Hz)) | 4 (ppm, (Hz)) | ||
|---|---|---|---|---|---|
| 1H | 19F | 1H | 19F | ||
| 5 | 1:1 | 0.036 (18.1) | 0.12 (58.5) | 0.0018 (0.9) | - |
| 2:1 | 0.042 (21.6) | 0.15 (71.7) | - | - | |
| 6 | 1:1 | - | - | 0.030 (15.0) | 0.10 (48.3) |
| 2:1 | - | - | 0.026 (13.1) | 0.12 (54.2) | |
| 7a | 1:1 | - | - | - | - |
| 2:1 | - | - | - | - | |
| 7b | 1:1 | - | - | - | - |
| 2:1 | - | - | - | - | |
| 7c | 1:1 | 0.027 (1.3) | - | - | - |
| 2:1 | - | - | - | - | |
| 8a | 1:1 | - | - | - | - |
| 2:1 | - | - | - | 0.015 (6.8) | |
| 8b | 1:1 | 0.0015 (0.8) | - | - | - |
| 2:1 | - | - | - | - | |
| 8c | 1:1 | 0.0017 (0.9) | - | 0.0037 (1.9) | - |
| 2:1 | - | - | 0.010 (5.1) | 0.018 (8.3) | |
| 9a | 1:1 | - | - | - | - |
| 2:1 | - | - | - | - | |
| 9b | 1:1 | - | - | - | - |
| 2:1 | - | - | - | - | |
| 9c | 1:1 | 0.0017 (0.9) | - | 0.0052 (2.6) | - |
| 2:1 | - | - | 0.016 (8.2) | - | |



| Cmpd. | Racemic Carboxylic Acid | Δδ | Cmpd. | n-Bu4N Salt of Racemic Carboxylic Acid | Δδ | ||||
|---|---|---|---|---|---|---|---|---|---|
| ppm | Hz | ppm | Hz | ||||||
| 10a |  | Me | 0.0023 | 1.1 | 10b |  | Me | - | - |
| CH | - | - | CH | - | - | ||||
| 11a |  | Me | 0.0059 | 3.0 | 11b |  | Me | 0.0041 | 2.0 |
| CH | 0.0047 | 2.4 | CH | 0.0021 | 1.0 | ||||
| 12a |  | CH | - | - | 12b |  | CH | - | - |
| 13a |  | CH | 0.0081 | 4.1 | 13b |  | CH | 0.0068 | 3.4 |
| Me | 0.022 | 11.1 | Me | 0.017 | 8.6 | ||||
| 14a |  | CH | 0.012 | 6.1 | 14b |  | CH | 0.012 | 6.0 |
| NH | 0.013 | 6.5 | NH | 0.014 | 6.8 | ||||
| Me | 0.0028 | 1.4 | Me | 0.0039 | 1.9 | ||||
| Ha | 0.072 | 36.2 | Ha (a) | 0.065 | 32.5 | ||||
| Hb | 0.027 | 13.4 | Hb (a) | 0.036 | 17.8 | ||||
| 15a |  | CH | - | - | 15a |  | CH | - | - |
| Me | 0.023 | 11.5 | Me | 0.002 | 0.9 | ||||
| 16a |  | CH | 0.016 | 7.9 | 16b |  | CH | 0.003 | 1.4 |
| NH | 0.020 | 10.2 | NH | 0.020 | 10.0 | ||||
3. Experimental Section
3.1. General Methods
3.2. Synthesis of Chiral Solvating Agents
3.2.1. Purification of (+)-Dehydroabietylamine (2)
3.2.2. Synthesis of (+)-Dehydroabietyl-N,N-dimethylmethanamine (5)
3.2.3. Synthesis of (+)-Dehydroabietyl-N,N-dimethylmethanaminium bis(trifluoromethanesulfon-imide) (6)
3.2.4. Synthesis of (+)-Dehydroabietyl-N,N,N-trimethylmethanaminium iodide (7a)
3.2.5. Synthesis of N-Benzyl-1-(+)-dehydroabietyl-N,N-dimethylmethanaminium bromide (8a)
3.2.6. Synthesis of 2-Hydroxy-N-(+)-dehydroabietyl-N,N-dimethylethanaminium bromide (9a)
3.2.7. General Procedure for Anion Exchange
3.2.8. Synthesis of Guests
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nuopponen, M.; Willfor, S.; Jääskeläinen, A.; Sundberg, A.; Vuorinen, T. A UV resonance Raman (UVRR) spectroscopic study on the extractable compounds of Scots pine (Pinus sylvestris) wood Part I: Lipophilic compounds. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2004, 60, 2953–2961. [Google Scholar] [CrossRef] [PubMed]
- Qin, M.; Hannuksela, T.; Holmbom, B. Physico-chemical characterization of TMP resin and related model mixtures. Colloids Surfaces A Physicochem. Eng. Asp. 2003, 221, 243–254. [Google Scholar] [CrossRef]
- Cheney, L.C. Penicillin Salts of Amines Derived from Rosin. U.S. Patent 2585436, 12 February 1952. [Google Scholar]
- Nolan, M.O.; Bond, H.W. Results of laboratory screening tests of chemical compounds for molluscicidal activity. III. Derivatives of abietic acid. Am. J. Trop. Med. Hyg. 1955, 4, 152–155. [Google Scholar] [PubMed]
- Rosscup, R.J.; Liehe, H.J. Lubricant Comprising a Lubricating Oil and a Ureido Compound. U.S. Patent 3015625, 2 January 1962. [Google Scholar]
- Gottstein, W.J.; Cheney, L.C. Dehydroabietylamine. A new resolving agent. J. Org. Chem. 1965, 30, 2072–2073. [Google Scholar] [CrossRef]
- Bolchi, C.; Fumagalli, L.; Moroni, B.; Pallavicini, M.; Valoti, E. A short entry to enantiopure 2-substituted 1,4-benzodioxanes by efficient resolution methods. Tetrahedron Asymmetry 2003, 14, 3779–3785. [Google Scholar] [CrossRef]
- Laaksonen, T.; Heikkinen, S.; Wähälä, K. Synthesis and applications of secondary amine derivatives of (+)-dehydroabietylamine in chiral molecular recognition. Org. Biomol. Chem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, H.; Grosch, B.; Sitterberg, S.; Bach, T. An enantiomerically pure 1,5,7-trimethyl-3-azabicyclo[3.3.1]nonan-2-one as 1H-NMR shift reagent for the ee determination of chiral lactams, quinolones, and oxazolidinones. J. Org. Chem. 2004, 69, 970–973. [Google Scholar] [CrossRef] [PubMed]
- Heo, K.S.; Hyun, M.H.; Cho, Y.J.; Ryoo, J.J. Determination of optical purity of 3,5-dimethoxybenzoyl-leucine diethylamide by chiral chromatography and 1H- and 13C-NMR spectroscopy. Chirality 2011, 23, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Grimm, S.H.; Allmendinger, L.; Hoefner, G.; Wanner, K.T. Enantiopurity Determination of the Enantiomers of the Triple Reuptake Inhibitor Indatraline. Chirality 2013, 25, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Deschamps, J.R.; Jacobson, A.E.; Rice, K.C. Chiral Resolution and Absolute Configuration of the Enantiomers of the Psychoactive “Designer Drug” 3,4-Methylenedioxypyrovalerone. Chirality 2015, 27, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Uccello-Barretta, G.; Balzano, F. Chiral NMR Solvating Additives for Differentiation of Enantiomers. Top. Curr. Chem. 2013, 341, 69–131. [Google Scholar] [PubMed]
- Wenzel, T.J. Discrimination of Chiral Compounds Using NMR Spectroscopy; John Wiley & Sons: Hoboken, NJ, USA, 2007; p. 549. [Google Scholar]
- Kumar, V.; Olsen, C.E.; Schäffer, S.J.C.; Parmar, V.S.; Malhotra, S.V. Synthesis and Applications of Novel Bis(ammonium) Chiral Ionic Liquids Derived from Isomannide. Org. Lett. 2007, 9, 3905–3908. [Google Scholar] [CrossRef] [PubMed]
- De Rooy, S.L.; Li, M.; Bwambok, D.K.; El-Zahab, B.; Challa, S.; Warner, I.M. Ephedrinium-based protic chiral ionic liquids for enantiomeric recognition. Chirality 2011, 23, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Satishkumar, S.; Periasamy, M. Chiral recognition of carboxylic acids by Troeger’s base derivatives. Tetrahedron Asymmetry 2009, 20, 2257–2262. [Google Scholar] [CrossRef]
- Jain, N.; Mandal, M.B.; Bedekar, A.V. Roof shape amines: synthesis and application as NMR chiral solvating agents for discrimination of α-functionalized acids. Tetrahedron 2014, 70, 4343–4354. [Google Scholar] [CrossRef]
- Yang, X.; Wang, G.; Zhong, C.; Wu, X.; Fu, E. Novel NMR chiral solvating agents derived from (1R,2R)-diaminocyclohexane: Synthesis and enantiodiscrimination for chiral carboxylic acids. Tetrahedron Asymmetry 2006, 17, 916–921. [Google Scholar] [CrossRef]
- Winkel, A.; Wilhelm, R. New chiral ionic liquids based on imidazolinium salts. Tetrahedron Asymmetry 2009, 20, 2344–2350. [Google Scholar] [CrossRef]
- Periasamy, M.; Dalai, M.; Padmaja, M. Chiral trans-1,2-diaminocyclohexane derivatives as chiral solvating agents for carboxylic acids. J. Chem. Sci. 2010, 122, 561–569. [Google Scholar] [CrossRef]
- Liu, L.; Ye, M.; Hu, X.; Yu, X.; Zhang, L.; Lei, X. Chiral solvating agents for carboxylic acids based on the salen moiety. Tetrahedron Asymmetry 2011, 22, 1667–1671. [Google Scholar] [CrossRef]
- Kumar, V.; Pei, C.; Olsen, C.E.; Schaeffer, S.J.C.; Parmar, V.S.; Malhotra, S.V. Novel carbohydrate-based chiral ammonium ionic liquids derived from isomannide. Tetrahedron Asymmetry 2008, 19, 664–671. [Google Scholar] [CrossRef]
- Heckel, T.; Winkel, A.; Wilhelm, R. Chiral ionic liquids based on nicotine for the chiral recognition of carboxylic acids. Tetrahedron Asymmetry 2013, 24, 1127–1133. [Google Scholar] [CrossRef]
- Gonzalez, L.; Altava, B.; Bolte, M.; Burguete, M.I.; Garcia-Verdugo, E.; Luis, S.V. Synthesis of Chiral Room Temperature Ionic Liquids from Amino Acids—Application in Chiral Molecular Recognition. Eur. J. Org. Chem. 2012, 26, 4996–5009. [Google Scholar] [CrossRef]
- Chaudhary, A.R.; Yadav, P.; Bedekar, A.V. Application of optically active aminonaphthols as NMR solvating agents for chiral discrimination of mandelic acid. Tetrahedron Asymmetry 2014, 25, 767–774. [Google Scholar] [CrossRef]
- Ashraf, S.A.; Pornputtkul, Y.; Kane-Maguire, L.A.P.; Wallace, G.G. Facile synthesis of a chiral ionic liquid derived from 1-phenylethylamine. Aust. J. Chem. 2007, 60, 64–67. [Google Scholar] [CrossRef]
- Altava, B.; Barbosa, D.S.; Isabel Burguete, M.; Escorihuela, J.; Luis, S.V. Synthesis of new chiral imidazolium salts derived from amino acids: Their evaluation in chiral molecular recognition. Tetrahedron Asymmetry 2009, 20, 999–1003. [Google Scholar] [CrossRef]
- Kannappan, J.; Jain, N.; Bedekar, A.V. Synthesis and applications of exo N-((1R,2R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-yl)benzamides as NMR solvating agents for the chiral discrimination of 1,1'-binaphthyl-2,2'-diyl hydrogenphosphates and α-substituted acids. Tetrahedron Asymmetry 2015, 26, 1102–1107. [Google Scholar] [CrossRef]
- Gospodarowicz, K.; Holynska, M.; Paluch, M.; Lisowski, J. Novel chiral hexaazamacrocycles for the enantiodiscrimination of carboxylic acids. Tetrahedron 2012, 68, 9930–9935. [Google Scholar] [CrossRef]
- Tabassum, S.; Gilani, M.A.; Wilhelm, R. Imidazolinium sulfonate and sulfamate zwitterions as chiral solvating agents for enantiomeric excess calculations. Tetrahedron Asymmetry 2011, 22, 1632–1639. [Google Scholar] [CrossRef]
- Foreiter, M.B.; Gunaratne, H.Q.N.; Nockemann, P.; Seddon, K.R.; Stevenson, P.J.; Wassell, D.F. Chiral thiouronium salts: Synthesis, characterisation and application in NMR enantio-discrimination of chiral oxoanions. New J. Chem. 2013, 37, 515–533. [Google Scholar] [CrossRef]
- Bozkurt, S.; Durmaz, M.; Naziroglu, H.N.; Yilmaz, M.; Sirit, A. Amino alcohol based chiral solvating agents: Synthesis and applications in the NMR enantiodiscrimination of carboxylic acids. Tetrahedron Asymmetry 2011, 22, 541–549. [Google Scholar] [CrossRef]
- Wang, W.; Ma, F.; Shen, X.; Zhang, C. New chiral auxiliaries derived from (S)-α-phenylethylamine as chiral solvating agents for carboxylic acids. Tetrahedron Asymmetry 2007, 18, 832–837. [Google Scholar] [CrossRef]
- Pena, C.; Gonzalez-Sabin, J.; Alfonso, I.; Rebolledo, F.; Gotor, V. Cycloalkane-1,2-diamine derivatives as chiral solvating agents. Study of the structural variables controlling the NMR enantiodiscrimination of chiral carboxylic acids. Tetrahedron 2008, 64, 7709–7717. [Google Scholar] [CrossRef]
- Gualandi, A.; Grilli, S.; Savoia, D.; Kwit, M.; Gawronski, J. C-hexaphenyl-substituted trianglamine as a chiral solvating agent for carboxylic acids. Org. Biomol. Chem. 2011, 9, 4234–4241. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Fukuda, N. “Calixarene-like” chiral amine macrocycles as novel chiral shift reagents for carboxylic acids. Tetrahedron Asymmetry 2009, 20, 111–114. [Google Scholar] [CrossRef]
- Perez-Trujillo, M.; Castanar, L.; Monteagudo, E.; Kuhn, L.T.; Nolis, P.; Virgili, A.; Williamson, R.T.; Parella, T. Simultaneous 1H- and 13C-NMR enantiodifferentiation from highly-resolved pure shift HSQC spectra. Chem. Commun. 2014, 50, 10214–10217. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, J.A.; Faulkner, S.; Nilsson, M.; Morris, G.A. Pure shift 1H-NMR: A resolution of the resolution problem? Angew. Chem. Int. Ed. Engl. 2010, 49, 3901–3903. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, S.R.; Suryaprakash, N. Chiral discrimination and the measurement of enantiomeric excess from a severely overcrowded NMR spectrum. Chem. Phys. Lett. 2013, 555, 286–290. [Google Scholar] [CrossRef]
- Lokesh, N.; Chaudhari, S.R.; Suryaprakash, N. RES-TOCSY: A simple approach to resolve overlapped 1H-NMR spectra of enantiomers. Org. Biomol. Chem. 2014, 12, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Cheney, L.C. Purification of Dehydroabietylamine. U.S. Patent 2787637, 2 April 1957. [Google Scholar]
- Su, G.; Huo, L.; Huang, W.; Wang, H.; Pan, Y. Synthesis and crystal structure of 2-dehydroabietyl-5-ethylsulfanyl-1,2,3,4-tetrazole. Chin. J. Struct. Chem. 2009, 28, 693–698. [Google Scholar]
- Wang, H.; Tang, L.; Pan, Y.; Liang, M. Synthesis of novel chiral quaternary ammonium salt from rosin. Chem. J. Internet 2004, 6, 56. [Google Scholar]
- Stella, S.; Chadha, A. Resolution of N-protected amino acid esters using whole cells of Candida parapsilosis ATCC 7330. Tetrahedron Asymmetry 2010, 21, 457–460. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laaksonen, T.; Heikkinen, S.; Wähälä, K. Synthesis of Tertiary and Quaternary Amine Derivatives from Wood Resin as Chiral NMR Solvating Agents. Molecules 2015, 20, 20873-20886. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules201119732
Laaksonen T, Heikkinen S, Wähälä K. Synthesis of Tertiary and Quaternary Amine Derivatives from Wood Resin as Chiral NMR Solvating Agents. Molecules. 2015; 20(11):20873-20886. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules201119732
Chicago/Turabian StyleLaaksonen, Tiina, Sami Heikkinen, and Kristiina Wähälä. 2015. "Synthesis of Tertiary and Quaternary Amine Derivatives from Wood Resin as Chiral NMR Solvating Agents" Molecules 20, no. 11: 20873-20886. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules201119732