
Synthesis, Crystal Structure, Absolute Configuration and Antitumor Activity of the Enantiomers of 5-Bromo-2-chloro-N-(1-phenylethyl)pyridine-3-sulfonamide

Abstract
:
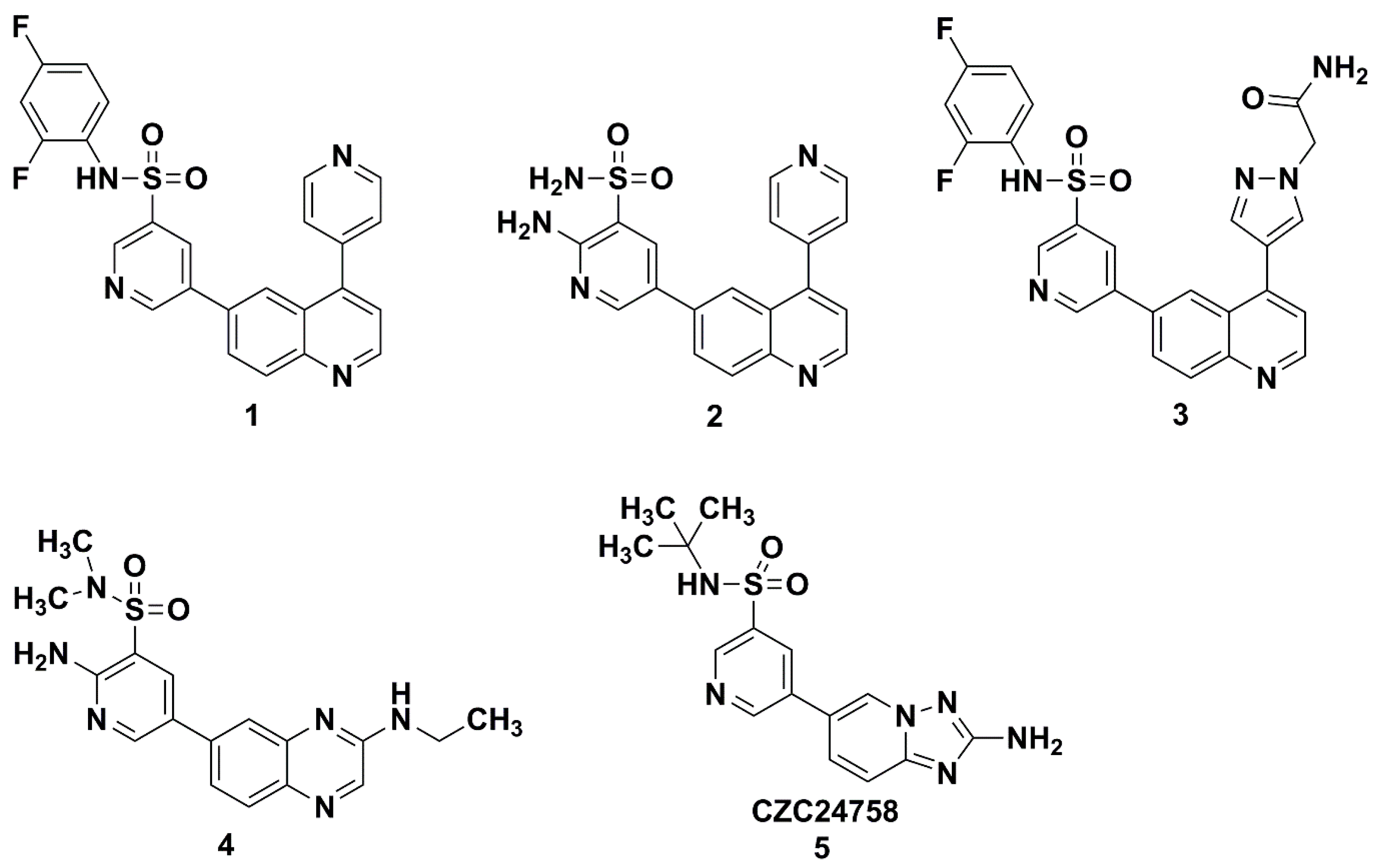
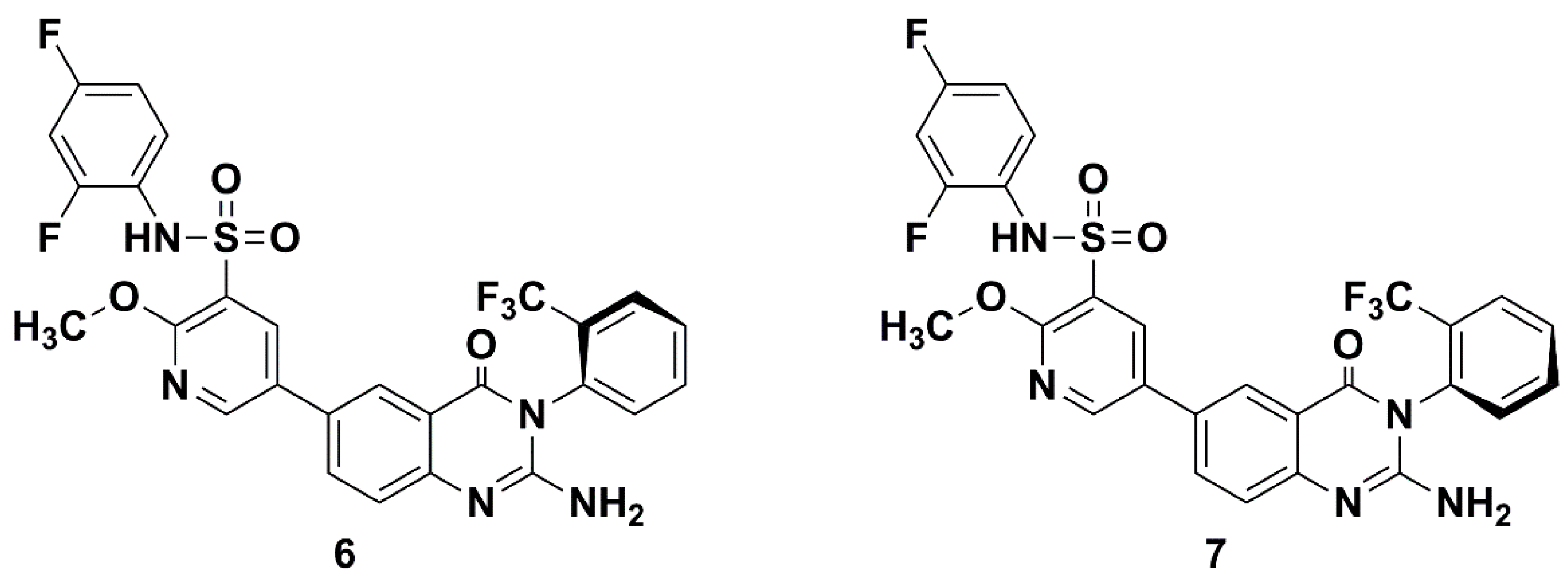
1. Introduction


2. Results and Discussion
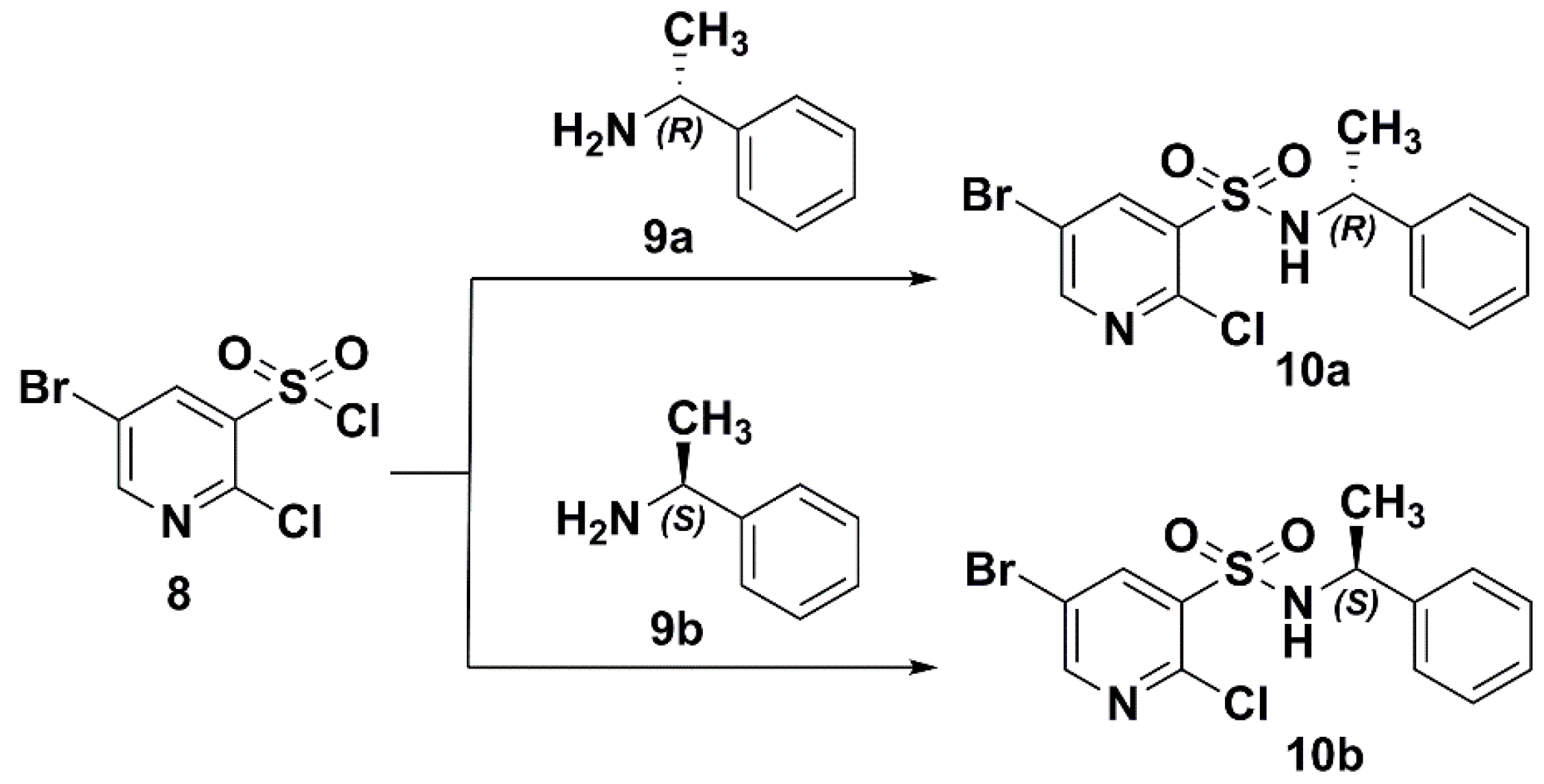
2.1. Preparation of the Target Compounds

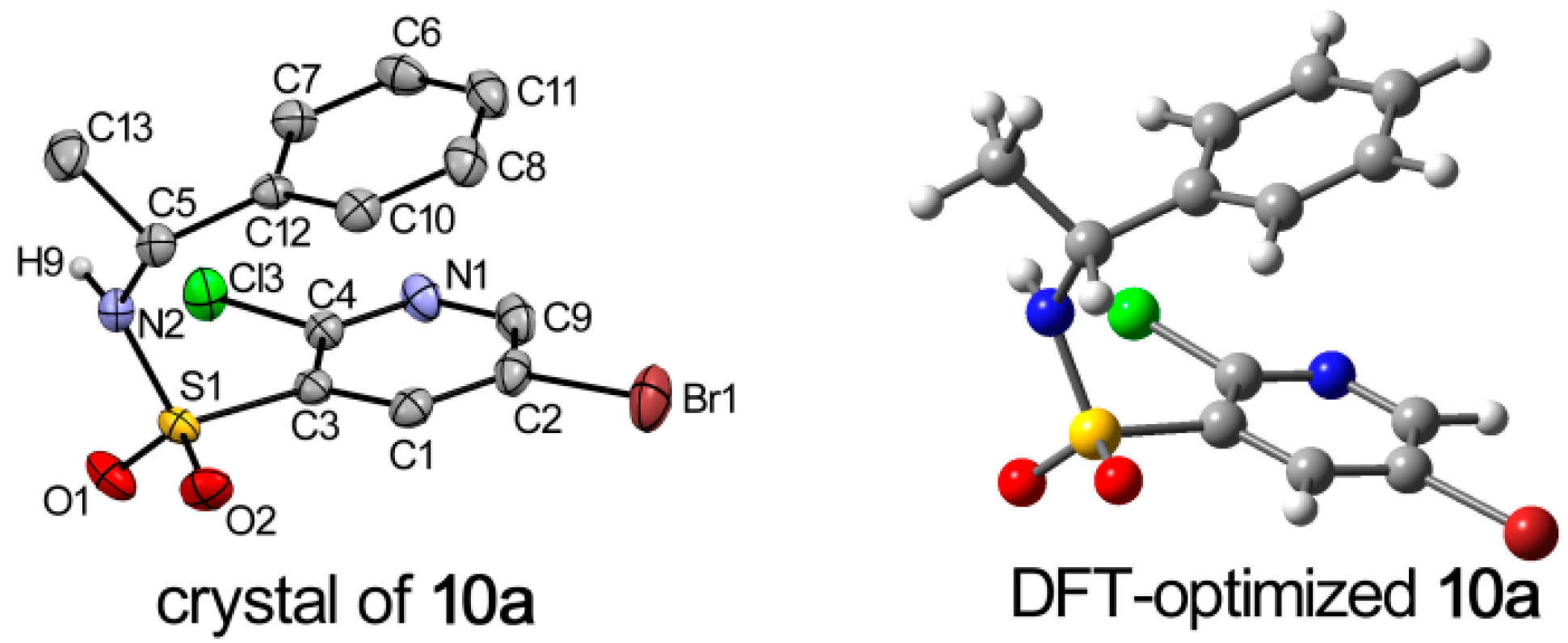
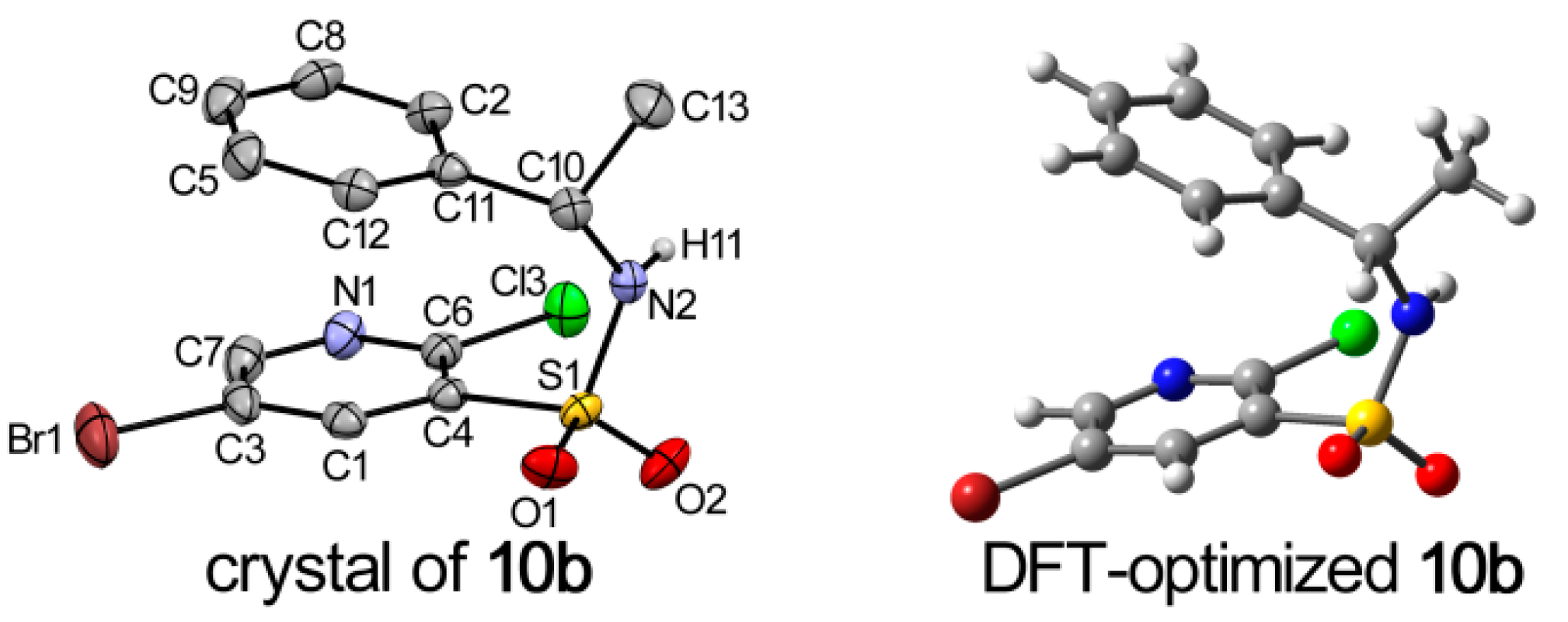
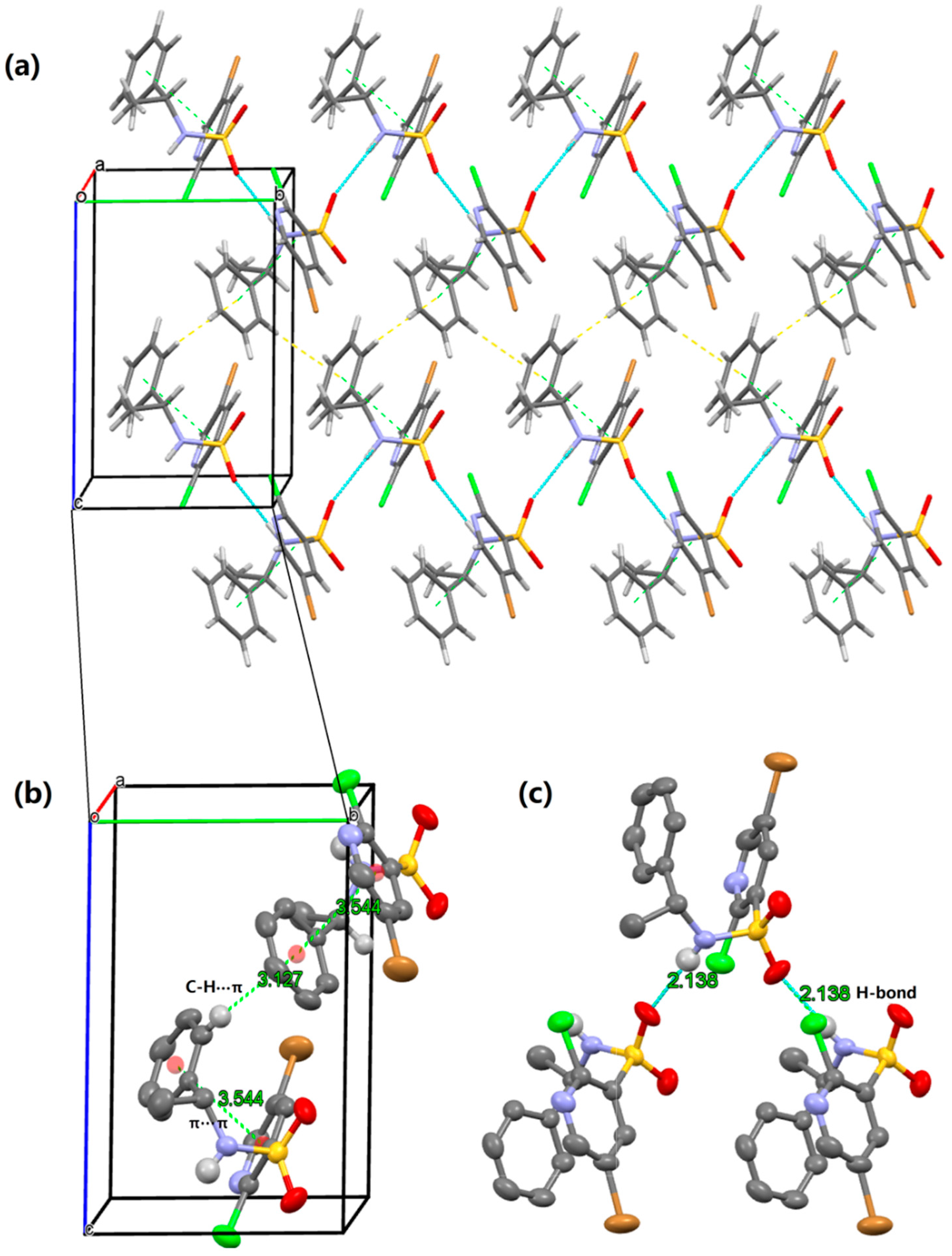
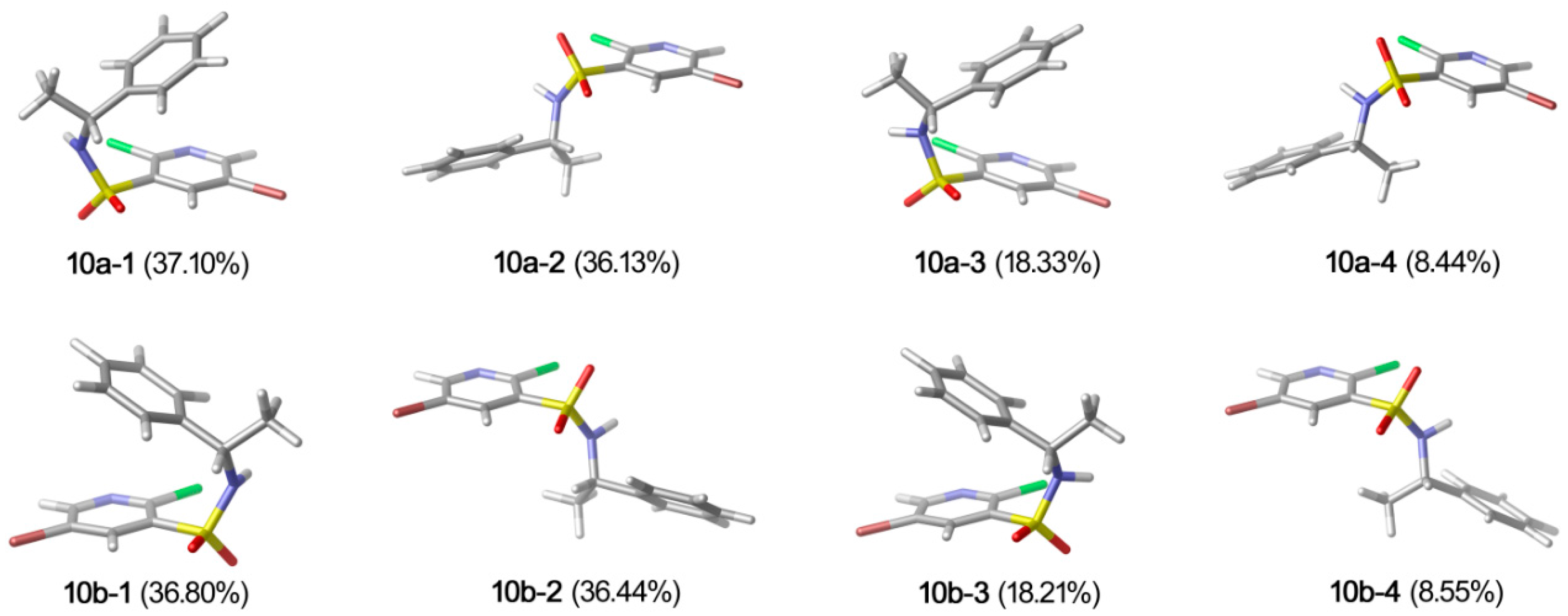
2.2. X-ray Structure Analysis and Conformational Analyses



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D—H···A | D—H | H···A | D···A | D—H···A |
|---|---|---|---|---|
| N2—H9···O1 i | 0.871 | 2.138 | 3.004 | 172.29 |
| O1···H9—N2 ii | 0.871 | 2.138 | 3.004 | 172.29 |

| Conformer | G(kcal·mol−1) | △G(kcal·mol−1) | Pi% |
|---|---|---|---|
| 10a-1 | −2632474.97 | 0.0000 | 37.10 |
| 10a-2 | −2632474.96 | 0.0157 | 36.13 |
| 10a-3 | −2632474.56 | 0.4135 | 18.33 |
| 10a-4 | −2632474.11 | 0.8678 | 8.44 |
| 10b-1 | −2632474.97 | 0.0000 | 36.80 |
| 10b-2 | −2632474.96 | 0.0056 | 36.44 |
| 10b-3 | −2632474.55 | 0.4123 | 18.21 |
| 10b-4 | −2632474.11 | 0.8553 | 8.55 |

| Bond Angle [°] | Exp. (10a) | Calcd. a | Difference |
|---|---|---|---|
| C(12)C(5)C(13) | 111.1 | 112.2 | 1.1 |
| C(12)C(5)N(2) | 113.6 | 114.9 | 1.3 |
| C(13)C(5)N(2) | 107.6 | 107.7 | 0.1 |
| C(5)N(2)S(1) | 121.1 | 123.4 | 2.3 |
| C(5)N(2)H(9) | 113 | 117.8 | 4.8 |
| H(9)N(2)S(1) | 120 | 112.3 | −7.7 |
| Bond angle [°] | Exp. (10b) | Calcd. b | Difference |
| C(13)C(10)C(11) | 111.0 | 112.2 | 1.2 |
| N(2)C(10)C(11) | 113.3 | 114.9 | 1.6 |
| C(13)C(10)N(2) | 107.7 | 107.7 | 0 |
| S(1)N(2)C(10) | 121.2 | 123.4 | 2.2 |
| H(11)N(2)C(10) | 119 | 117.8 | −1.2 |
| H(11)N(2)S(1) | 114 | 112.3 | −1.7 |
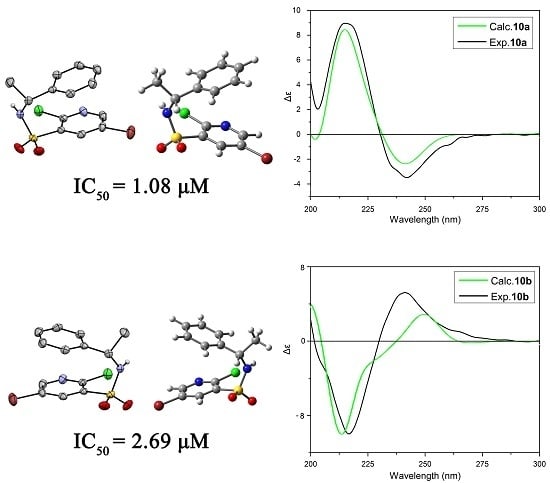
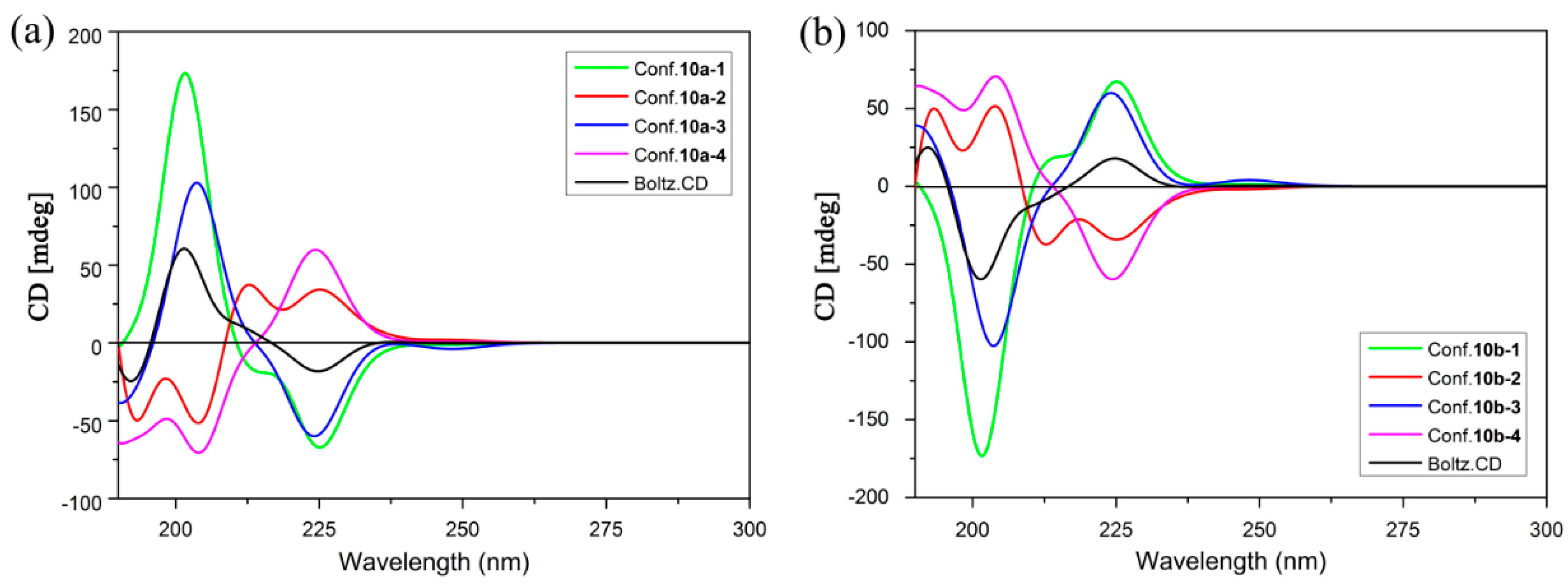
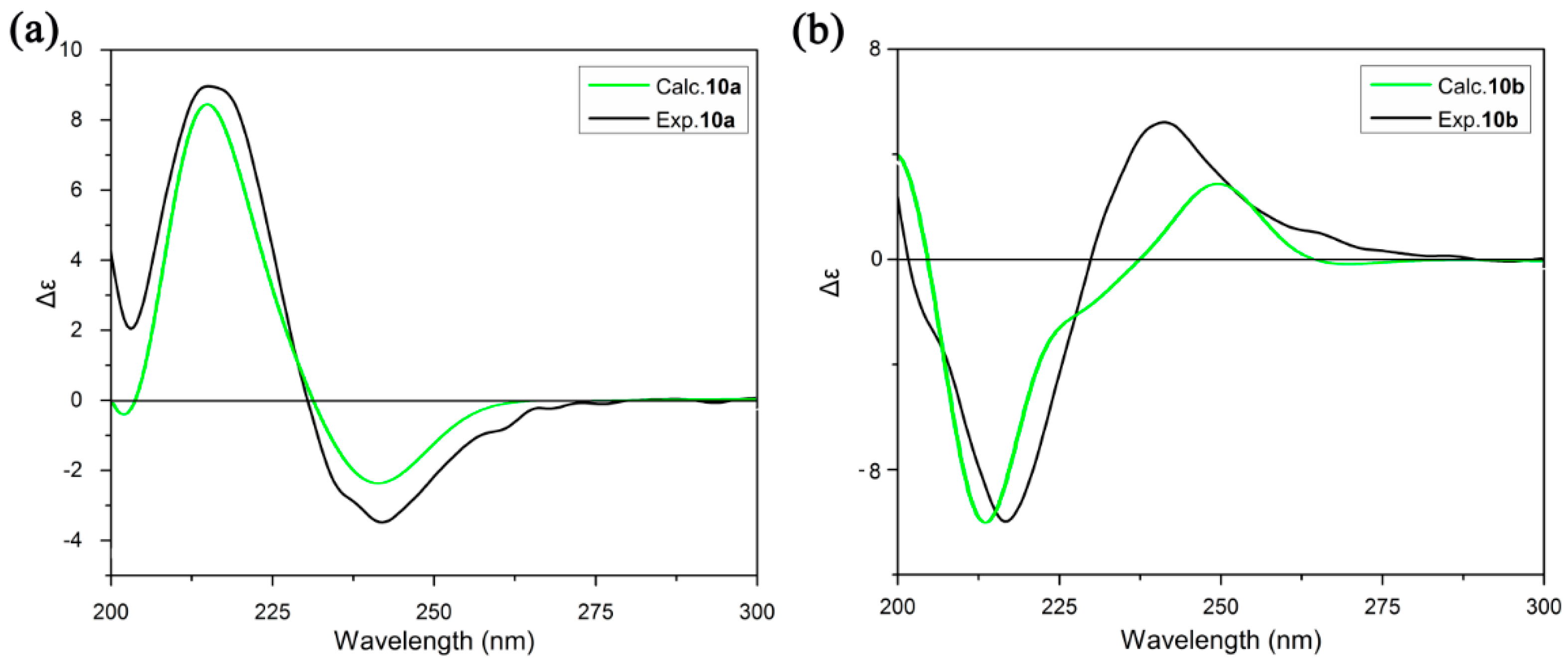
2.3. ECD Analysis



2.4. Optical Rotation Analysis
| No. | Theoretical OR/° | Experimental OR/° |
|---|---|---|
| 10a | 73.0 | 58.2 |
| 10b | −89.9 | −65.9 |
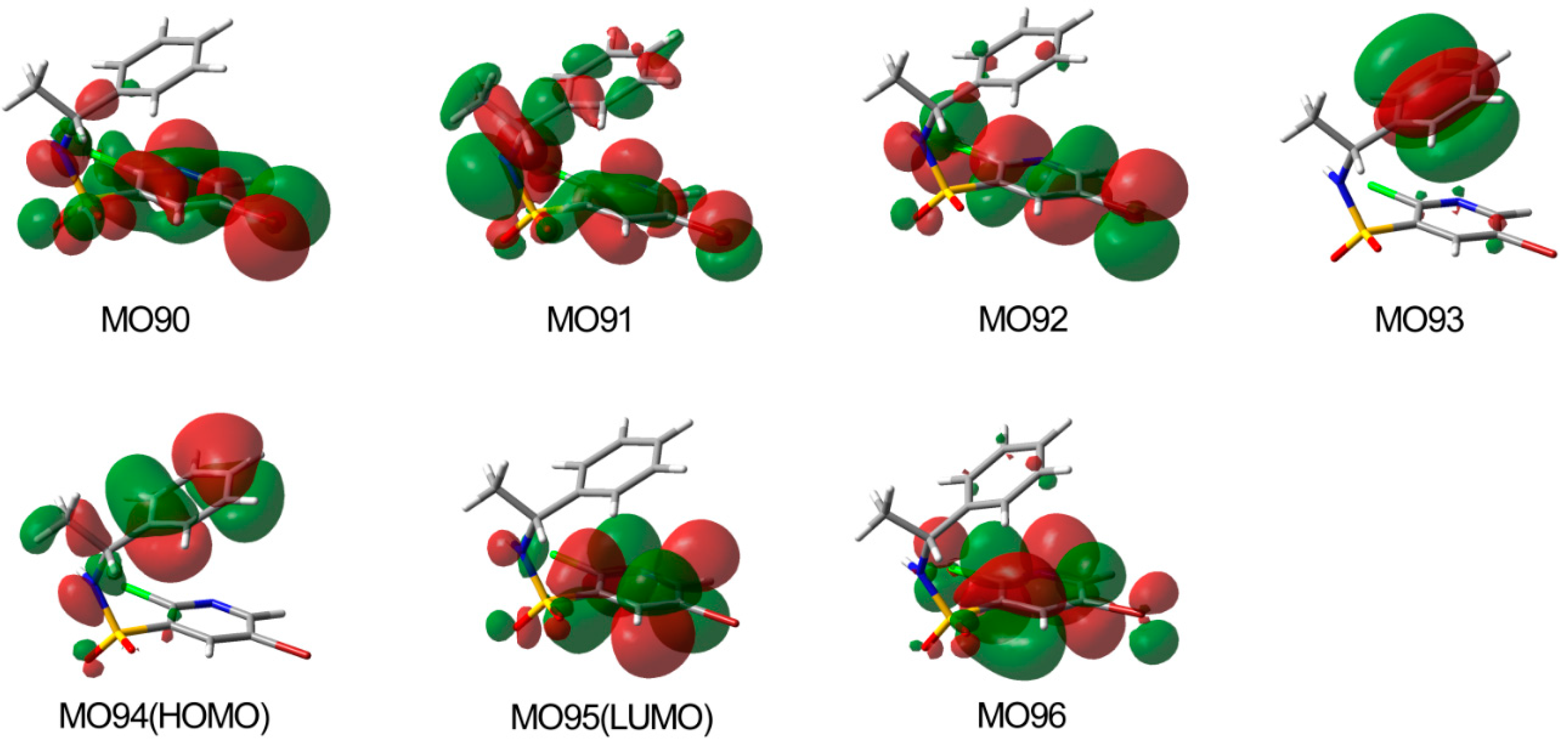
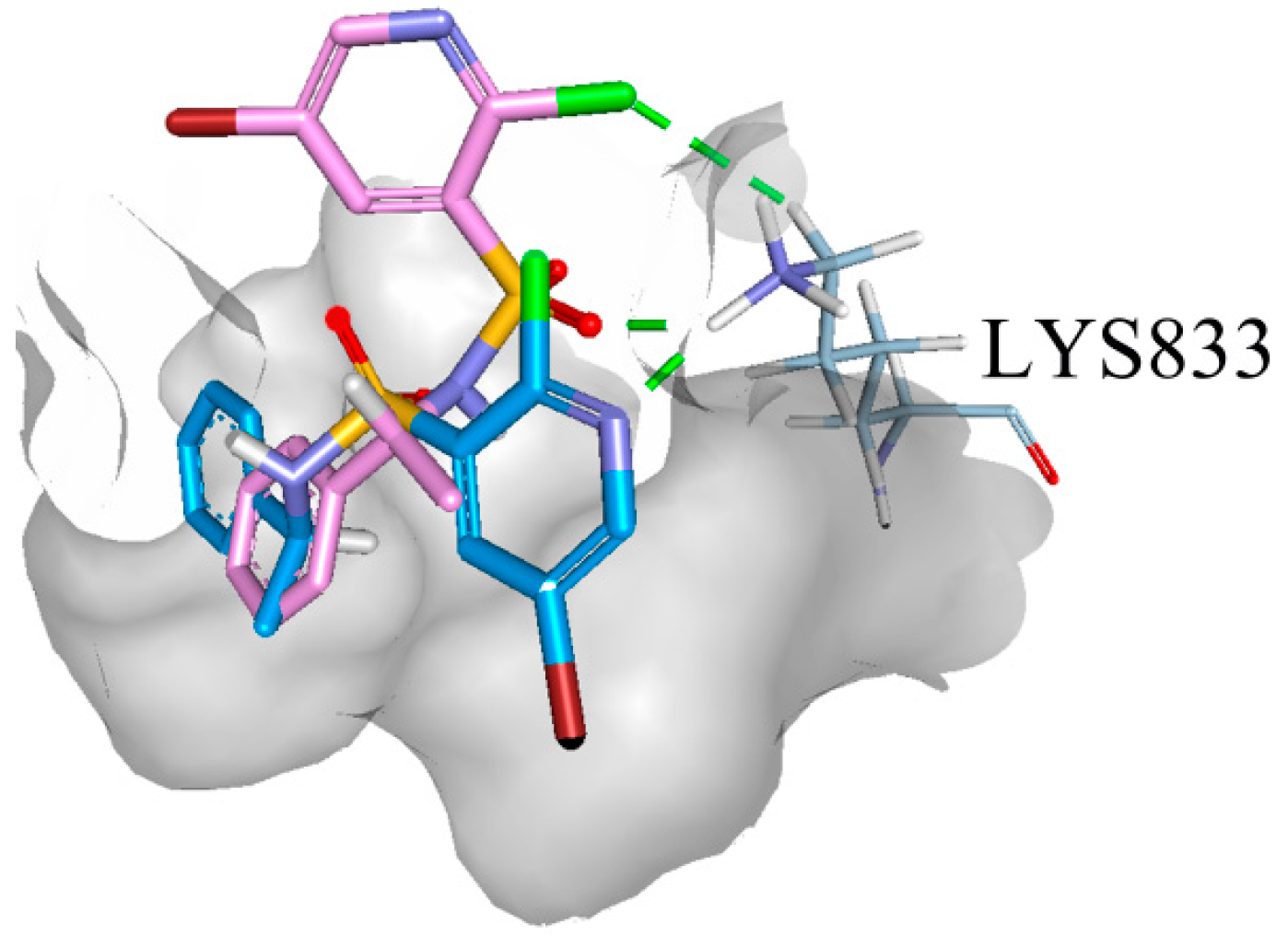
2.5. Binding Model Analysis

2.6. PI3Kα Kinase Assay
2.7. In Vitro Cytotoxicity
3. Experimental Section
3.1. General Remarks
3.2. Materials
3.3. Synthesis of (R)-5-Bromo-2-chloro-N-(1-phenylethyl)pyridine-3-sulfonamide (10a)
3.4. Synthesis of (S)-5-Bromo-2-chloro-N-(1-phenylethyl)pyridine-3-sulfonamide (10b)
3.5. Computational Details
3.6. PI3Kα Kinase Assay
3.7. MTT Assay in Vitro
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mohan, R.; Banerjee, M.; Ray, A.; Manna, T.; Wilson, L.; Owa, T.; Bhattacharyya, B.; Panda, D. Antimitotic sulfonamides inhibit microtubule assembly dynamics and cancer cell proliferation. Biochemistry 2006, 45, 5440–5449. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, H.; Ueda, N.; Niijima, J.; Sugumi, H.; Kotake, Y.; Koyanagi, N.; Yoshimatsu, K.; Asada, M.; Watanabe, T. Novel sulfonamides as potential, systemically active antitumor agents. J. Med. Chem. 1992, 35, 2496–2497. [Google Scholar] [CrossRef] [PubMed]
- Leivers, A.L.; Tallant, M.; Shotwell, J.B.; Dickerson, S.; Leivers, M.R.; McDonald, O.B.; Gobel, J.; Creech, K.L.; Strum, S.L.; Mathis, A. Discovery of selective small molecule type III phosphatidylinositol 4-kinase alpha (PI4KIIIα) inhibitors as anti hepatitis C (HCV) agents. J. Med. Chem. 2014, 57, 2091–2106. [Google Scholar] [CrossRef] [PubMed]
- Sunose, M.; Bell, K.; Ellard, K.; Bergamini, G.; Neubauer, G.; Werner, T.; Ramsden, N. Discovery of 5-(2-amino-[1,2,4] triazolo [1,5-a] pyridin-7-yl)-N-(tert-butyl) pyridine-3-sulfonamide (CZC24758), as a potent, orally bioavailable and selective inhibitor of PI3K for the treatment of inflammatory disease. Bioorg. Med. Chem. Lett. 2012, 22, 4613–4618. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Lin, S.; Liu, P.; Liu, X.; Tao, J.; Deng, X.; Yi, C.; Xu, H. Discovery of a novel series of thienopyrimidine as highly potent and selective PI3K inhibitors. ACS Med. Chem. Lett. 2015, 6, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Stec, M.M.; Andrews, K.L.; Booker, S.K.; Caenepeel, S.; Freeman, D.J.; Jiang, J.; Liao, H.; McCarter, J.; Mullady, E.L.; San Miguel, T. Structure–activity relationships of phosphoinositide 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) dual inhibitors: Investigations of various 6, 5-heterocycles to improve metabolic stability. J. Med. Chem. 2011, 54, 5174–5184. [Google Scholar] [CrossRef] [PubMed]
- Knight, S.D.; Adams, N.D.; Burgess, J.L.; Chaudhari, A.M.; Darcy, M.G.; Donatelli, C.A.; Luengo, J.I.; Newlander, K.A.; Parrish, C.A.; Ridgers, L.H.; et al. Discovery of GSK2126458, a highly potent inhibitor of PI3K and the mammalian target of rapamycin. ACS Med. Chem. Lett. 2010, 1, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, N.; Siegmund, A.; Liu, L.; Yang, K.; Bryan, M.C.; Andrews, K.L.; Bo, Y.; Booker, S.K.; Caenepeel, S.; Freeman, D. Phospshoinositide 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) dual inhibitors: discovery and structure–activity relationships of a series of quinoline and quinoxaline derivatives. J. Med. Chem. 2011, 54, 4735–4751. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, N.D.; Kim, T.-S.; Andrews, K.; Booker, S.K.; Caenepeel, S.; Chen, K.; D’Amico, D.; Freeman, D.; Jiang, J.; Liu, L. Discovery and optimization of a series of benzothiazole phosphoinositide 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) dual inhibitors. J. Med. Chem. 2011, 54, 1789–1811. [Google Scholar] [CrossRef] [PubMed]
- Suhonen, A.; Morgan, I.S.; Nauha, E.; Helttunen, K.; Tuononen, H.M.; Nissinen, M. The effect of a rigid sulfonamide bond on molecular folding: A case study. Cryst. Growth Des. 2015, 15, 2602–2608. [Google Scholar] [CrossRef]
- Chaudhari, A.; Dhanak, D.; Donatelli, C.A.; Faitg, T.H.; Feng, Y.; Knight, S.D.; Parrish, C.A.; Ralph, J.M. Quinoxaline Derivatives as pi3 Kinase Inhibitors. U.S. 2008293706, 27 November 2008. [Google Scholar]
- Adams, N.D.; Burgess, J.L.; Darcy, M.G.; Donatelli, C.A.; Knight, S.D.; Newlander, K.A.; Ridgers, L.; Sarpong, M.A.; Schmidt, S.J. Quinoline Derivatives as pi3 Kinase Inhibitors. WO 2008144463, 27 November 2008. [Google Scholar]
- Wang, Z.; Zhao, L.; Chen, Y.; Xu, W.; Sun, T. Determination of absolute configurations of bedaquiline analogs by quantum chemical electronic circular dichroism calculations and an X-ray diffraction study. Eur. J. Org. Chem. 2014, 2014, 3814–3821. [Google Scholar] [CrossRef]
- Mori, T.; Inoue, Y.; Grimme, S. Time dependent density functional theory calculations for electronic circular dichroism spectra and optical rotations of conformationally flexible chiral donor-acceptor dyad. J. Org. Chem. 2006, 71, 9797–9806. [Google Scholar] [CrossRef] [PubMed]
- Marchesan, D.; Coriani, S.; Forzato, C.; Nitti, P.; Pitacco, G.; Ruud, K. Optical rotation calculation of a highly flexible molecule: the case of paraconic acid. J. Phys. Chem. A 2005, 109, 1449–1453. [Google Scholar] [CrossRef] [PubMed]
- Kwit, M.; Sharma, N.D.; Boyd, D.R.; Gawronski, J. Determination of absolute configuration of conformationally flexible cis-dihydrodiol metabolites: Effect of diene substitution pattern on the circular dichroism spectra and optical rotations. Chirality 2008, 20, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Deppmeier, B.; Driessen, A.; Hehre, T.; Hehre, W.; Johnson, J.; Klunzinger, P.; Leonard, J.; Pham, I.; Pietro, W.; Yu, J. Spartan’08; Wavefunction, Inc.: Irvine, CA, USA, 2008. [Google Scholar]
- Bosnich, B. Molecular mechanics force fields for cyclopentadienyl complexes. Chem. Soc. Rev. 1994, 23, 387–395. [Google Scholar] [CrossRef]
- Huang, N.; Kalyanaraman, C.; Bernacki, K.; Jacobson, M.P. Molecular mechanics methods for predicting protein–ligand binding. Phys. Chem. Chem. Phys. 2006, 8, 5166–5177. [Google Scholar] [CrossRef] [PubMed]
- Koch, W.; Holthausen, M.C.; Holthausen, M.C. A Chemist's Guide to Density Functional Theory; Wiley-Vch: Weinheim, Germany, 2001; Volume 2. [Google Scholar]
- Geerlings, P.; de Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef] [PubMed]
- Gaussian 09, Revision C. 01; Gaussian: Wallingford, CT, USA, 2010.
- Bruhn, T.; Hemberger, Y.; Schaumloffel, A.; Bringmann, G. SpecDis, Version 1.51, University of Wuerzburg: Wuerzburg, Germany, 2011.
- Bruker, A. APEX2-Software Suite for Crystallographic Programs, Bruker AXS Inc.: Madison, WI, USA, 2009.
- Bruker, A. SMART, SAINT and SADABS; Bruker AXS Inc.: Madison, WI, USA, 2000. [Google Scholar]
- Sheldrick, G. shelxs-97 and shelxl-97 Programs for Structure Resolution and for Structure Refinement; University of Göttingen: Gottingen, Germany, 1997. [Google Scholar]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; Streek, J.V.; Wood, P.A. Mercury CSD 2.0–new features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Miertus, S.; Tomasi, J. Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes. Chem. Phys. 1982, 65, 239–245. [Google Scholar] [CrossRef]
- Kong, D.; Yamori, T. ZSTK474 is an ATP-competitive inhibitor of class I phosphatidylinositol 3 kinase isoforms. Cancer Sci. 2007, 98, 1638–1642. [Google Scholar] [CrossRef] [PubMed]
- Dugar, S.; Hollinger, F.P.; Kuila, B.; Arora, R.; Sen, S.; Mahajan, D. Synthesis and evaluation of pyrrolotriazine based molecules as PI3 kinase inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 3142–3146. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds reported herein are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Z.; Li, L.; Yan, N.; Du, L.; Sun, C.; Sun, T. Synthesis, Crystal Structure, Absolute Configuration and Antitumor Activity of the Enantiomers of 5-Bromo-2-chloro-N-(1-phenylethyl)pyridine-3-sulfonamide. Molecules 2015, 20, 20926-20938. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules201119740
Zhou Z, Li L, Yan N, Du L, Sun C, Sun T. Synthesis, Crystal Structure, Absolute Configuration and Antitumor Activity of the Enantiomers of 5-Bromo-2-chloro-N-(1-phenylethyl)pyridine-3-sulfonamide. Molecules. 2015; 20(11):20926-20938. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules201119740
Chicago/Turabian StyleZhou, Zhixu, Linwei Li, Ning Yan, Lei Du, Changshan Sun, and Tiemin Sun. 2015. "Synthesis, Crystal Structure, Absolute Configuration and Antitumor Activity of the Enantiomers of 5-Bromo-2-chloro-N-(1-phenylethyl)pyridine-3-sulfonamide" Molecules 20, no. 11: 20926-20938. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules201119740