
Chelation-Assisted Substrate-Controlled Asymmetric Lithiation-Allylboration of Chiral Carbamate 1,2,4-Butanetriol Acetonide


Abstract
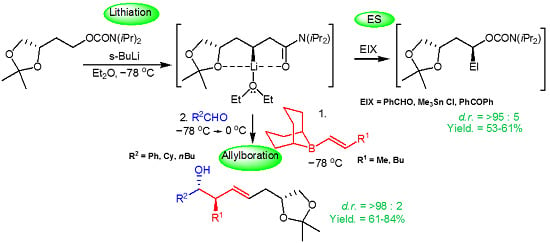
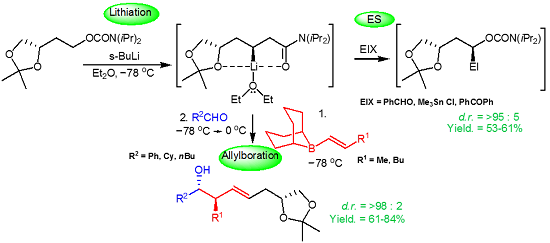
:
1. Introduction
2. Results and Discussion
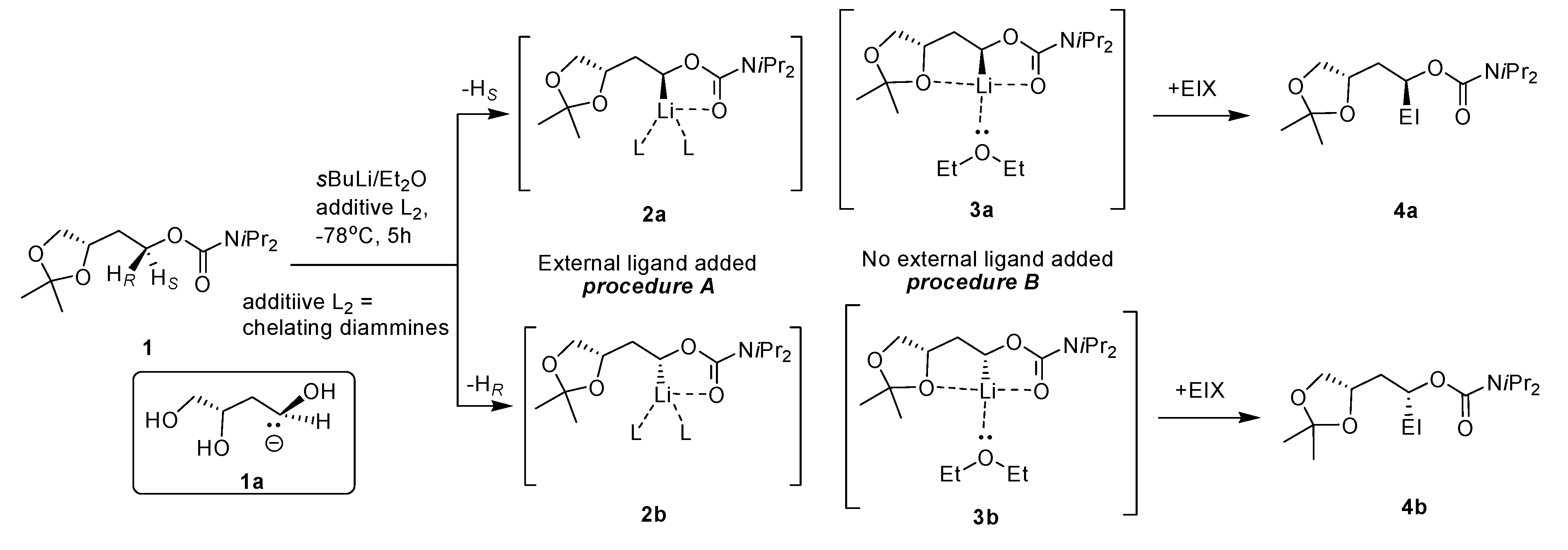
2.1. Chemistry



| Entry | EIX | Additive L2 | Yield a (%) | d.r. b 4a:4b | (conc., CH3OH) |
|---|---|---|---|---|---|
| I | PhCHO | TMEDA | 69 | 51:49 | nd c |
| (−)-Sp | 74 | 98:2 | −17.4 (1.0) | ||
| (+)-Sp surrogate | 53 | 20:80 | nd | ||
| None | 84 | 98:2 | −17.2 (1.0) | ||
| II | Me3SnCl | TMEDA | 80 | 53:43 | nd |
| (−)-Sp | 75 | 97:3 | +13.0 (1.2) | ||
| (+)-Sp surrogate | nd | nd | nd | ||
| None | 78 | 98:2 | +11.3 (1.1) | ||
| III | PhCOPh | TMEDA | 72 | 50:50 | nd |
| (−)-Sp | 80 | 95:5 | +23.6 (1.0) | ||
| (+)-Sp surrogate | 61 | 29:71 | nd | ||
| None | 84 | 95:5 | +27.3 (1.5) |

{kind=link}
{kind=link}
| Entry | R2 | R3 | Yield (%) | d.r. a 8:8a | (conc., CH3OH) |
|---|---|---|---|---|---|
| I | Me | Ph | 56 | 90:10 | +10.4 |
| II | Me | Cy | 54 | 95:5 | −19.5 |
| III | Me | Bu | 60 | 92:8 | +21.9 |
| IV | Bu | Ph | 48 | 90:10 | −14.5 |
| V | Bu | Cy | 52 | 94:6 | −18.4 |
3. Experimental Section
3.1. General Information
3.2. (S)-2-(2,2-Dimethyl-1,3-dioxolan-4-yl)ethyl Diisopropylcarbamate (1)
3.3. 9-((E)-Prop-1-enyl)-9-borabicyclo[3.3.1]nonane (5a)
3.4. B-(trans-1-Hexenyl)-9-borabicyclo(3,3,1)nonane (5b)
3.5. (R)-3-((S)-2,2-Dimethyl-1,3-dioxolan-4-yl)-1-hydroxy-1-phenylpropan-2-yl Diisopropylcarbamate (4a-I)
3.6. (R)-3-((S)-2,2-Dimethyl-1,3-dioxolan-4-yl)-1-hydroxy-1,1-diphenylpropan-2-yldiisopropyl-carbamate (4a-III)
3.7. (S)-2-((S)-2,2-Dimethyl-1,3-dioxolan-4-yl)-1-(trimethylstannyl)ethyl Diisopropylcarbamate (4a-II)
3.8. (1R,2R,Z)-5-((R)-2,2-Dimethyl-1,3-dioxalan-4-yl)-2-methyl-1-phenylpent-3-en-1-ol (8-I)
3.9. (1R,2R,Z)-1-Cyclohexyl-5-((R)-2,2-dimethyl-1,3-dioxalan-4-yl)-2-methylpent-3-en-1-ol (8-II).
3.10. (4R,5S,Z)-1-((R)-2,2-Dimethyl-1,3-dioxolan-4-yl)-4-methylnon-2-en-5-ol (8-III)
3.11. (1R,2R)-2-((Z)-3-((R)-2,2-Dimethyl-1,3-dioxolan-4-yl)prop-1-en-1-yl)-1-phenylhexan-1-ol (8-IV).
3.12. (1S,2R)-1-Cyclohexyl-2-((Z)-3-((R)-2,2-dimethyl-1,3-dioxolan-4-yl)prop-1-en-1-yl)hexan-1-ol (8-V)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hoppe, D.; Hintze, F.; Tebben, P.; Paetow, M.; Ahrens, H.; Schwerdtfeger, J.; Sommerfeld, P.; Haller, J.; Guarnieri, S.; Kolczewski, S.; et al. Enantioslective synthesis via sparteine-induced asymmetric deprotonation. Pure Appl. Chem. 1994, 66, 1479–1486. [Google Scholar] [CrossRef]
- Hoppe, D.; Hintze, F.; Tebben, P. Chirale Lithium-1-oxyalkanide durch asymmetrische Deprotonierung; enantioselective Synthese von 2-Hydroxyalkansäuren und sekundären Alkanolen. Angew. Chem. Int. Ed. 1990, 102, 1457–1459. [Google Scholar] [CrossRef]
- Helmke, H.; Hoppe, D. Chelation directed asymmetric lithiation and C-substituion of 1,2,4-butanetriol acetonide. Synlett 1995, 978–980. [Google Scholar] [CrossRef]
- Paulsen, H.; Graeve, C.; Hoppe, D. Convenient Synthesis of Enantiomerically Enriched (Oxybutenyl)stannanes and Their Lewis Acid Catalyzed Homoaldol Reactions. Synthesis 1996, 141–144. [Google Scholar] [CrossRef]
- Basu, A.; Thayumanavan, S. Configurational stability and transfer of stereochemical information in the reactions of enantioenriched organolithium reagents. Angew. Chem. Int. Ed. 2002, 41, 716–738. [Google Scholar] [CrossRef]
- Stymiest, J.L.; Dutheuil, G.; Mahmood, A.; Aggarwal, V.K. Lithiated carbamates: Iterative homologation of boranes and boronic esters. Angew. Chem. Int. Ed. 2007, 46, 7491–7495. [Google Scholar] [CrossRef] [PubMed]
- Sommerfeld, P.; Hoppe, D. Enantioselective generation of 3-amino-1-oxy-substituted carbanions by sparteine-induced deprotonation: Asymmetric synthesis of 3-hydroxy-alkylamines. Synlett 1992, 764–766. [Google Scholar] [CrossRef]
- Guarnieri, W.; Grehl, M.; Hoppe, D. Regio and stereoselective electrophilic C-substituion of 2-(N,N-dibenzylamino)-1,v-alkanediols by lithiation of the their carbamates. Angew. Chem. Int. Ed. 1994, 106, 1815–1818. [Google Scholar] [CrossRef]
- Stymiest, J.L.; Bagutski, V.; French, R.M.; Aggarwal, V.K. Highly enantioselective conversion of secondary alcohols into tertiary alcohols with broad scope. Nature 2008, 456, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Dutheuil, G.; Webster, M.P.; Worthington, P.A.; Aggarwal, V.K. Stereocontrolled synthesis of carbon chains bearing contiguous methyl groups by iterative boronic ester homologations: Application to the total synthesis of (+)-Faranal. Angew. Chem. Int. Ed. 2009, 48, 6179–6183. [Google Scholar]
- Bonet, A.; Odachowski, M.; Leonori, D.; Essafi, S.; Varinder Aggarwal, V.K. Enantiospecific sp2-sp3 coupling of secondary and tertiary boronic esters. Nat. Chem. 2014, 6, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Rasappan, R.; Aggarwal, V.K. Synthesis of hydroxyphthioceranic acid using a traceless lithiation-borylation-protodeboronation strategy. Nat. Chem. 2014, 6, 810–814. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.; Essafi, S.; Bame, J.R.; Bull, S.P.; Webster, M.P.; Balieu, S.; Dale, J.W.; Butts, C.P.; Harvey, J.N.; Aggarwal, V.K. Assembly-line synthesis of organic molecules with tailored shapes. Nature 2014, 513, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.L.-Y.; Scott, H.K.; Hesse, M.J.; Willis, C.L.; Aggarwal, V.K. Highly diastereo- and enantioselective allylboration of aldehydes using α-substituted allyl/crotylpinacolboronic esters via in situ generated borinic esters. J. Am. Chem. Soc. 2013, 135, 5316–5319. [Google Scholar] [CrossRef] [PubMed]
- Hesse, M.J.; Essafi, S.; Watson, C.G.; Harvey, J.N.; Hirst, D.; Willis, C.L.; Aggarwal, V.K. Highly selective allylborations of aldehydes using α,α-disubstituted allylic pinacolboronic esters. Angew. Chem. Int. Ed. 2014, 53, 6145–6149. [Google Scholar] [CrossRef] [PubMed]
- Pulis, A.P.; Fackler, P.; Aggarwal, V.K. Short stereoselective synthesis of the phytophthora universal mating hormone α1 using lithiation/borylation reactions. Angew. Chem. Int. Ed. 2014, 126, 4471–4474. [Google Scholar] [CrossRef]
- Roesner, S.; Brown, C.A.; Mohiti, M.M.; Pulis, A.P.; Rasappan, R.; Blair, B.J.; Essafi, S.; Leonori, D.; Aggarwal, V.K. Stereospecific conversion of alcohols into pinacolboronic esters using lithiation–borylation methodology with pinacolborane. Chem. Commun. 2014, 50, 4053–4055. [Google Scholar] [CrossRef] [PubMed]
- Pulis, A.P.; Blair, D.J.; Torres, E.; Aggarwal, V.K. Synthesis of enantioenriched tertiary boronic esters by the lithiation/borylation of secondary alkyl benzoates. J. Am. Chem. Soc. 2013, 135, 16054–16057. [Google Scholar] [CrossRef] [PubMed]
- Althaus, M.; Mahmood, A.; Suárez, J.R.; Thomas, S.P.; Aggarwal, V.K. Application of the lithiation/borylation reaction to the preparation of enantioenriched allylic boron-reagents and subsequent in situ conversion into 1,2,4-trisubstituted homolallylic alcohols with complete control over all elements of stereochemistry. J. Am. Chem. Soc. 2010, 132, 4025–4028. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, A.; Suárez, J.R.; Thomas, S.P.; Aggarwal, V.K. One-pot synthesis of 2,3,4,5,6-pentasubstituted tetrahydropyrans using lithiation/borylation, allylation and Prins cyclisation reactions. Tetrahedron Lett. 2013, 54, 49–51. [Google Scholar] [CrossRef]
- Dearden, M.J.; Firkin, C.R.; Hermet, J.P.R.; O’Brien, P. A readily-accessible (+)-sparteine surrogate. J. Am. Chem. Soc. 2002, 124, 11870–11871. [Google Scholar] [CrossRef] [PubMed]
- Dixon, A.J.; McGrath, M.J.; O’Brien, P. Synthesis of (+)-(1R,2S,9S)-11-methyl-7,11-diazatricyclo[7.3.1.02,7]tridecane, A (+)-sparteine surrogate. Org. Synth. 2006, 83, 141–144. [Google Scholar]
- Hoppe, D.; Hense, T. Enantioselective synthesis with lithium/(−)-sparteine carbanion pairs. Angew. Chem. Int. Ed. 1997, 36, 2282–2316. [Google Scholar] [CrossRef]
- Gallagher, D.J.; Du, H.; Long, S.A.; Beak, P. Chiral organolithium complexes: The structure of β-lithiated β-phenylcarboxamides and the mechanism of asymmetric substitution in the presence of (−)-sparteine. J. Am. Chem. Soc. 1996, 118, 11391–11398. [Google Scholar] [CrossRef]
- Ebner, T.; Eichelbaum, M.; Fischer, P.; Meese, C.O. Stereospecific hydroxylation of (+)-sparteine in the rat. Arch. Pharm. 1989, 322, 399–403. [Google Scholar] [CrossRef]
- Richardson, T.I.; Rychnovsky, S.D. Filipin III: Configuration assignment and confirmation by synthetic correlation. J. Org. Chem. 1996, 61, 4219–4231. [Google Scholar] [CrossRef] [PubMed]
- Rychnovsky, S.D. Oxo polyene macrolide antibiotics. Chem. Rev. 1995, 95, 2021–2040. [Google Scholar] [CrossRef]
- Denmark, S.E.; Almstead, N.G. Allylation of carbonyls: Methodology and stereochemistry. In Modern Carbonyl Chemistry; Otera, J., Ed.; Wiley-VCH: Weinheim, Germany, 2000; Volume 10, p. 299. [Google Scholar]
- Chemler, S.R.; Roush, W.R. Recent applications of the allylation reaction to the synthesis of natural products. In Modern Carbonyl Chemistry; Otera, J., Ed.; Wiley-VCH: Weinheim, Germany, 2000; Volume 11, p. 403. [Google Scholar]
- Lachance, H.; Hall, D.G. Allylboration of carbonyl compounds. In Organic Reactions; Denmark, S.E., Ed.; John Wiley & Sons: New York, NY, USA, 2008; Volume 73, p. 1. [Google Scholar]
- Denmark, S.E.; Fu, J. Catalytic Enantioselective addition of allylic organometallic reagents to aldehydes and ketones. Chem. Rev. 2003, 103, 2763–2793. [Google Scholar] [CrossRef] [PubMed]
- Roush, W.R. Methods of organic synthesis. In Stereoselective Synthesis; Helmchen, G., Hoffman, R.W., Muler, J., Schaumann, E., Eds.; Thieme Verlag: Stuttgart, Germany, 1996; Volume 3, p. 1410. [Google Scholar]
- Hoffmann, R.W.; Zeiss, H.J. Diastereoselective synthesis of β-methyl homoallyl alcohols. Angew. Chem. Int. Ed. 1979, 18, 306–309. [Google Scholar] [CrossRef]
- Brown, H.C.; Bhat, K.S. Enantiomeric Z- and E-crotyldiisopinocampheylboranes. Synthesis in high optical purity of all four possible stereoisomers of β-methylhomoallyl alcohols. J. Am. Chem. Soc. 1986, 108, 293–294. [Google Scholar] [CrossRef]
- Rauniyar, V.; Hall, D.G. Rationally improved chiral Brønsted acid for catalytic enantioselective allylboration of aldehydes with an expanded reagent scope. J. Org. Chem. 2009, 74, 4236–4241. [Google Scholar] [CrossRef] [PubMed]
- Penner, M.; Rauniyar, V.; Kaspar, L.T.; Hall, D.G. Catalytic asymmetric synthesis of palmerolide via organoboron methodology. J. Am. Chem. Soc. 2009, 131, 14216–14217. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Handa, M.; Roush, W.R. Enantioselective synthesis of 2-methyl-1,2-syn- and 2-methyl-1,2-anti-3-butenediols via allene hydroboration-aldehyde allylboration reaction sequences. J. Am. Chem. Soc. 2009, 131, 14602–14606. [Google Scholar] [CrossRef] [PubMed]
- Carosi, L.; Hall, D.G. Catalytic enantioselective preparation of alpha-substituted allylboronates. One-pot addition to functionalized aldehydes and a route to chiral allylic trifluoroborate salts. Angew. Chem. Int. Ed. 2007, 46, 5913–5918. [Google Scholar] [CrossRef] [PubMed]
- Kister, J.; DeBaillie, A.C.; Lira, R.; Roush, W.R. Stereoselective synthesis of γ-substituted (Z)-allylic boranes via kinetically controlled hydroboration of allenes with 10-TMS-9-borabicyclo[3.3.2]decane. J. Am. Chem. Soc. 2009, 131, 14174–14178. [Google Scholar] [CrossRef] [PubMed]
- González, A.Z.; Román, J.G.; Alicea, E.; Canales, E.; Soderquist, J.A. (E)-2-Boryl-1,3-butadiene derivatives of the 10-TMS-9-BBDs: Highly selective reagents for the asymmetric synthesis of anti-1,2-disubstituted-3,4-pentadien-1-ols. J. Am. Chem. Soc. 2009, 131, 1269–1273. [Google Scholar] [CrossRef] [PubMed]
- Corey, E.J.; Rhodes, J.J. The application of the formyl hydrogen bond postulate to the understanding of enantioselective reactions involving chiral boron lewis acids and aldehydes. Tetrahedron Lett. 1997, 38, 37–40. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Fjikawa, R.; Yamada, A.; Miyaura, N. Synthesis of B-trisubstituted borazines via the rhodium-catalyzed hydroboration of alkenes with N,N′,N″-trimethyl or N,N′,N″-triethylborazine. Chem. Lett. 1999, 1069–1071. [Google Scholar] [CrossRef]
- Andersen, M.W.; Hildebrndt, B.; Kosher, G.; Hoffmann, R.W. Stereoselective synthesis of alcohols, XXX: E- and Z-pentenylboronates, reagents for simple diastereoselection on addition to aldehydes. Chem. Ber. 1989, 122, 1777–1781. [Google Scholar] [CrossRef]
- Hoffmann, R.W.; Dirich, K.; Kosher, G.; Strürmer, R. Stereoselective synthesis of alcohols, XXXI: Stereoselective C-C bond formation using chiral Z-pentenylboronates. Chem. Ber. 1989, 122, 1783–1786. [Google Scholar] [CrossRef]
- Fang, G.Y.; Aggarwal, V.K. Asymmetric synthesis of α-substituted allylic boranes and their application in synthesis of iso-agatharesinol. Angew. Chem. Int. Ed. 2007, 46, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Howells, D.; Robiette, R.; Fang, G.Y.; Knowles, L.S.; Woodrow, M.D.; Harvey, J.N.; Aggarwal, V.K. Reactions of silyl-stabilised sulfur ylides with organoboranes: Enantioselectivity, mechanism, and understanding. Org. Biomol. Chem. 2008, 6, 1185–1189. [Google Scholar] [CrossRef] [PubMed]
- Soderquist, J.A.; Martinez, J.; Oyola, Y.; Dock, I. Novel route of carboxylic acids via the DCME reaction. Tetrahedron Lett. 2004, 45, 5541–5545. [Google Scholar] [CrossRef]
- Brown, H.C.; Kim, K.W.; Cole, T.A.; Singaram, B. Chiral synthesis via organoboranes. 8. Synthetic utility of boronic esters of essentially 100% optical purity. Synthesis of primary amines of very high enantiomeric purities. J. Am. Chem. Soc. 1986, 108, 6761–6764. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds (8I–8V) are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahmood, A.; Alkhathlan, H.Z.; Parvez, S.; Khan, M.; Shahzad, S.A. Chelation-Assisted Substrate-Controlled Asymmetric Lithiation-Allylboration of Chiral Carbamate 1,2,4-Butanetriol Acetonide. Molecules 2015, 20, 9890-9905. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules20069890
Mahmood A, Alkhathlan HZ, Parvez S, Khan M, Shahzad SA. Chelation-Assisted Substrate-Controlled Asymmetric Lithiation-Allylboration of Chiral Carbamate 1,2,4-Butanetriol Acetonide. Molecules. 2015; 20(6):9890-9905. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules20069890
Chicago/Turabian StyleMahmood, Adeem, Hamad Z. Alkhathlan, Saima Parvez, Merajuddin Khan, and Sohail A. Shahzad. 2015. "Chelation-Assisted Substrate-Controlled Asymmetric Lithiation-Allylboration of Chiral Carbamate 1,2,4-Butanetriol Acetonide" Molecules 20, no. 6: 9890-9905. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules20069890