HPLC Separation of Diastereomers: Chiral Molecular Tools Useful for the Preparation of Enantiopure Compounds and Simultaneous Determination of Their Absolute Configurations

Abstract
:1. Introduction
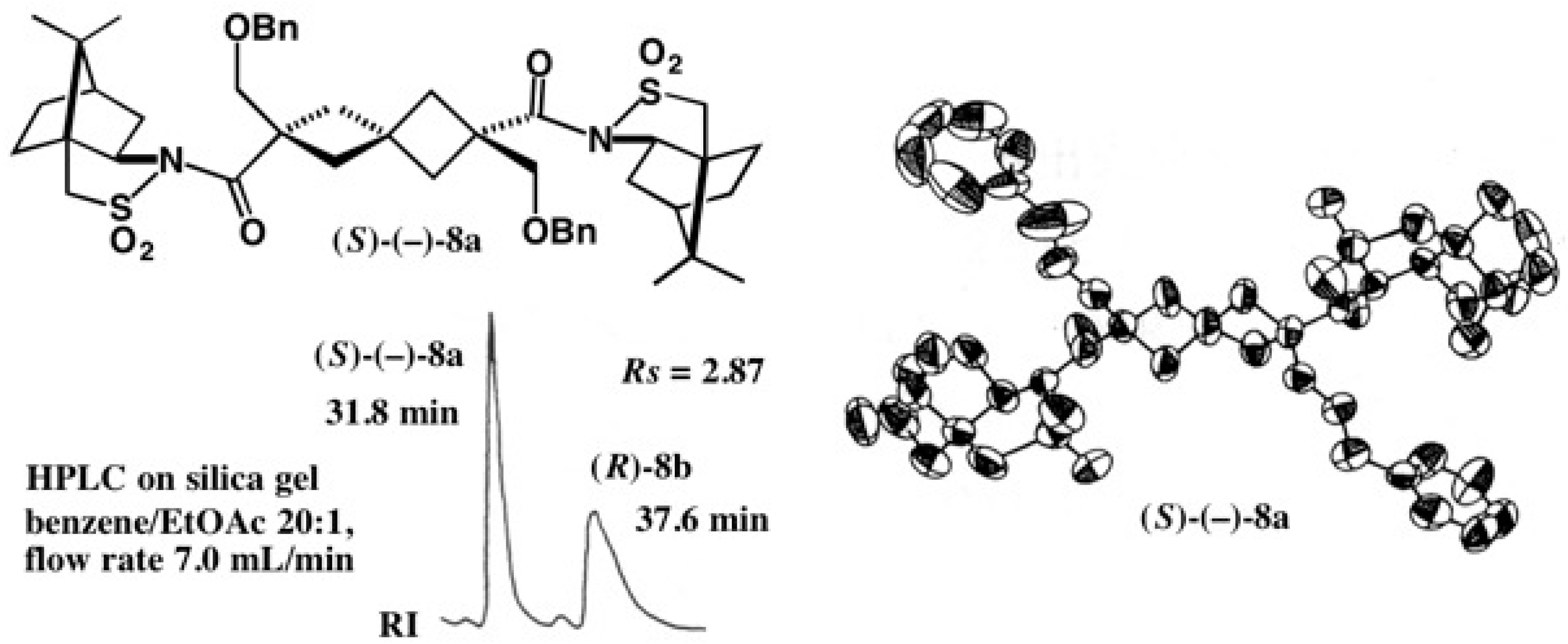
2. Use of (−)-Camphorsultam for Carboxylic Acids
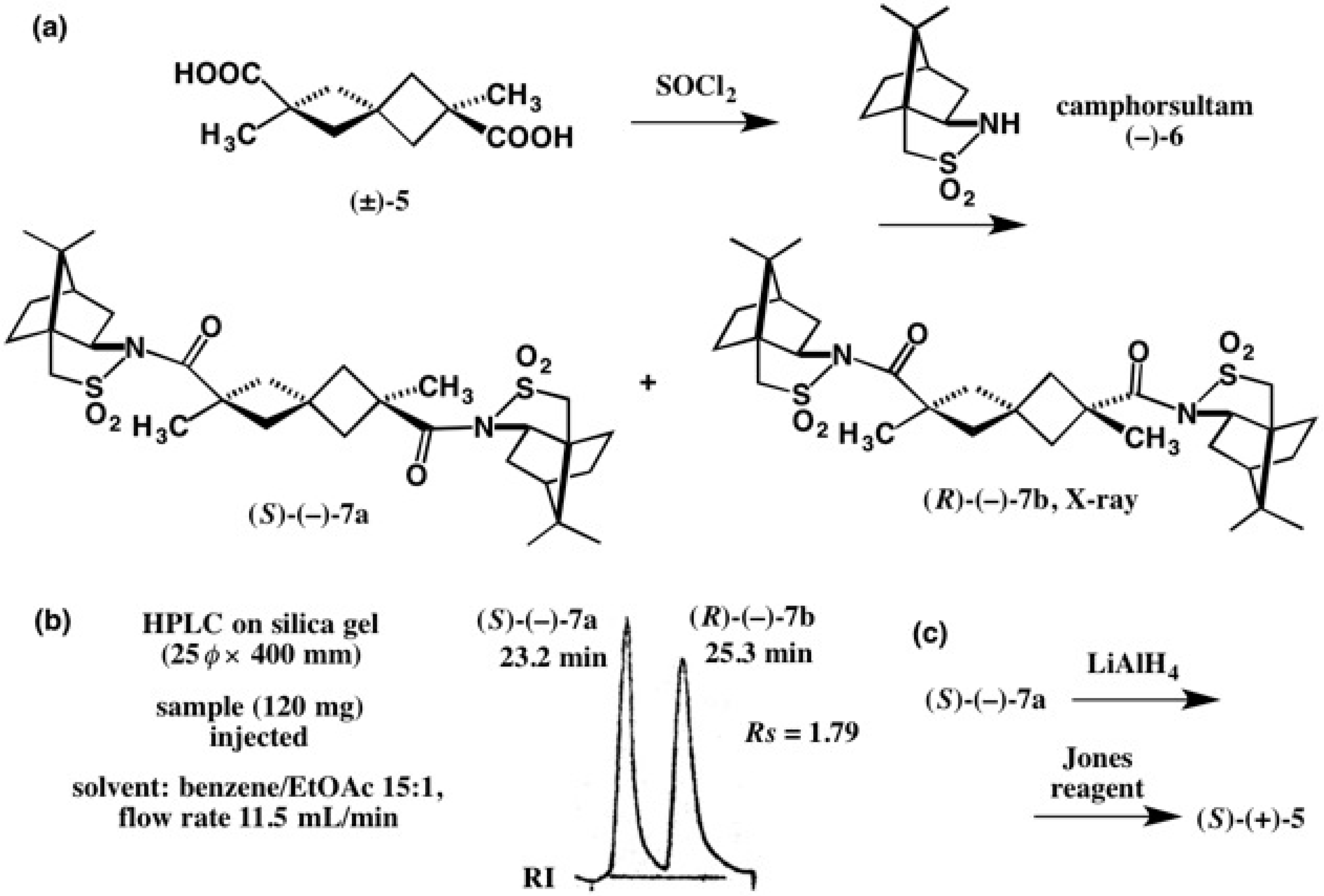
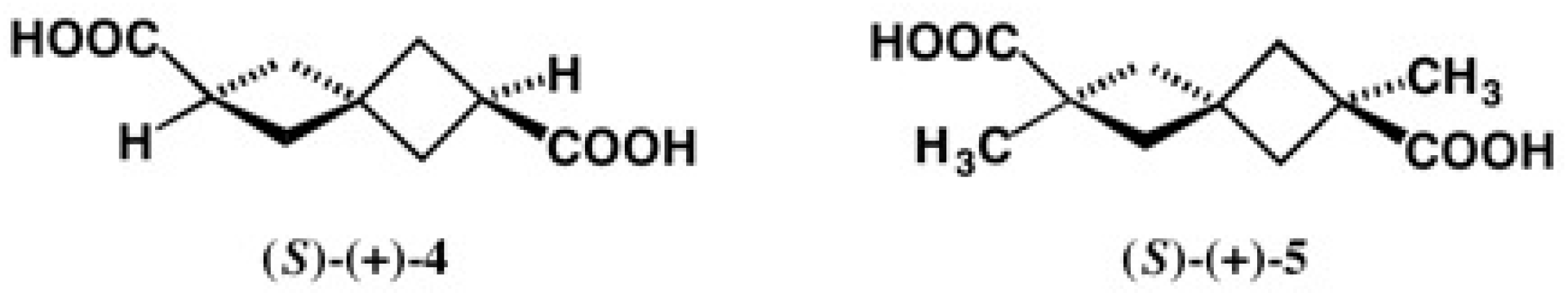
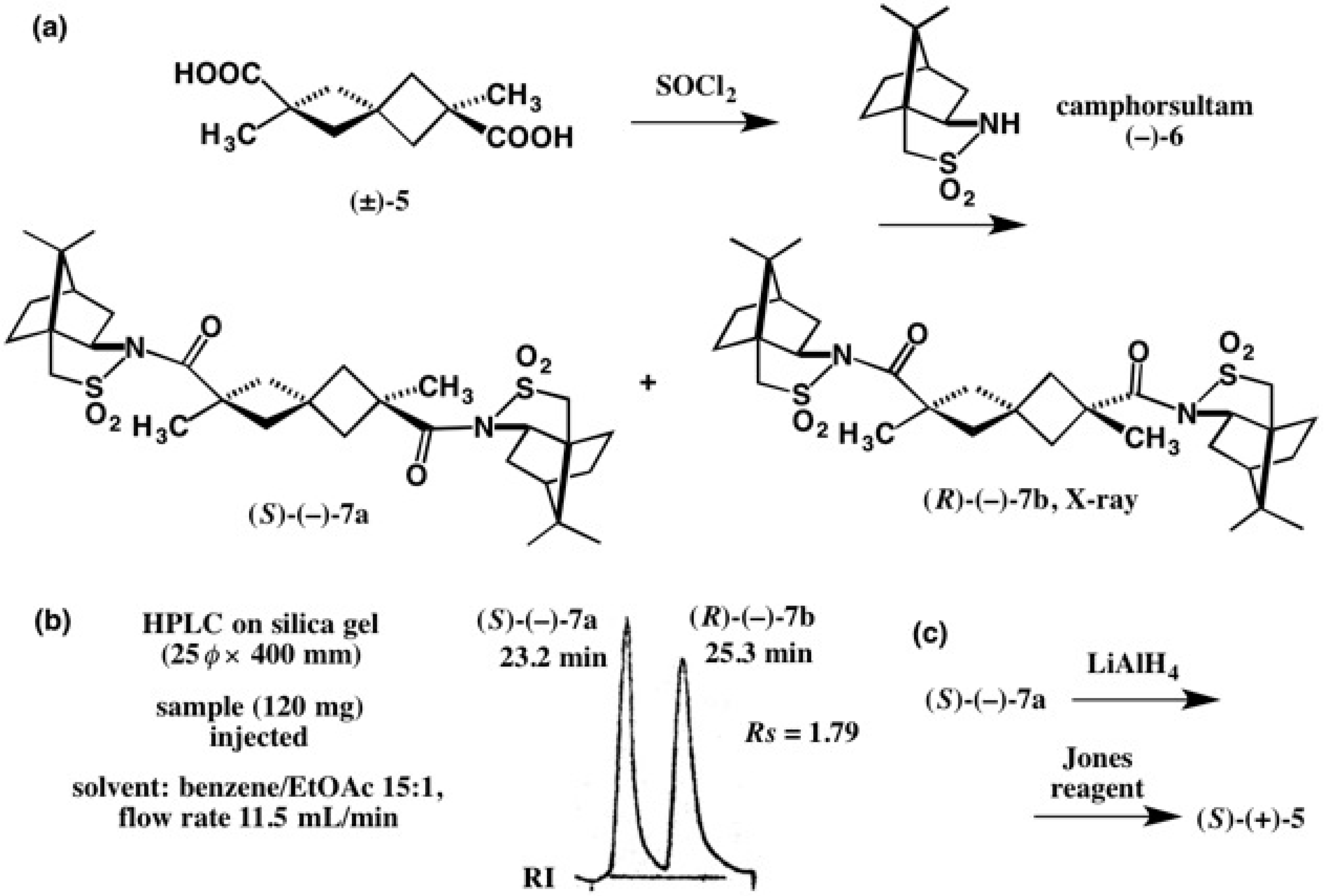
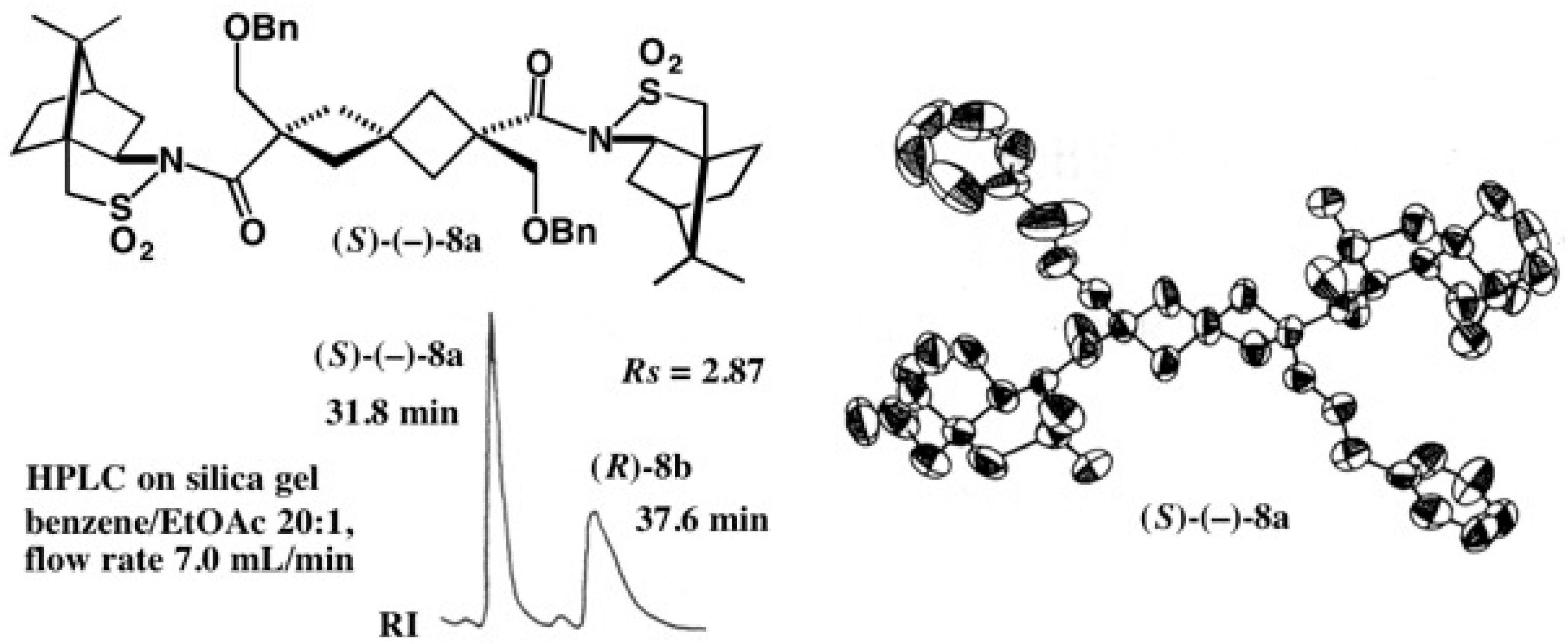
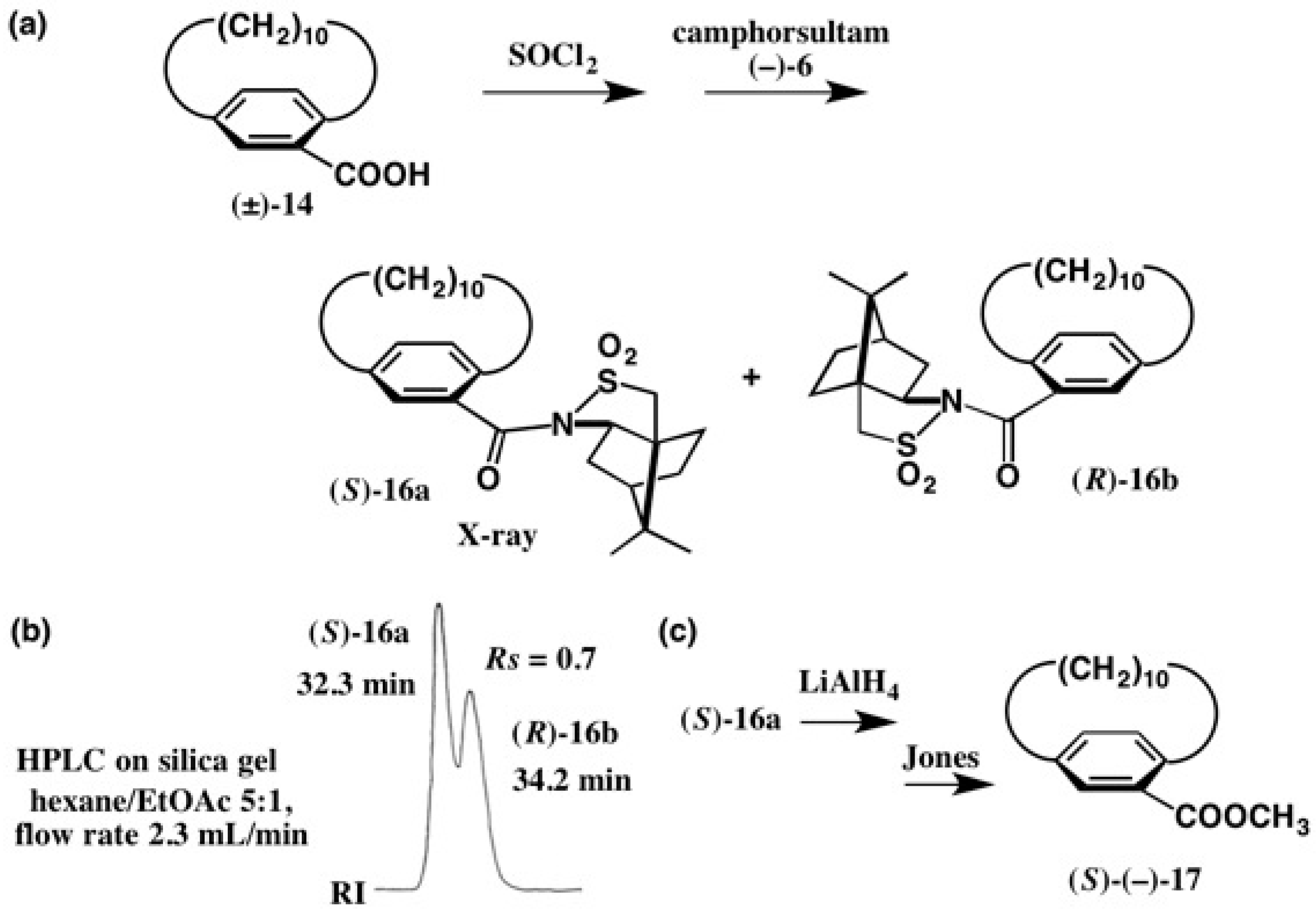
2.1. Application to Spiro[3.3]Heptane-Dicarboxylic Acids
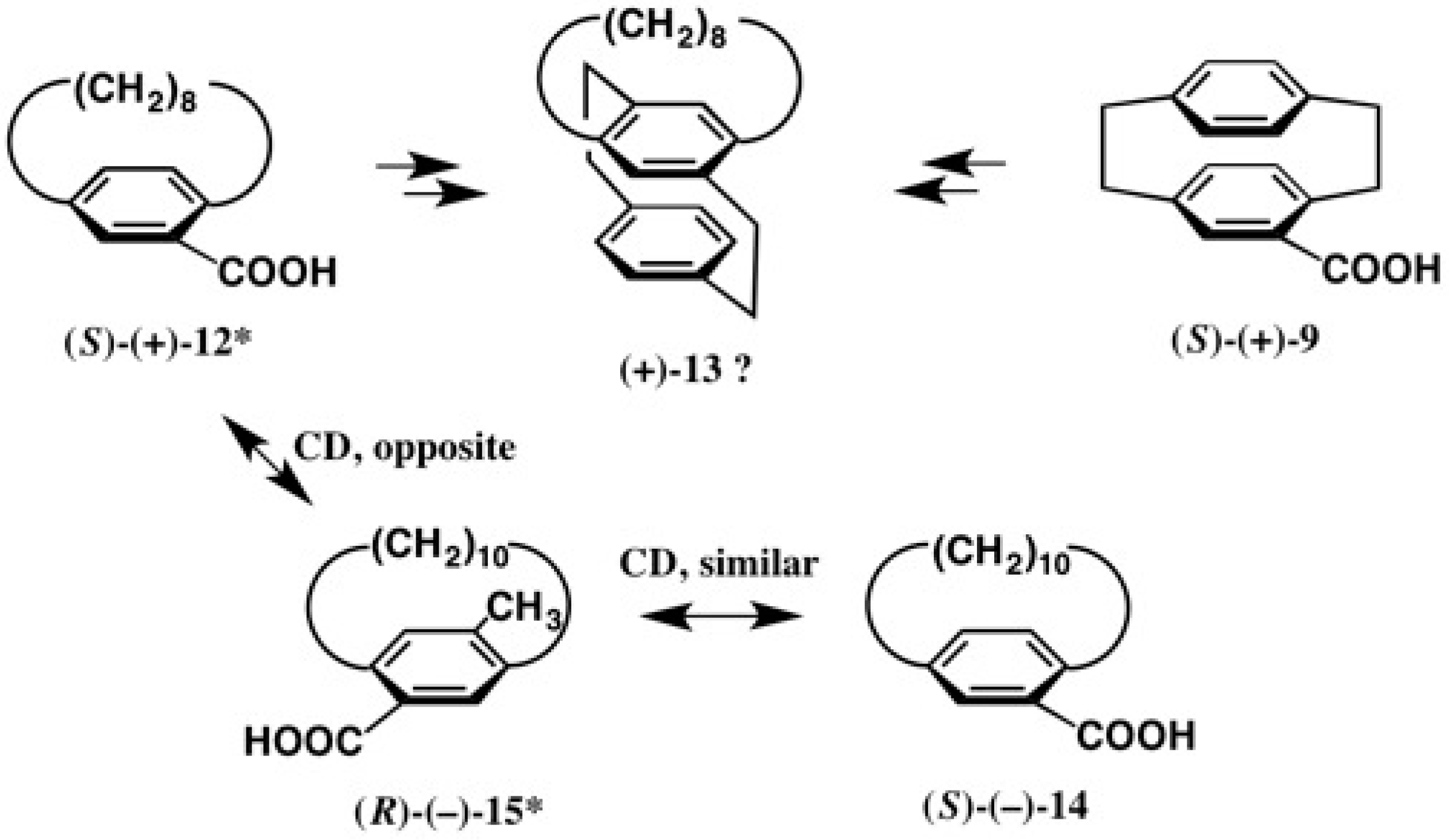
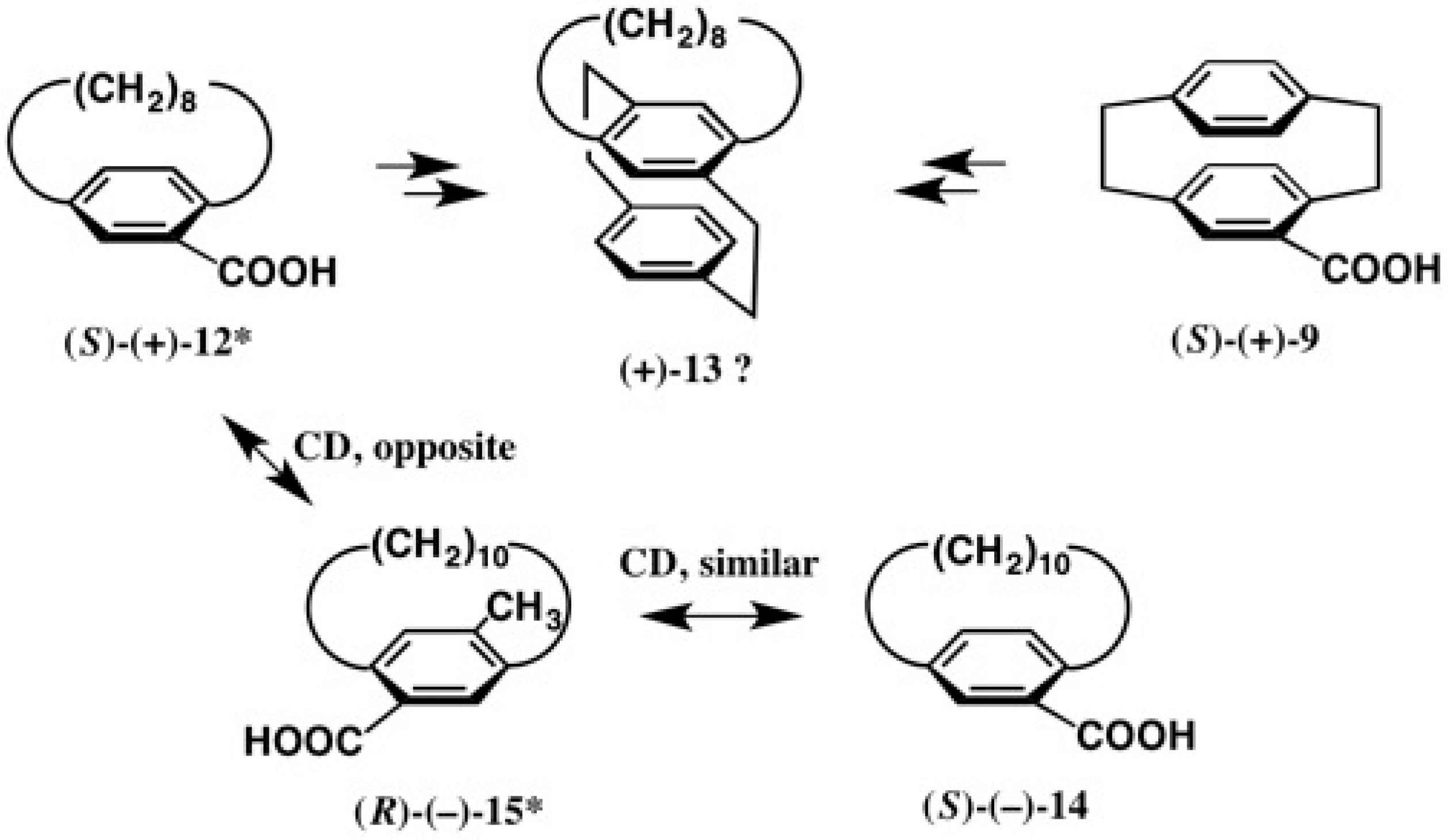
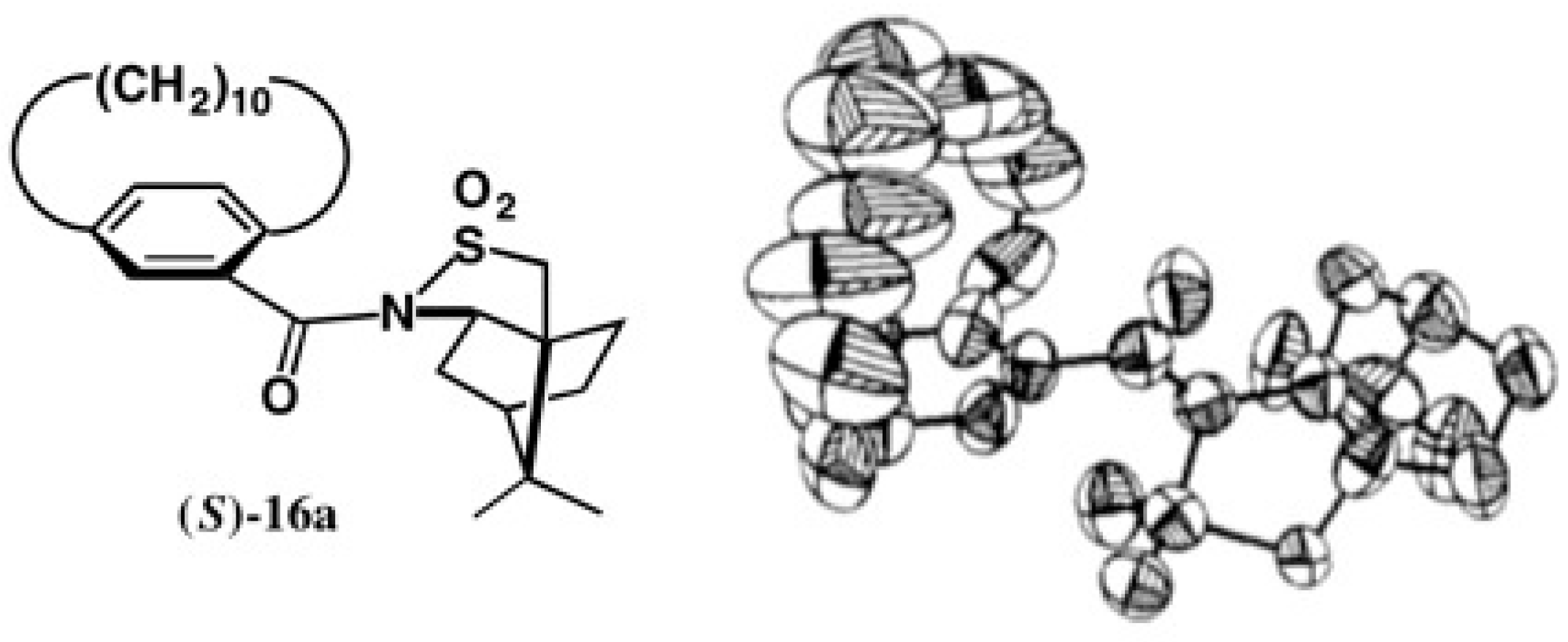
2.2. Application to Cyclophane-Carboxylic Acids
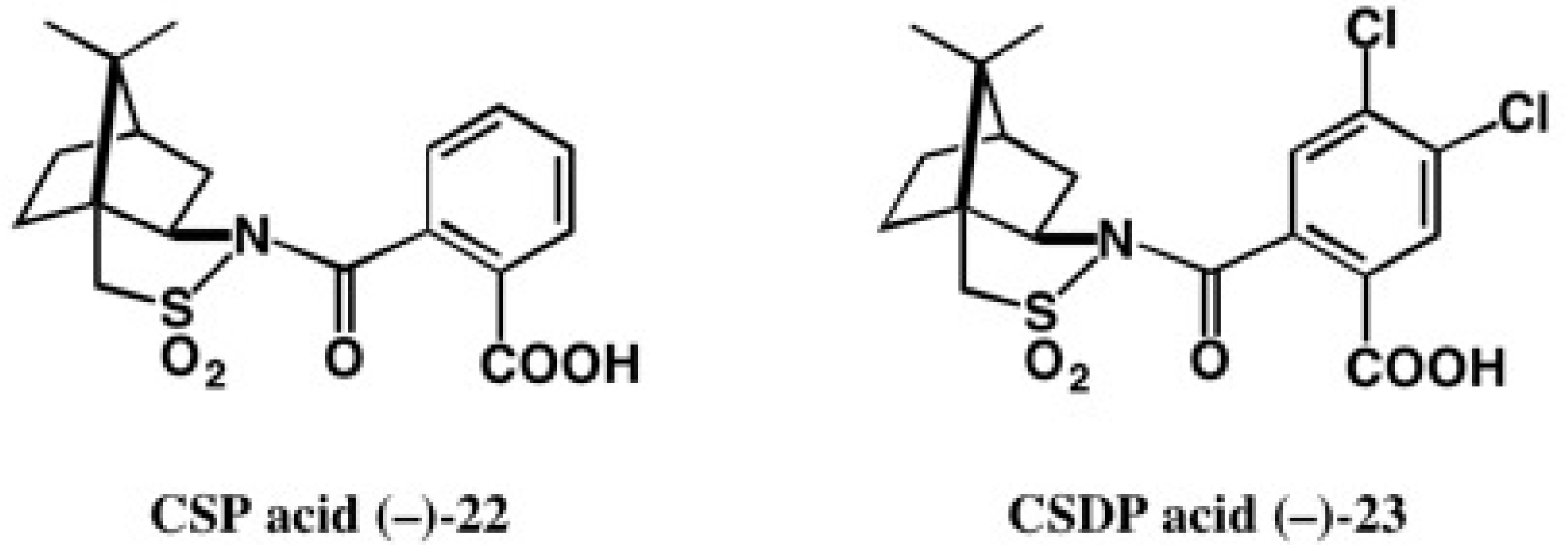
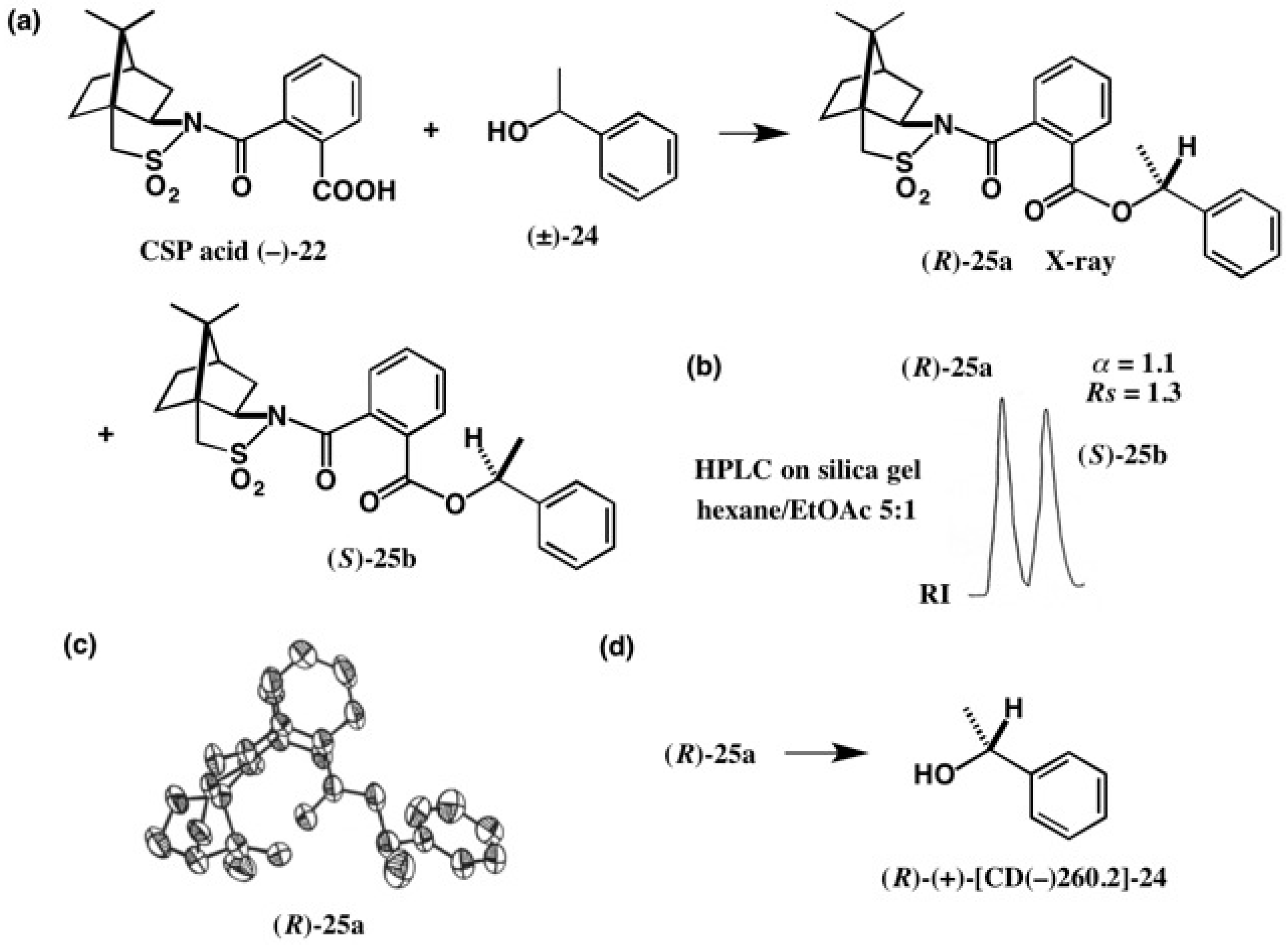
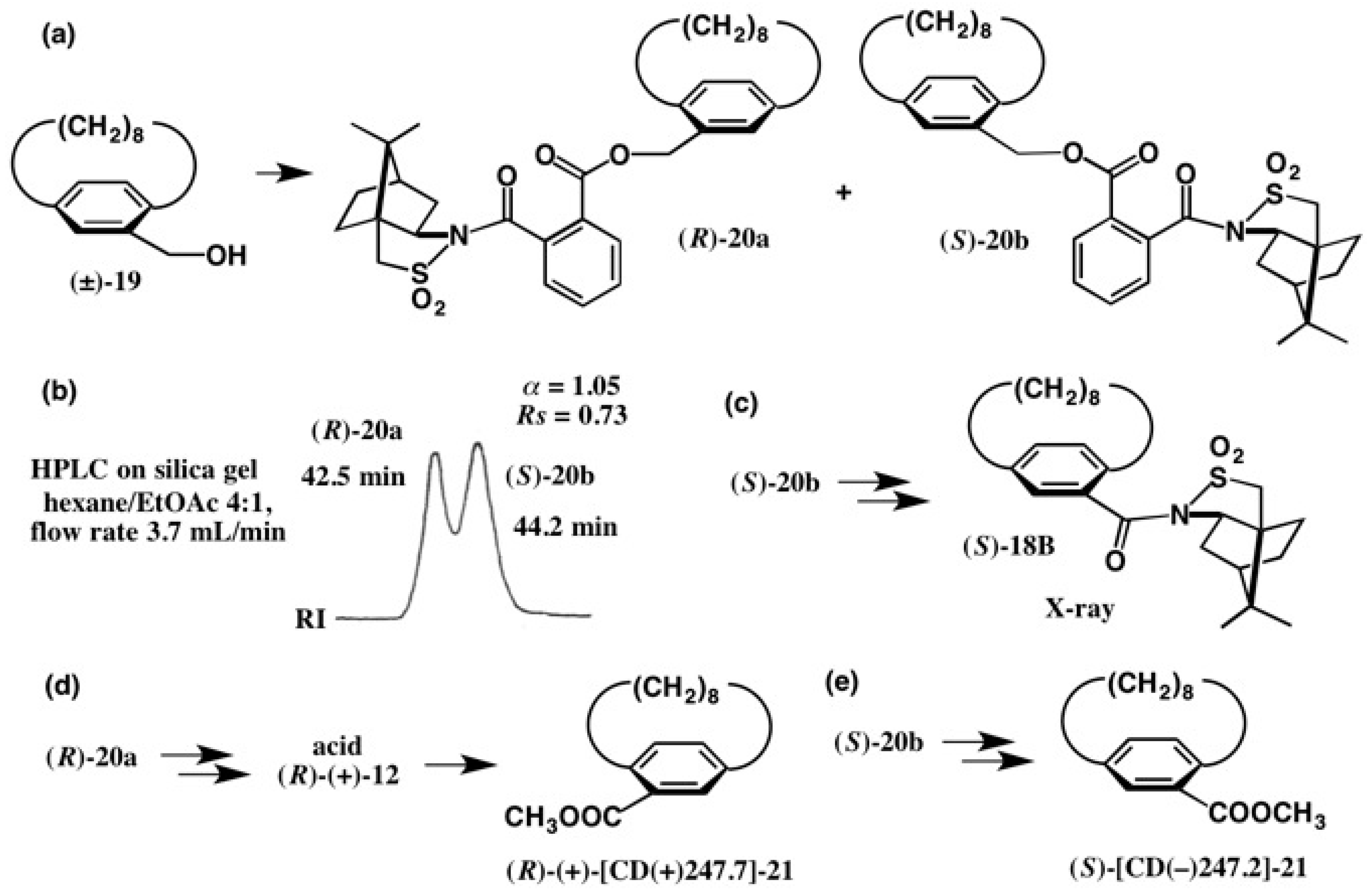
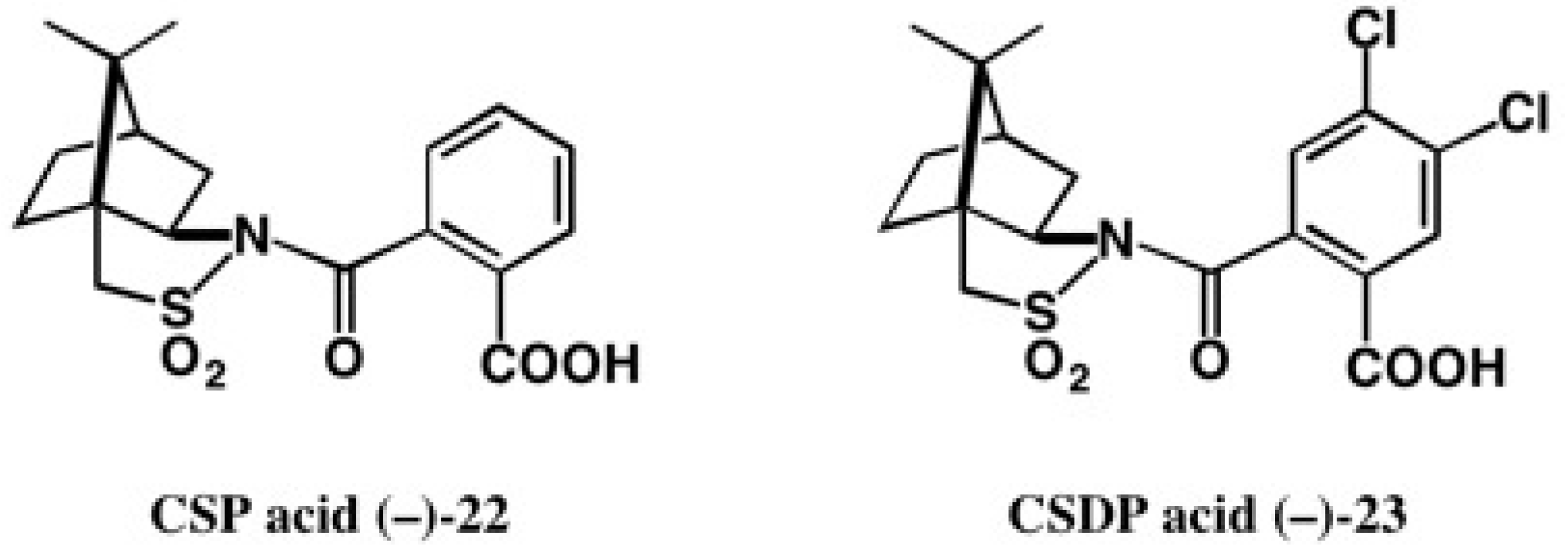
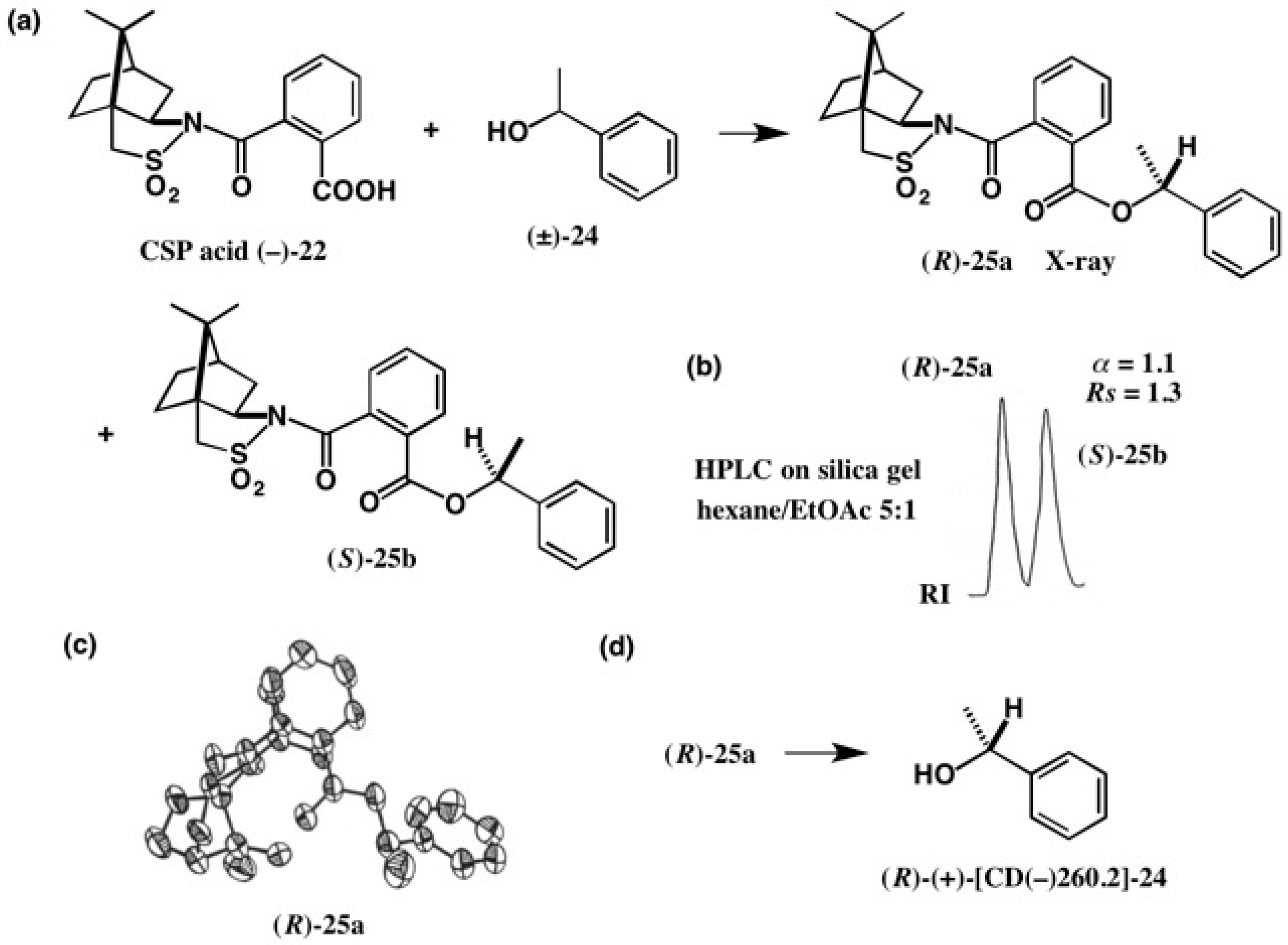
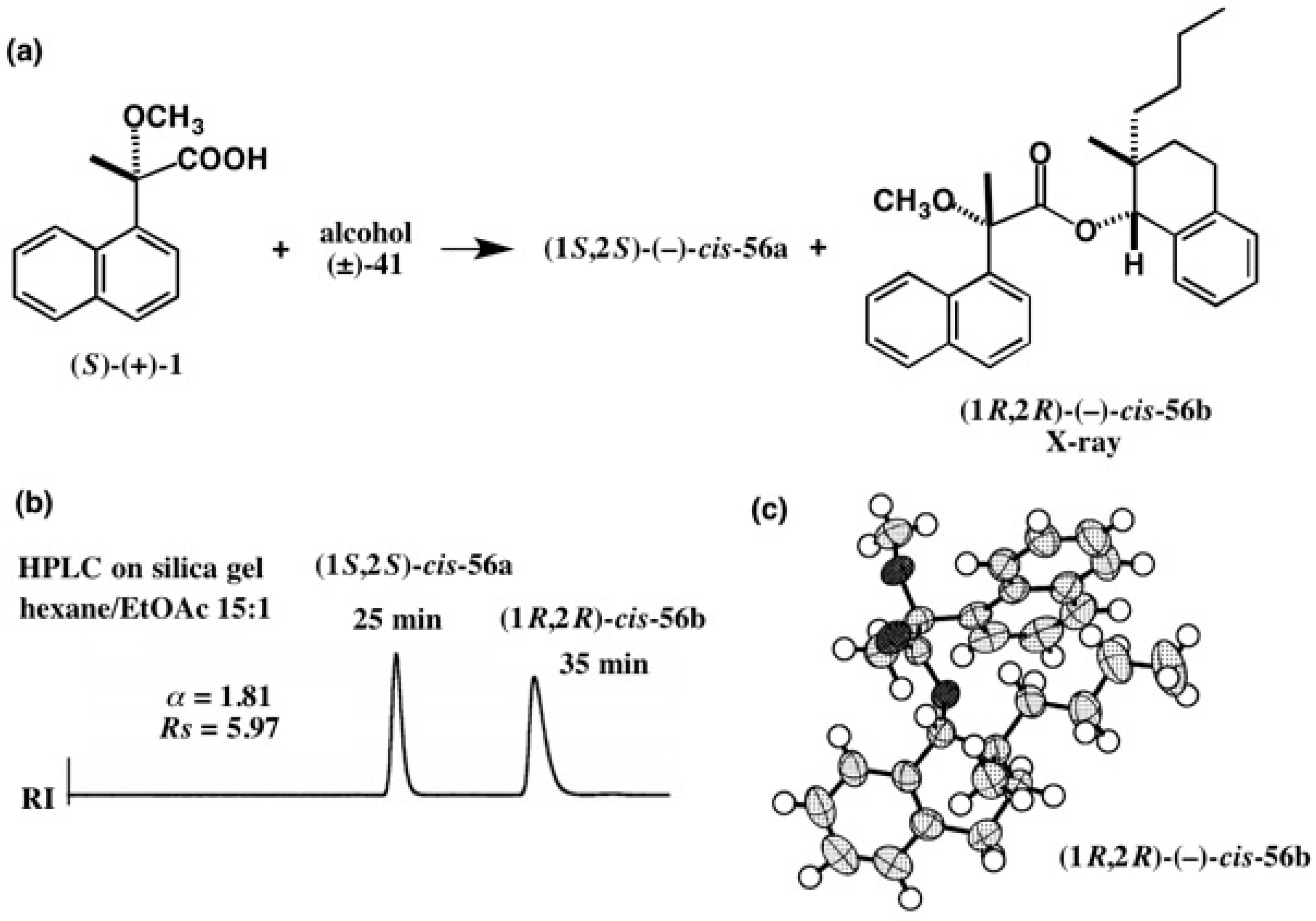
3. Use of Camphorsultam-Phthalic Acid (CSP Acid) for Alcohols—The CSP Acid Method
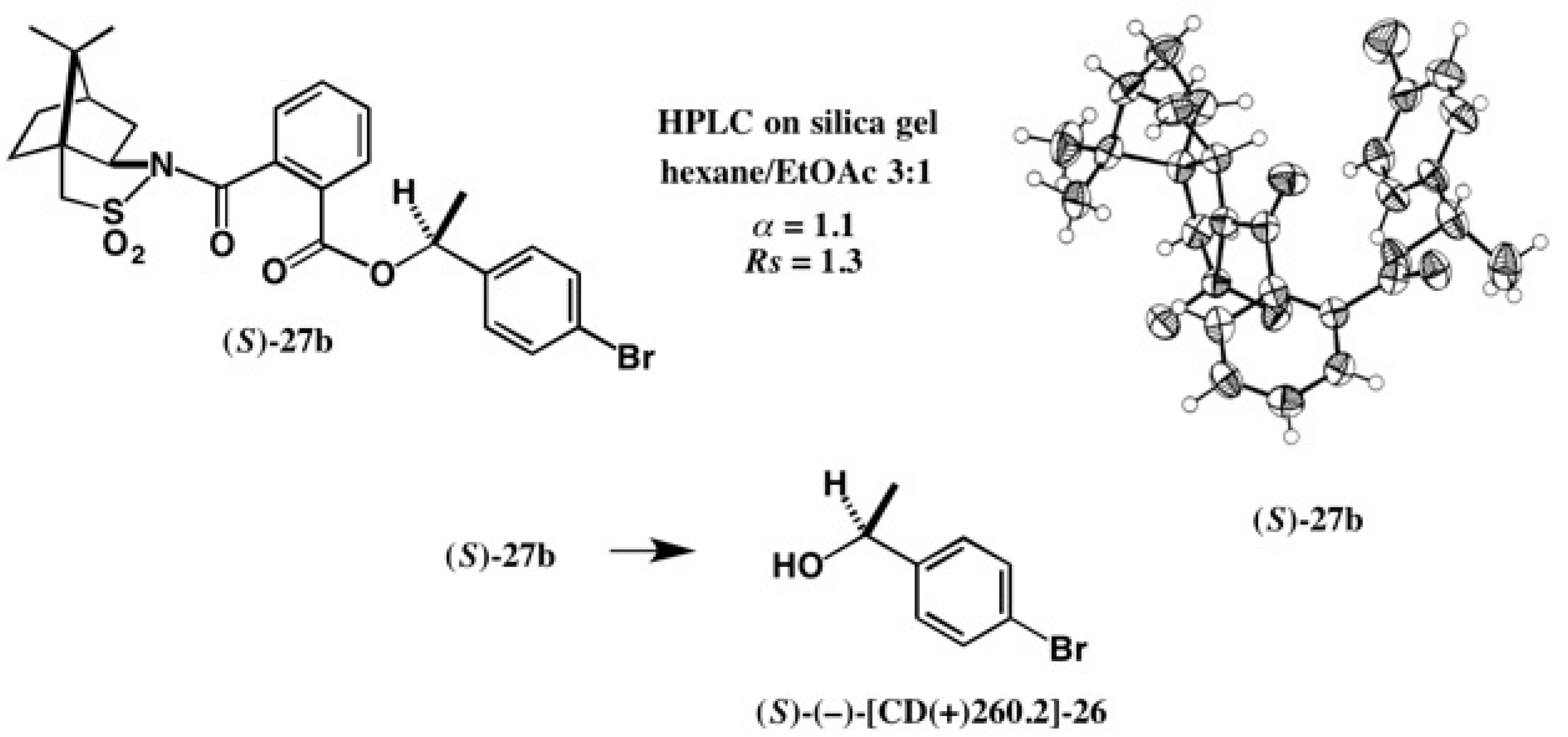
Application to Alcohols with an Aromatic Group
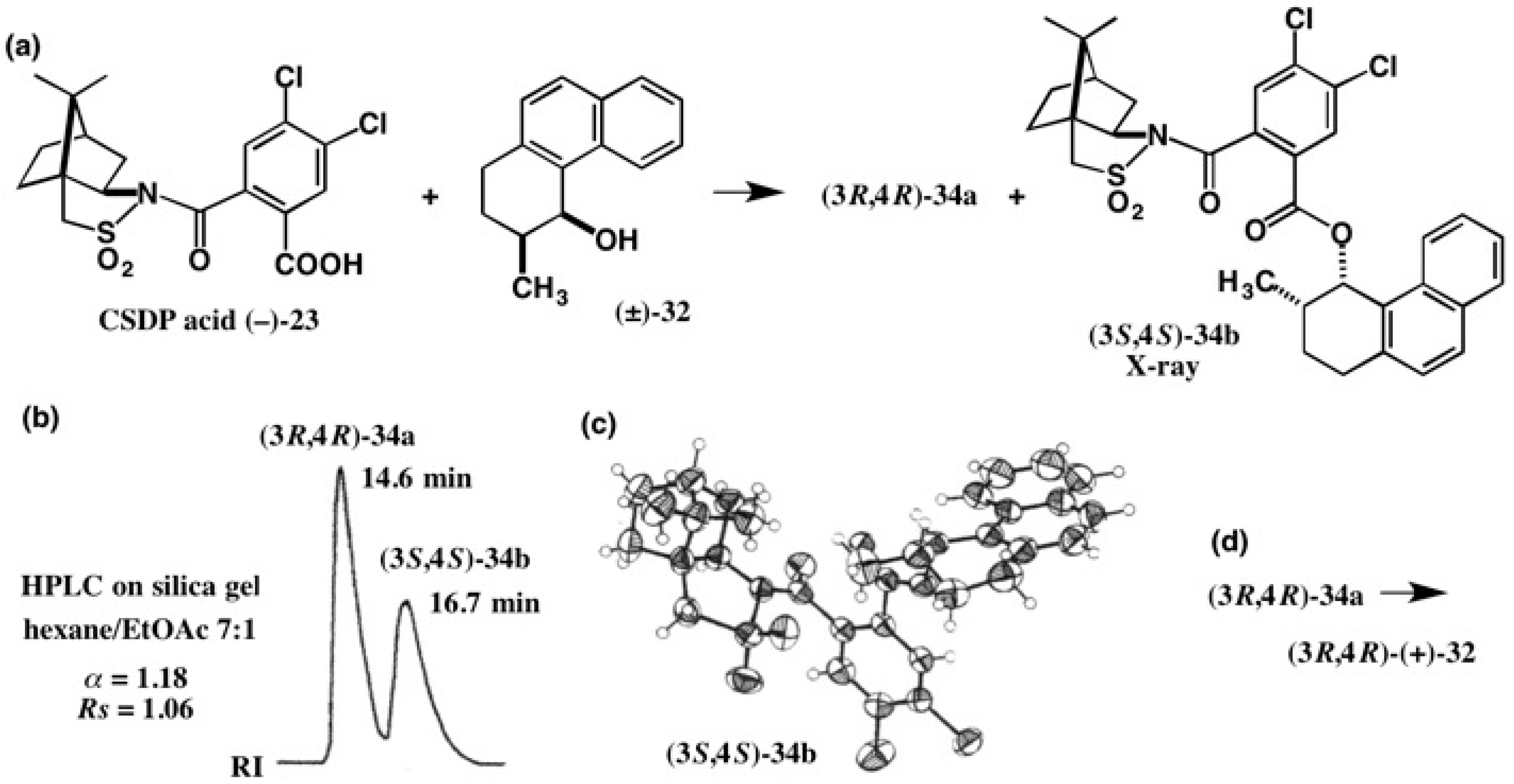
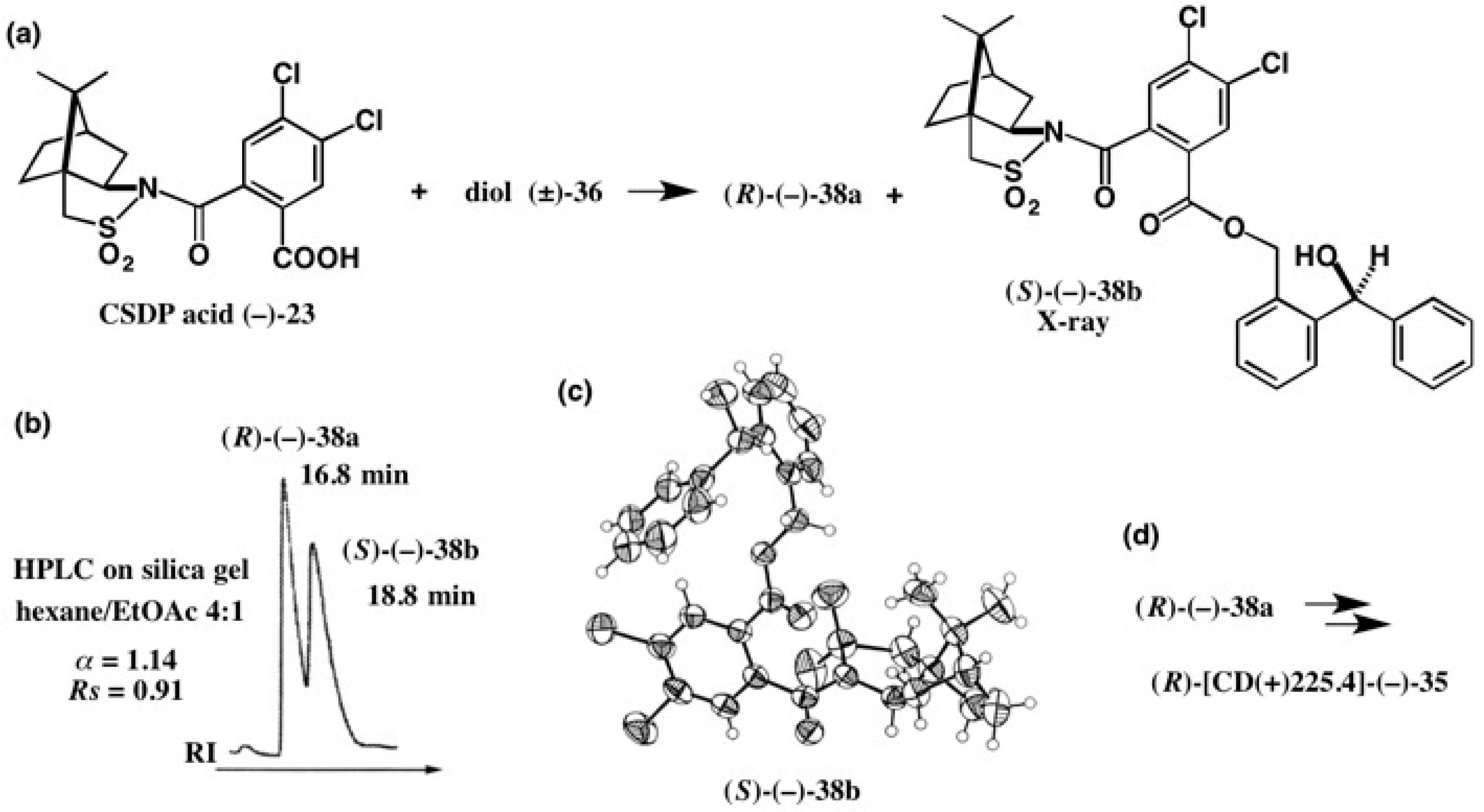
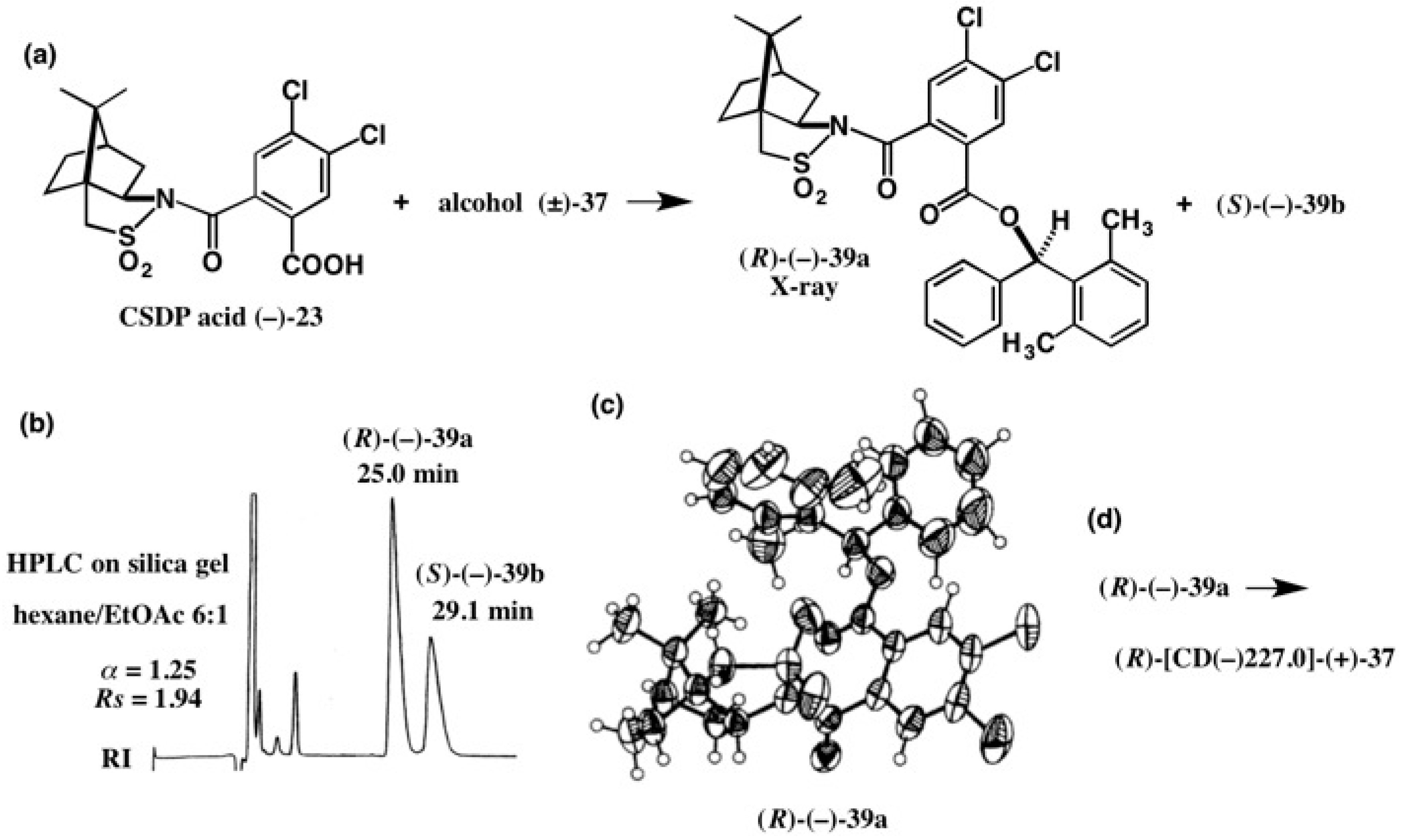
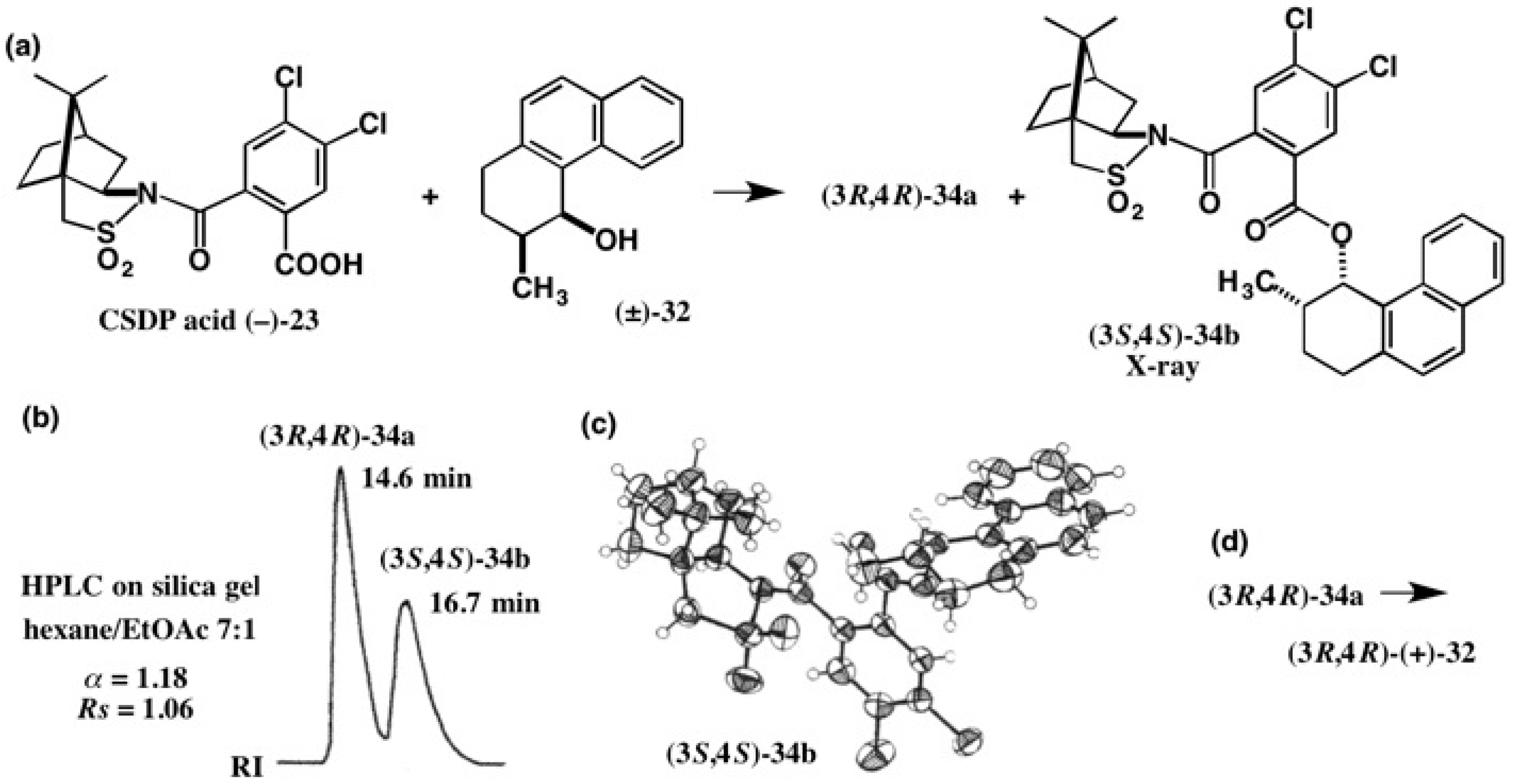
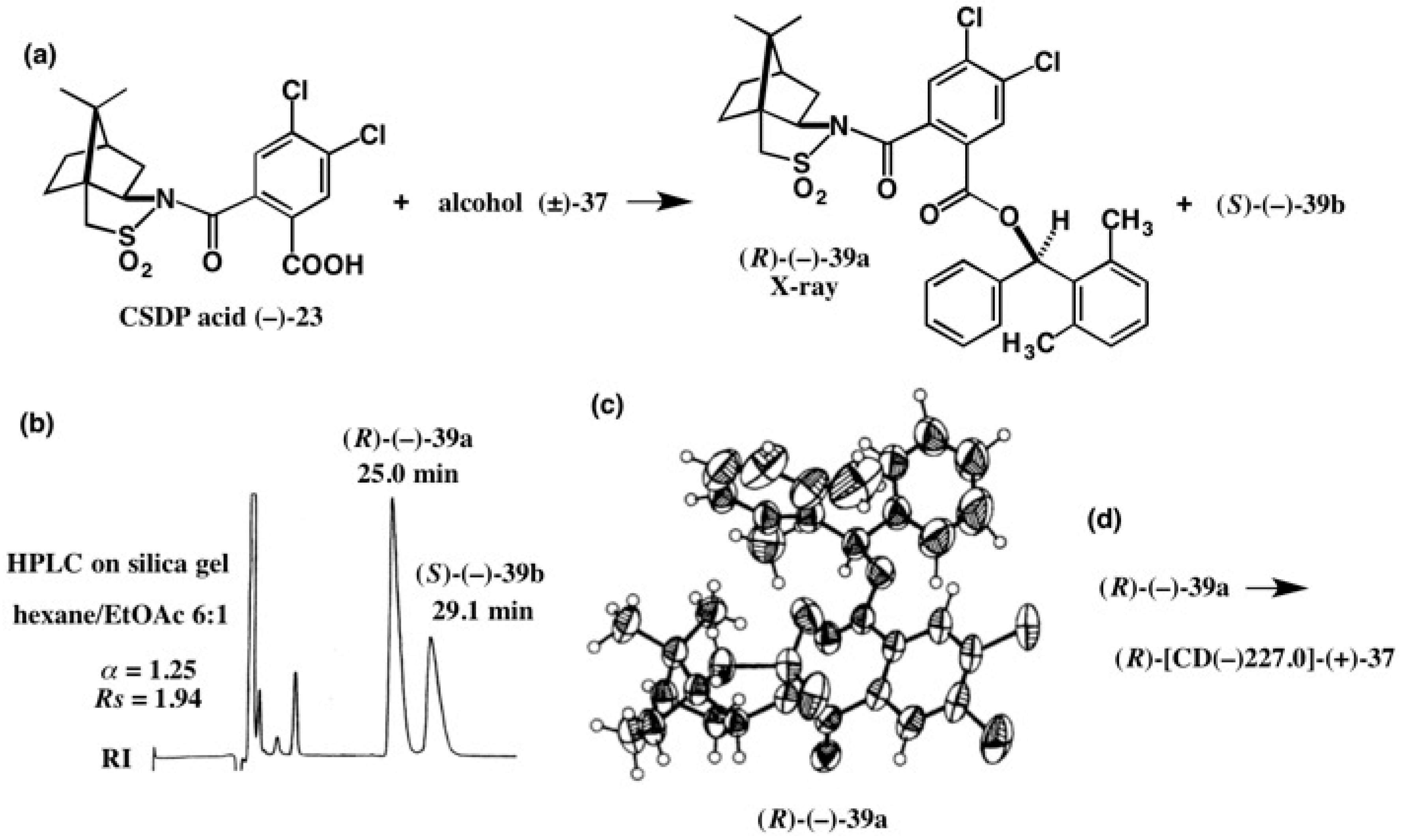
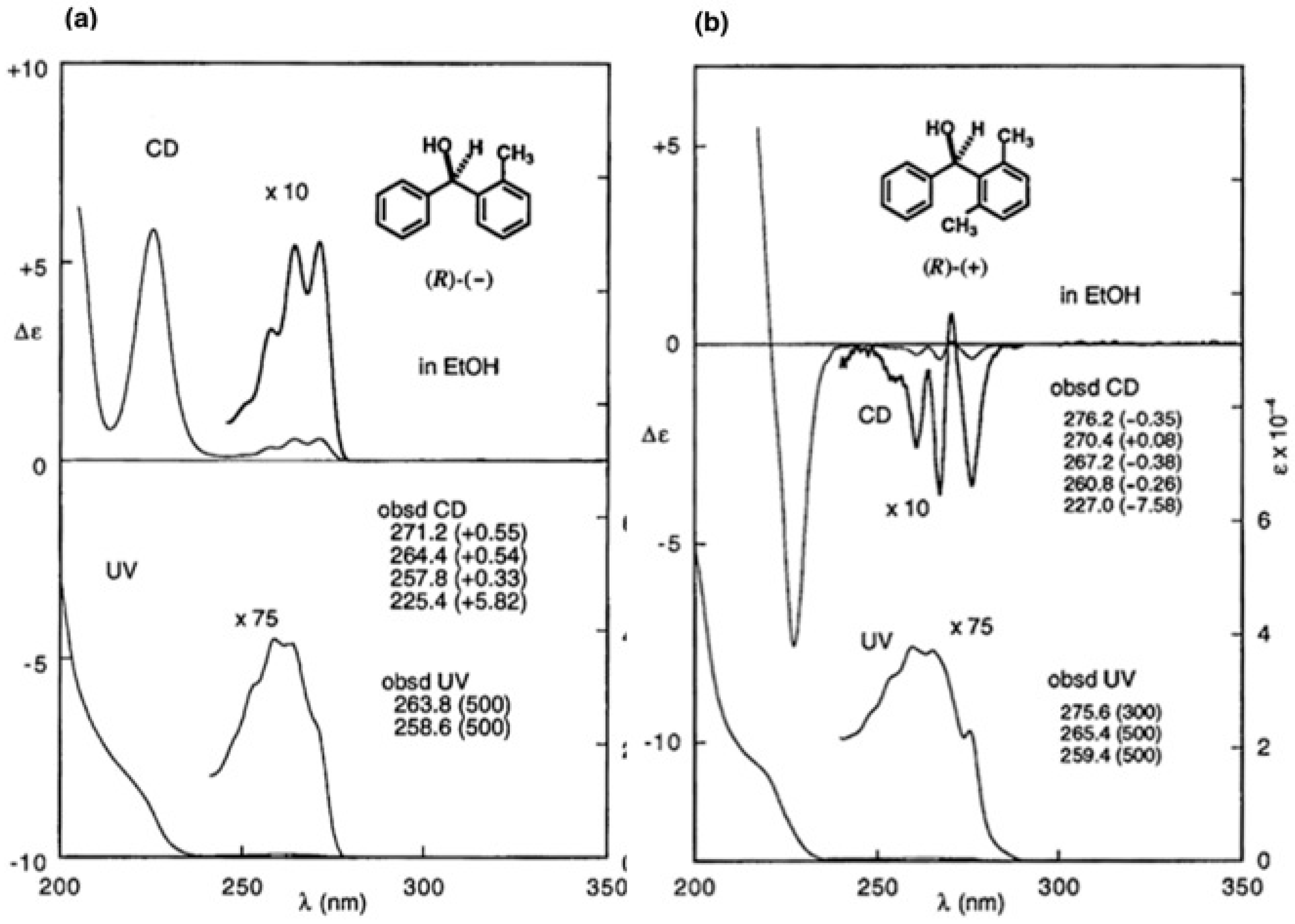
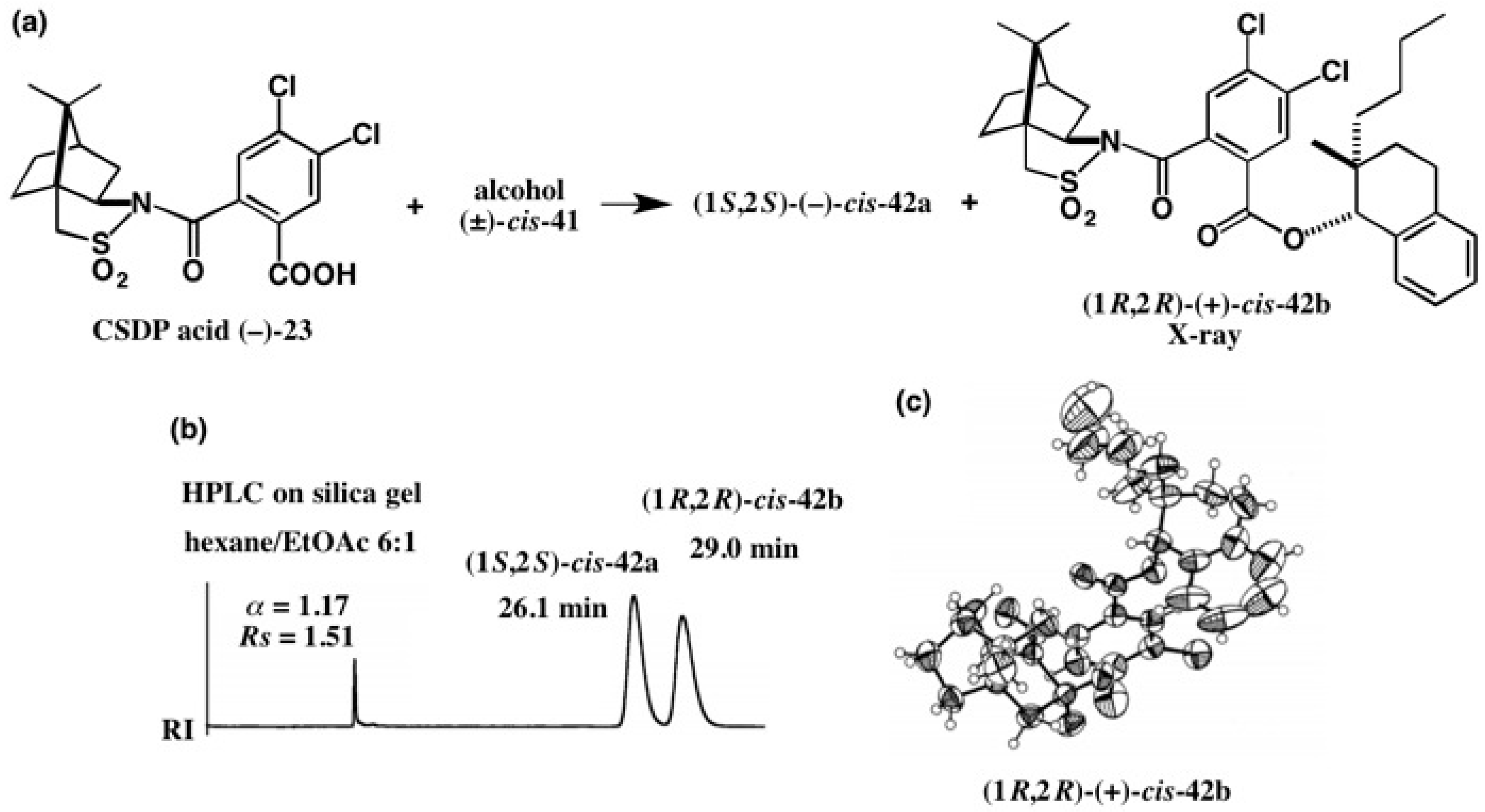
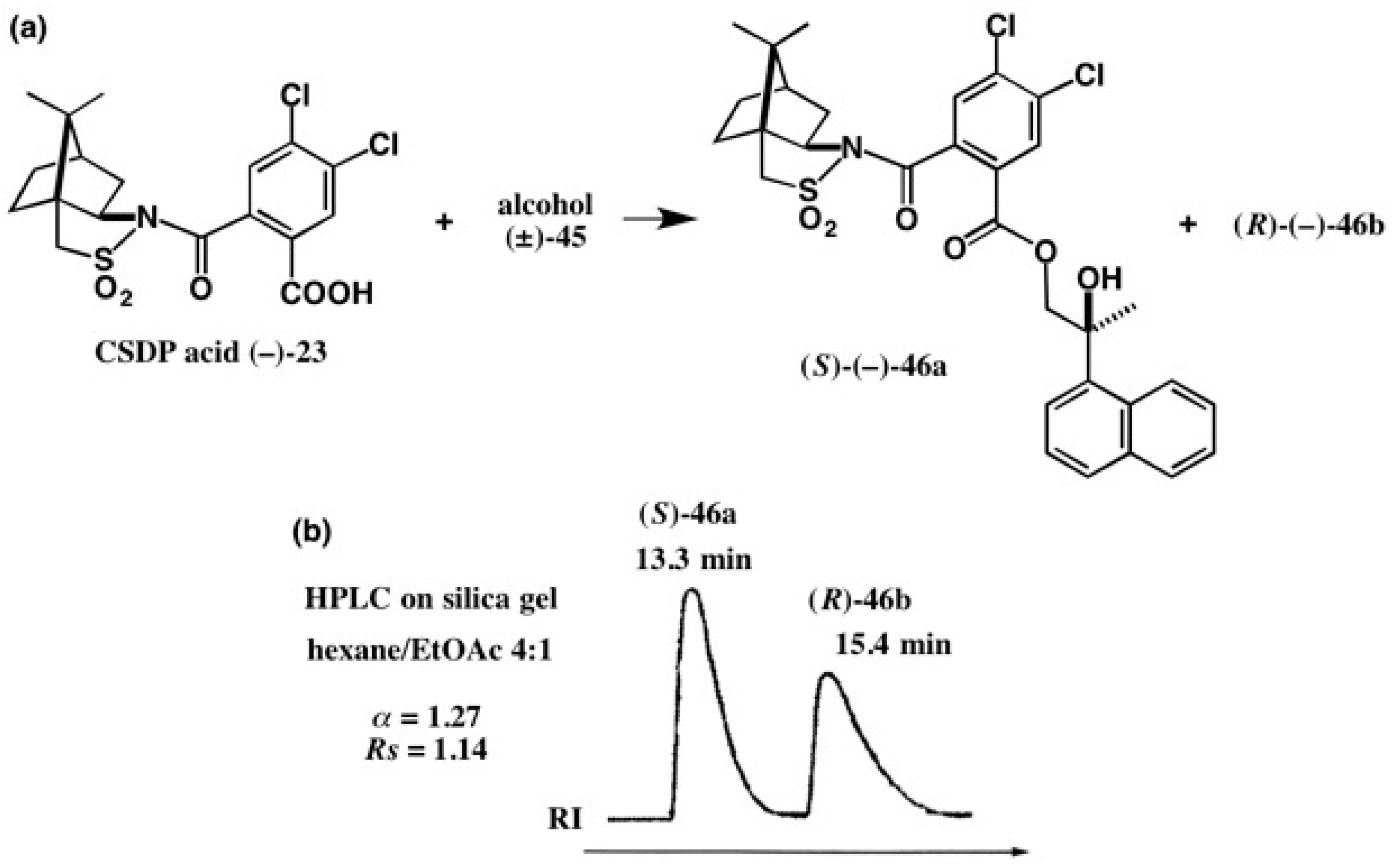
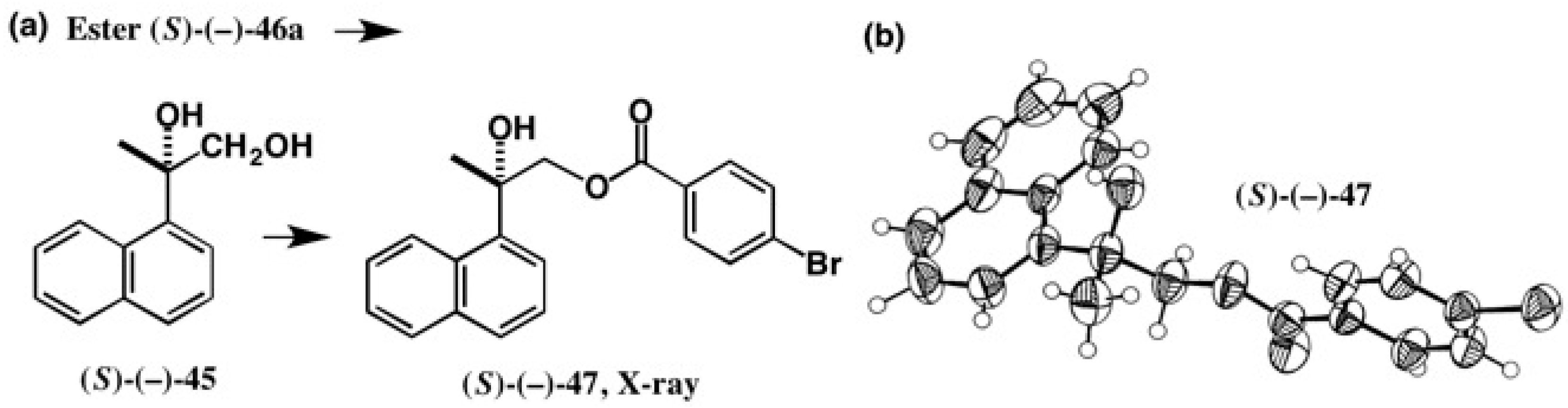
4. Camphorsultam-Dichlorophthalic Acid (CSDP Acid) for Alcohols—The CSDP Acid Method
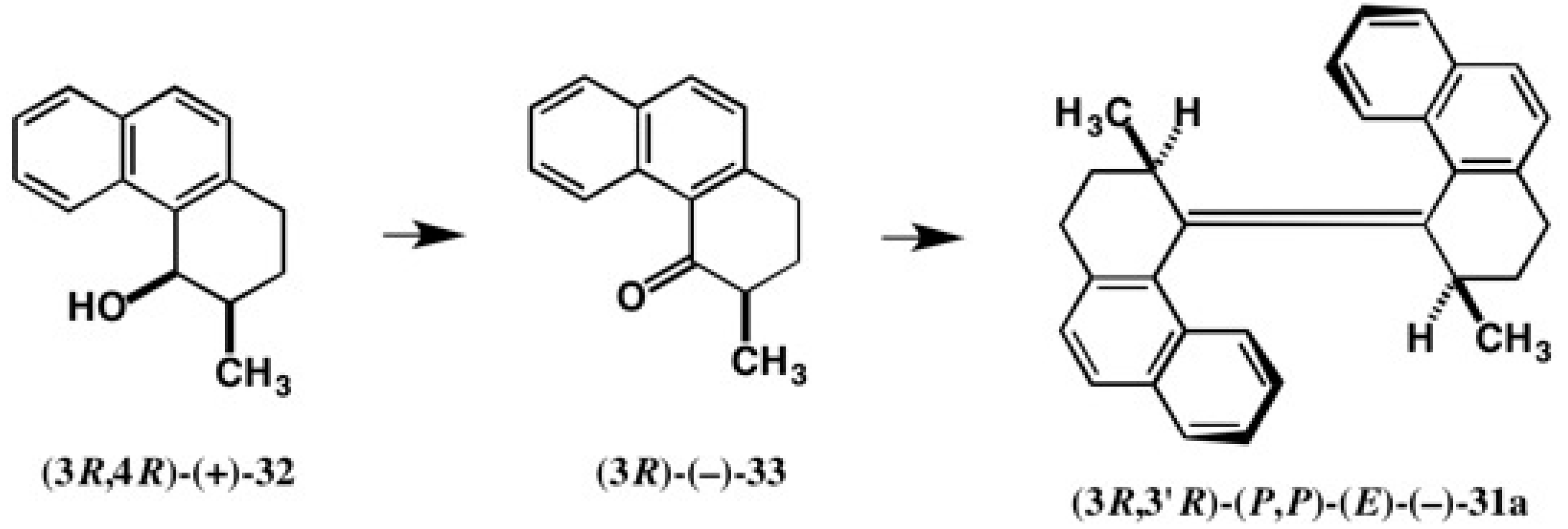
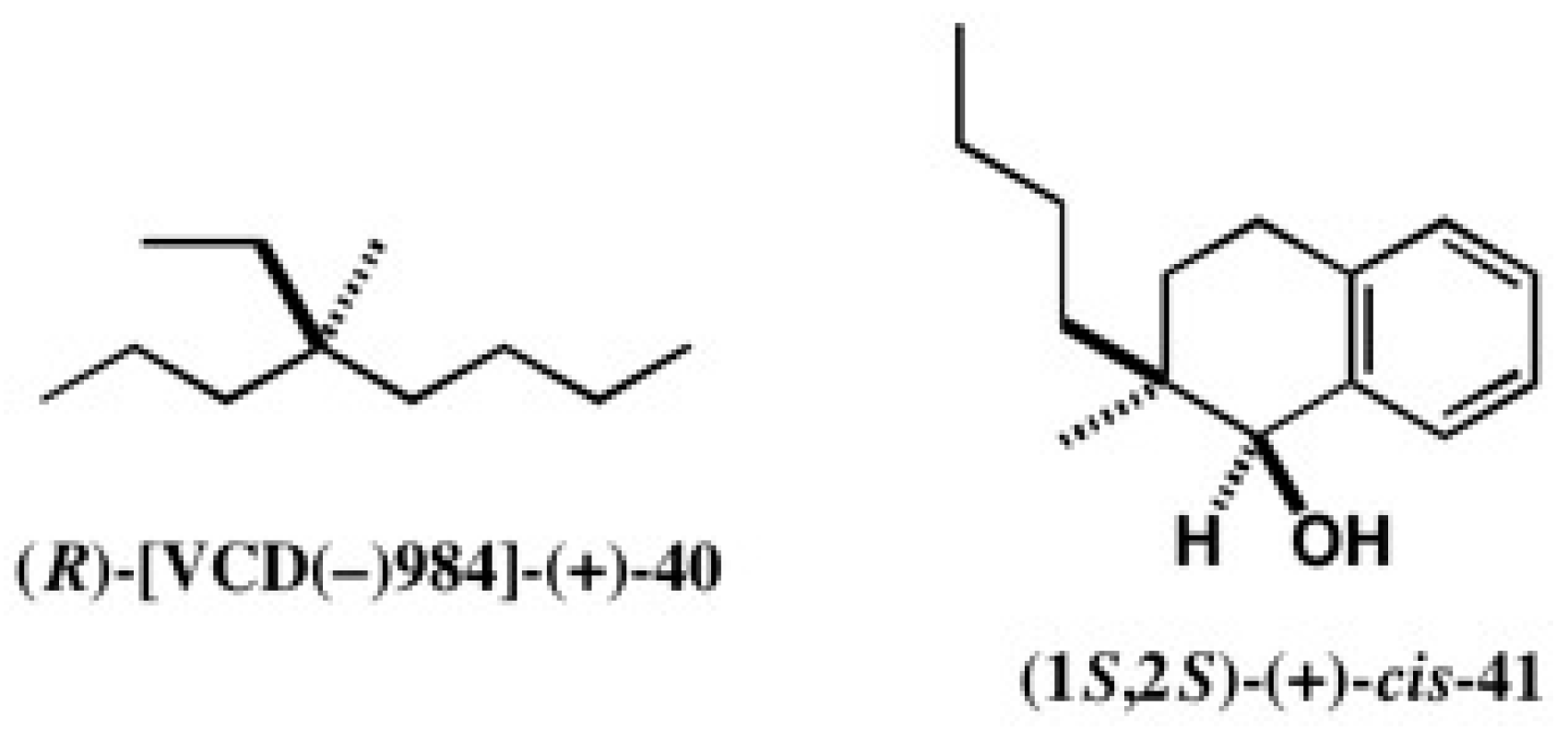
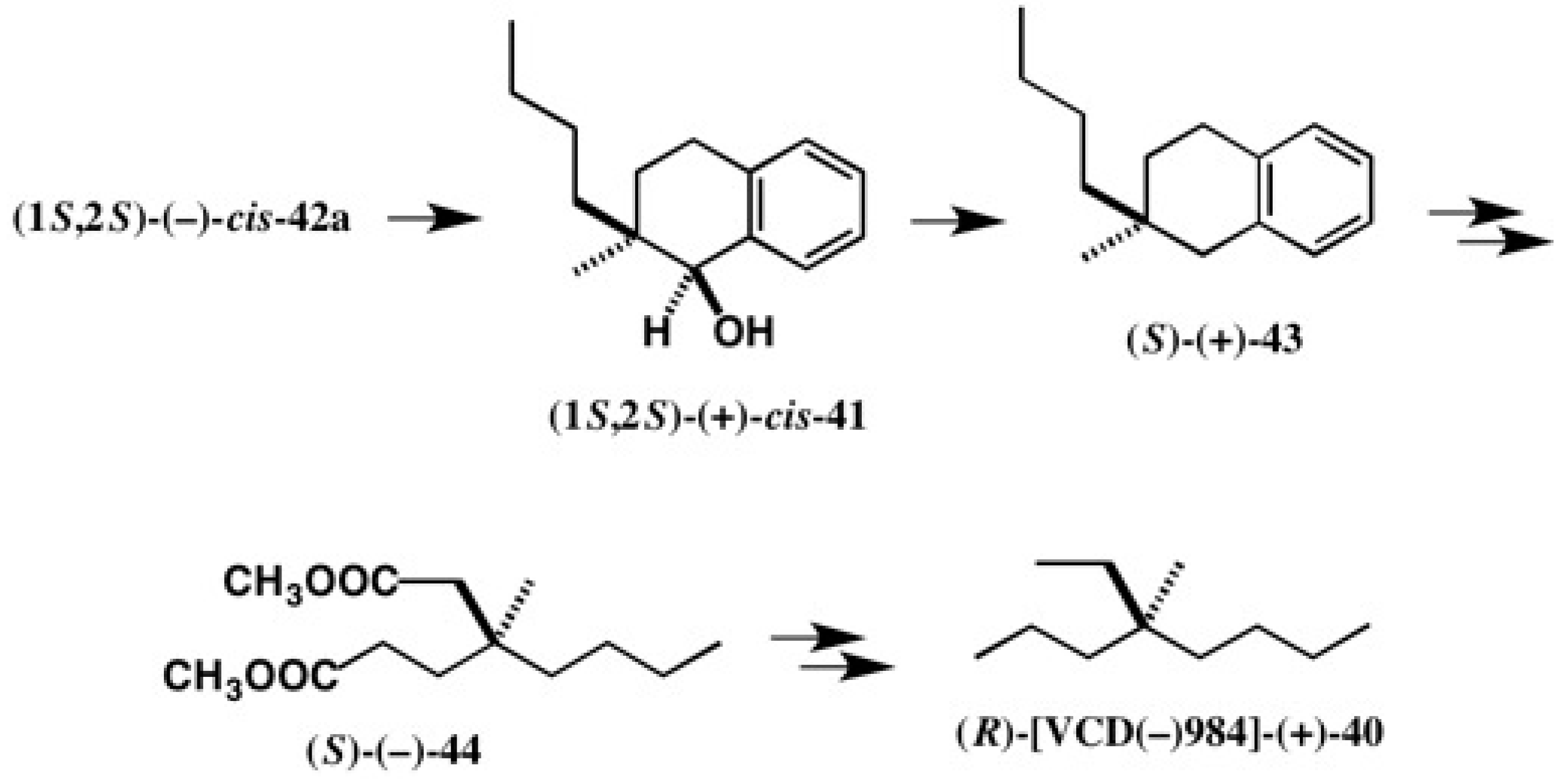
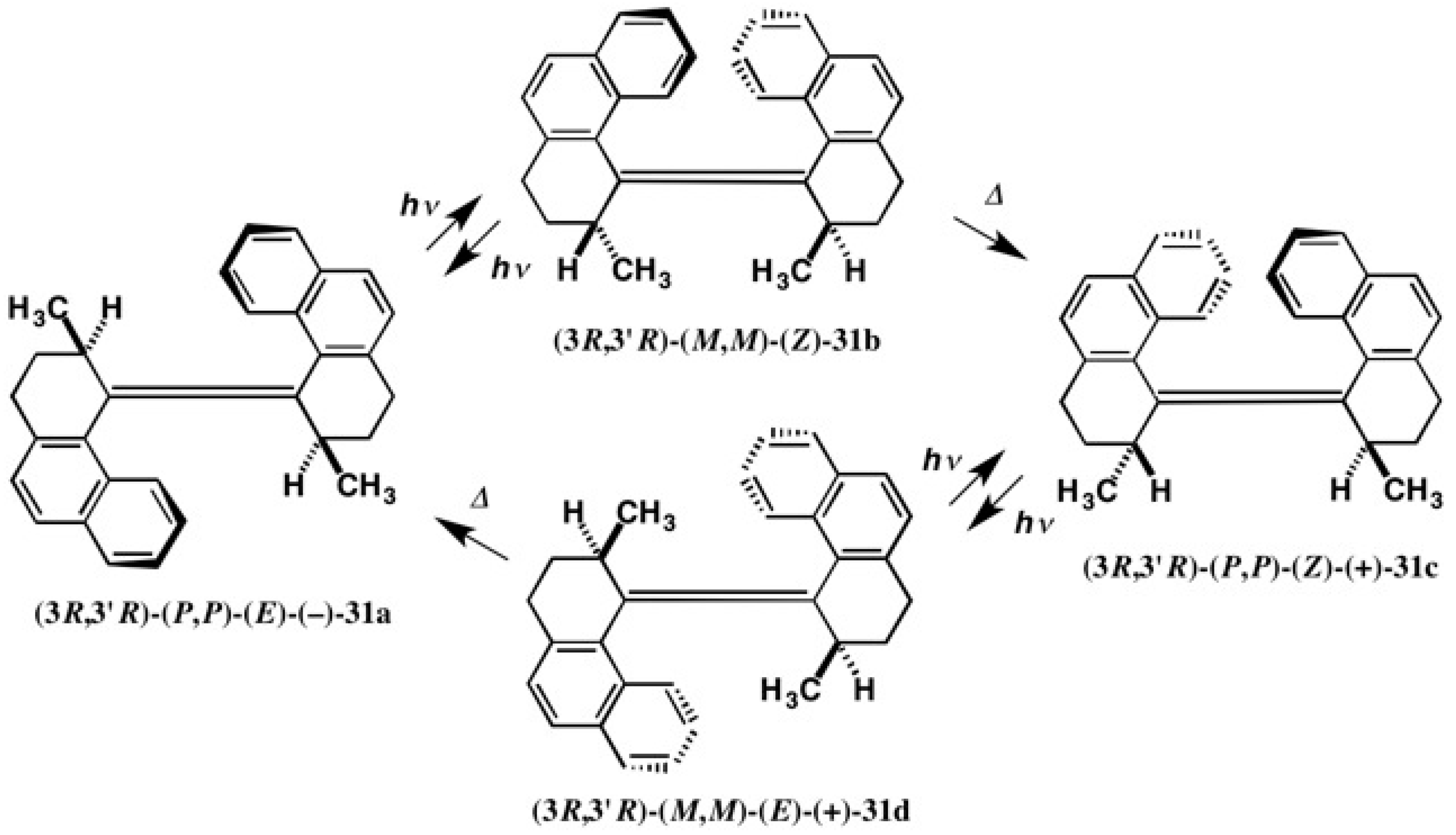
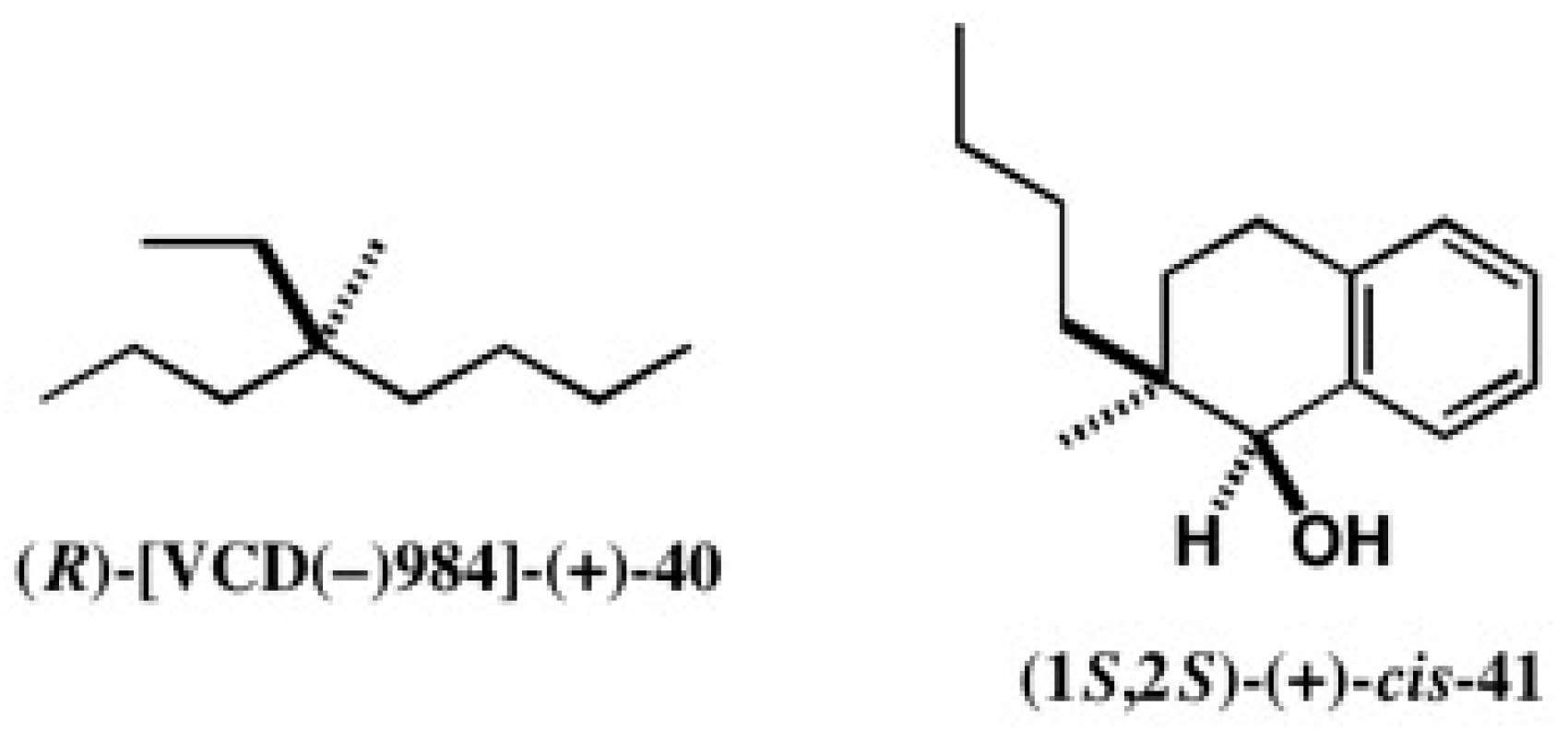
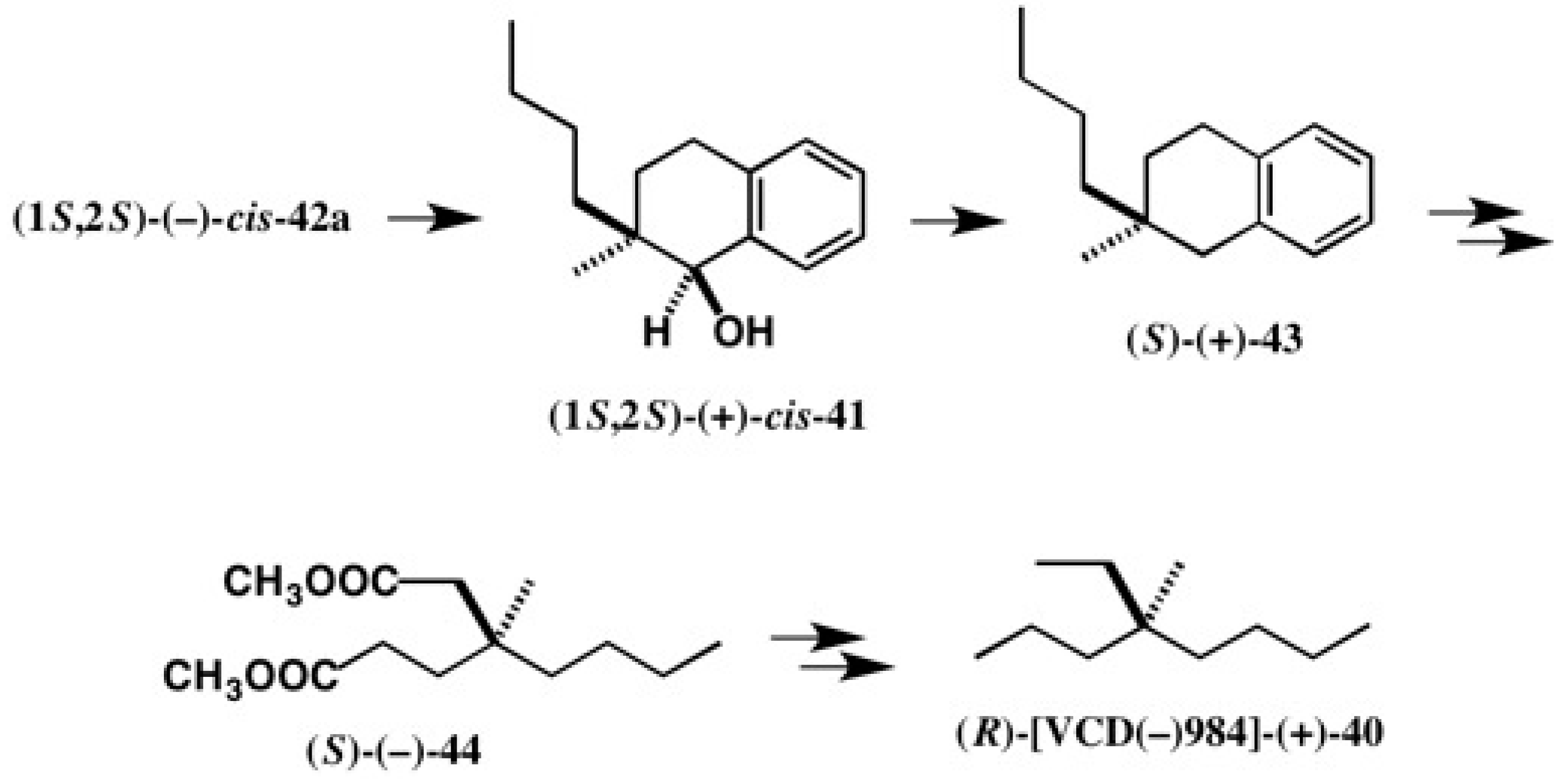
4.1. Synthesis of Chiral Molecular Motor
4.2. Application to Diphenyl Methanols
4.3. Synthesis of a Cryptochiral Hydrocarbon
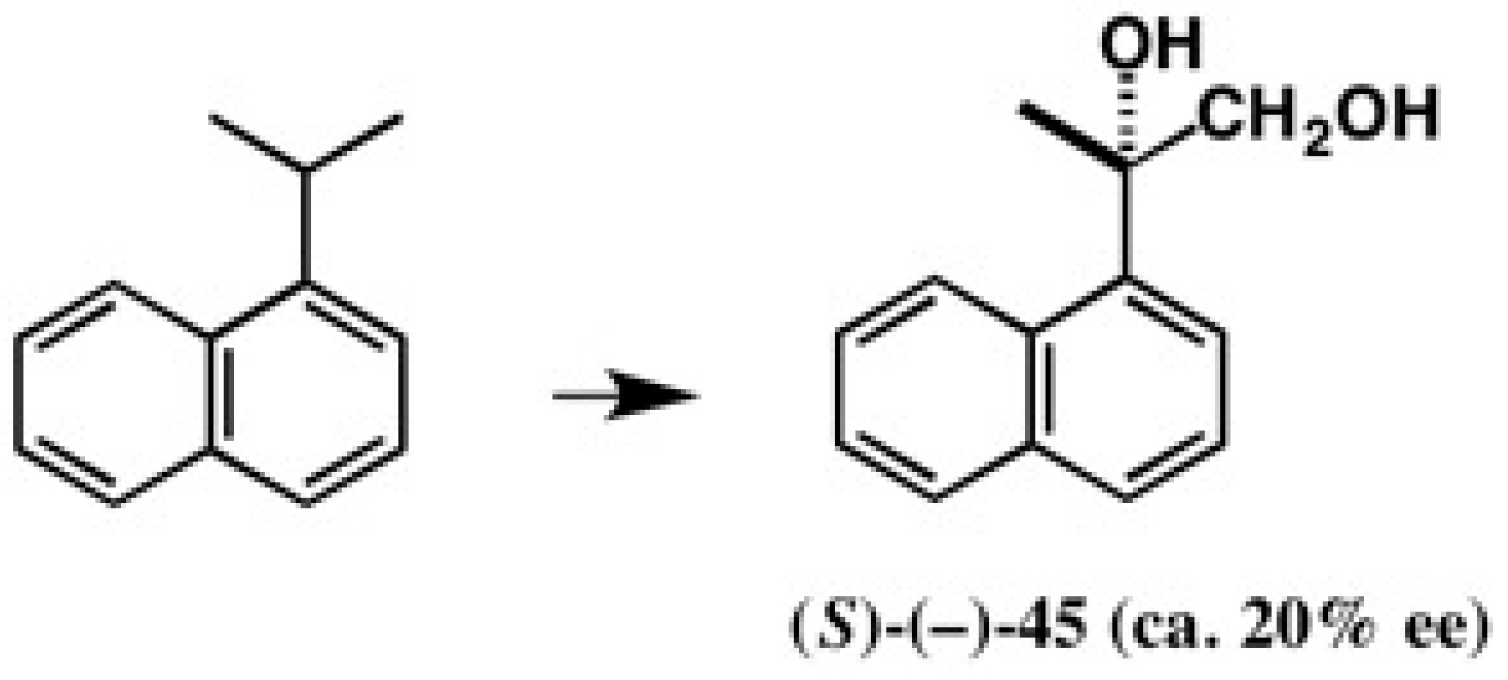
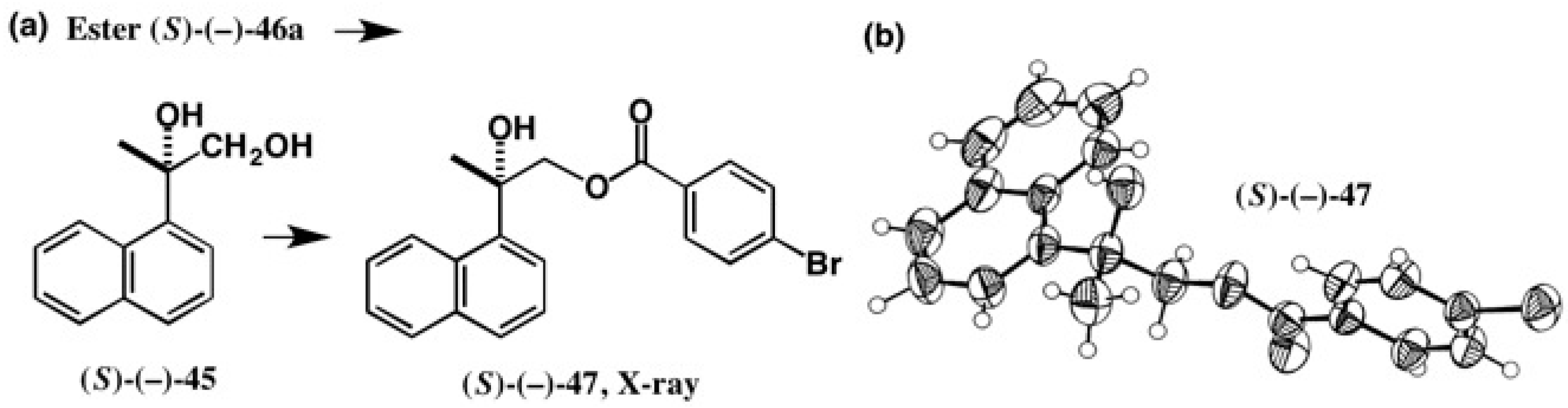
4.4. Application to 2-(1-Naphthyl)Propane-1,2-Diol
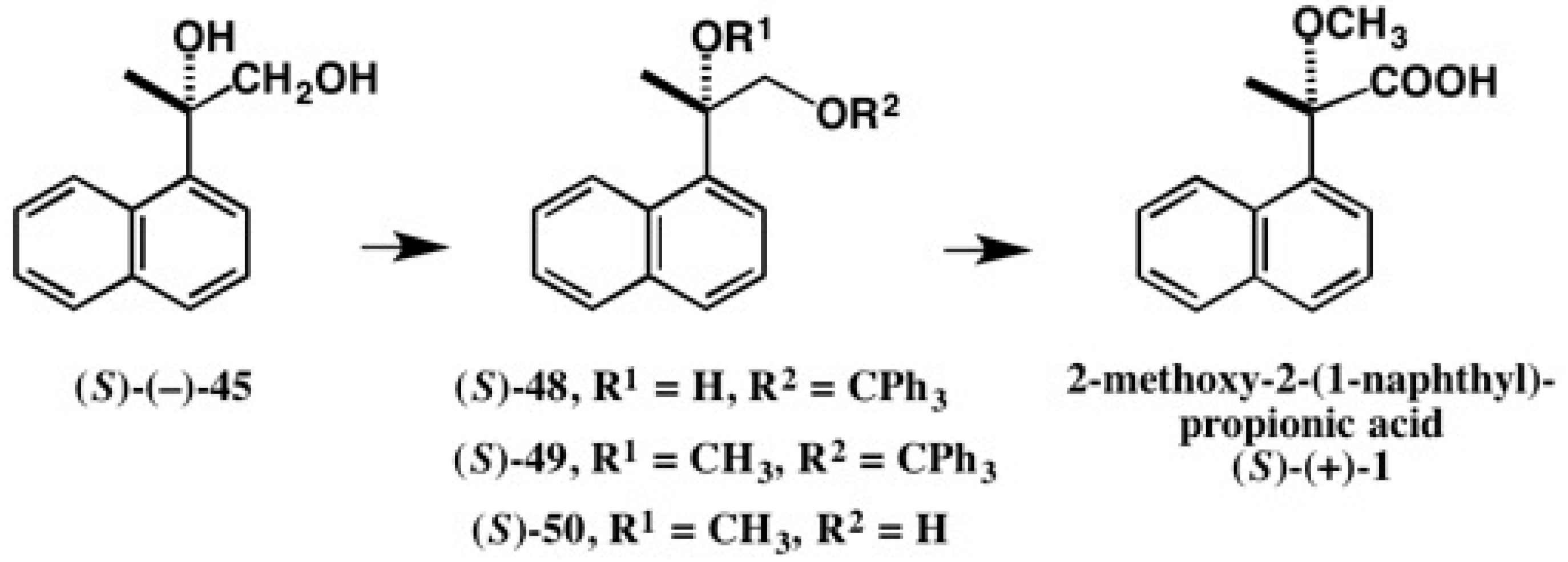
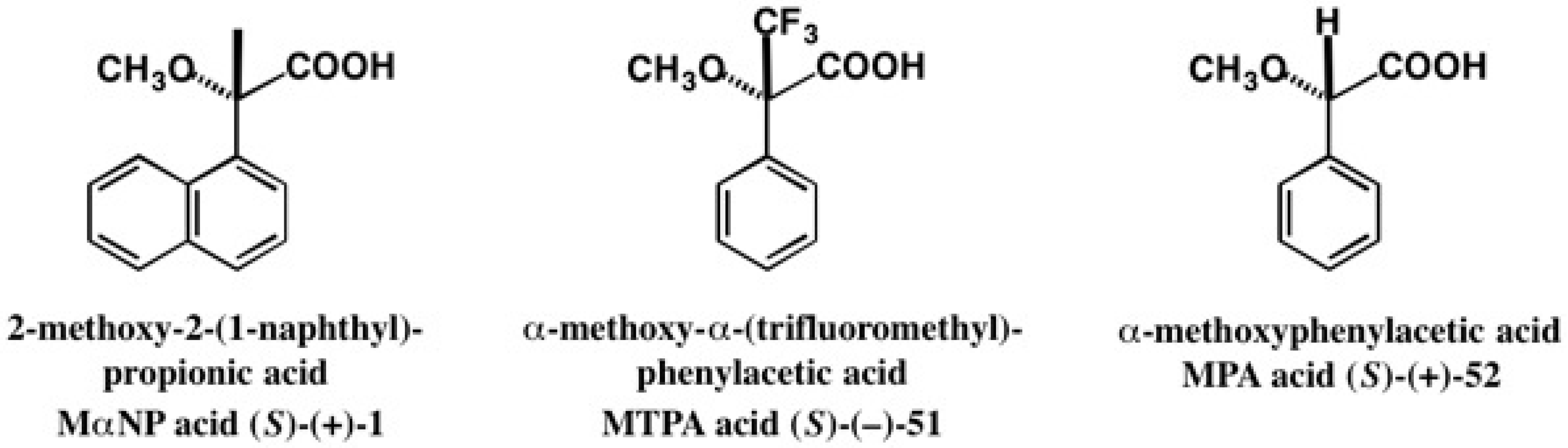
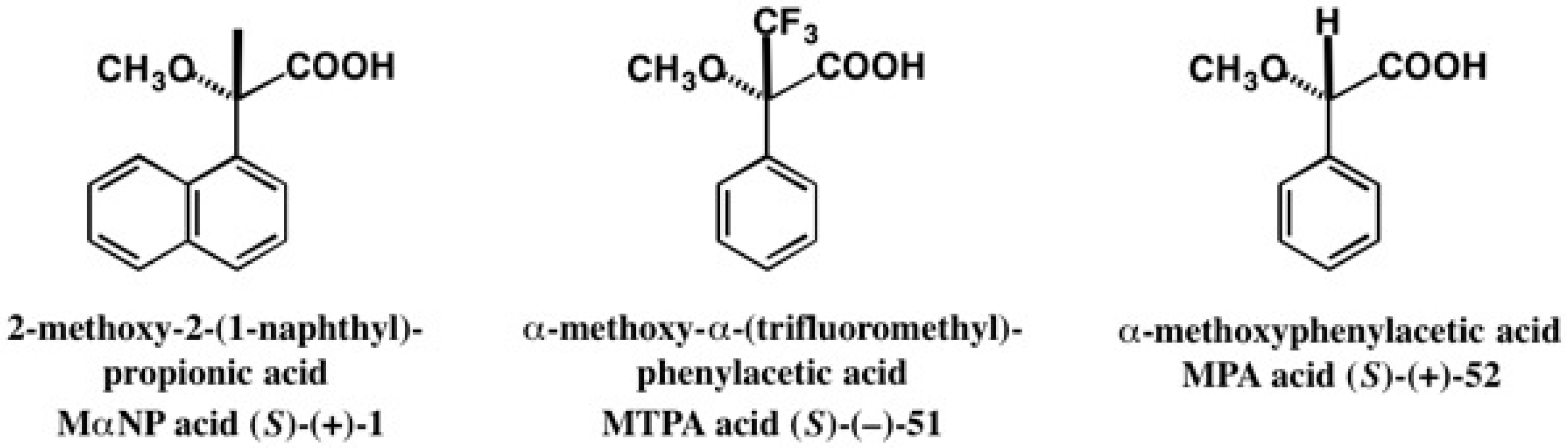
5. Use of 2-Methoxy-2-(1-Naphthyl)Propionic Acid (MαNP Acid) for Alcohols—The MαNP Acid Method
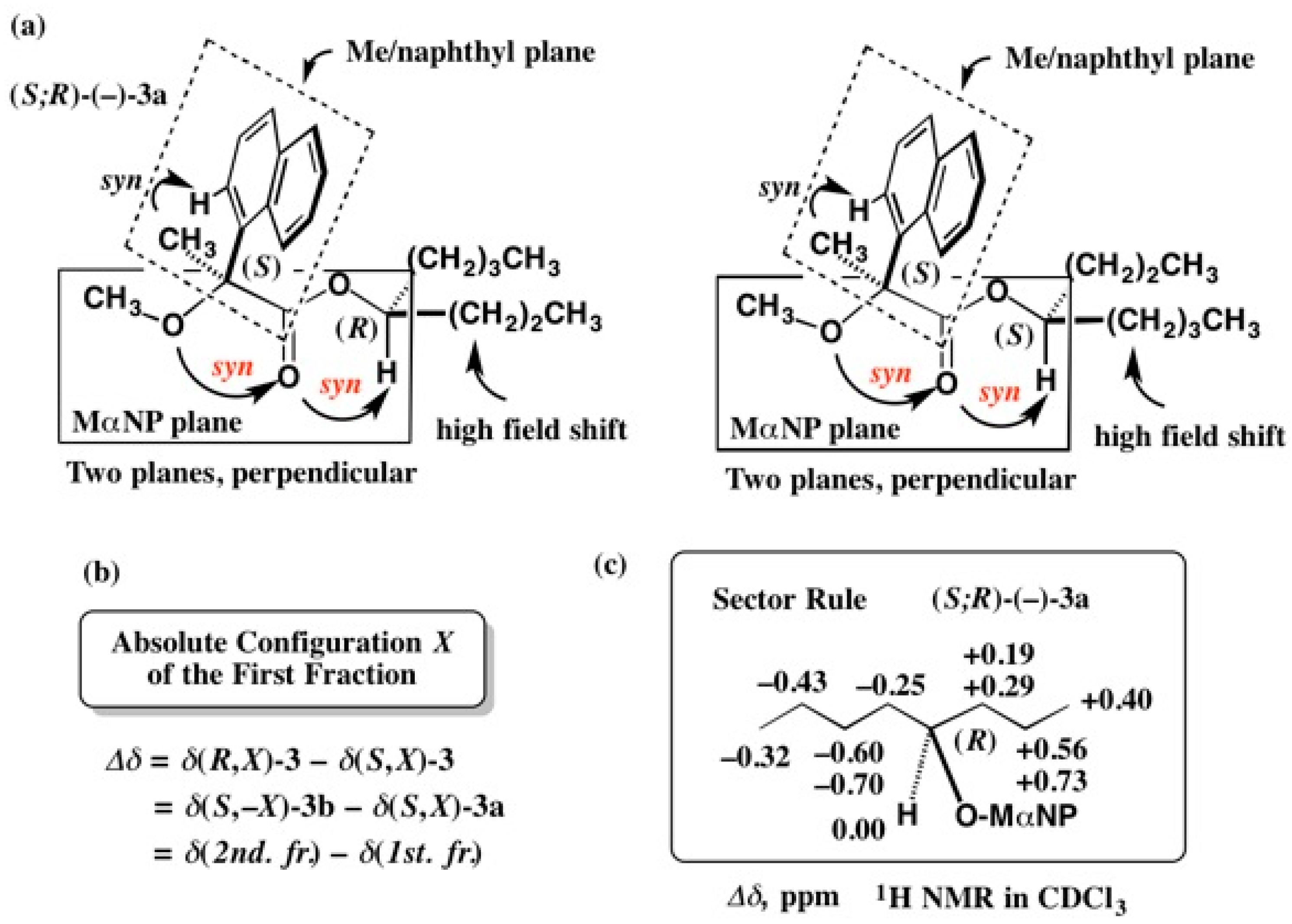
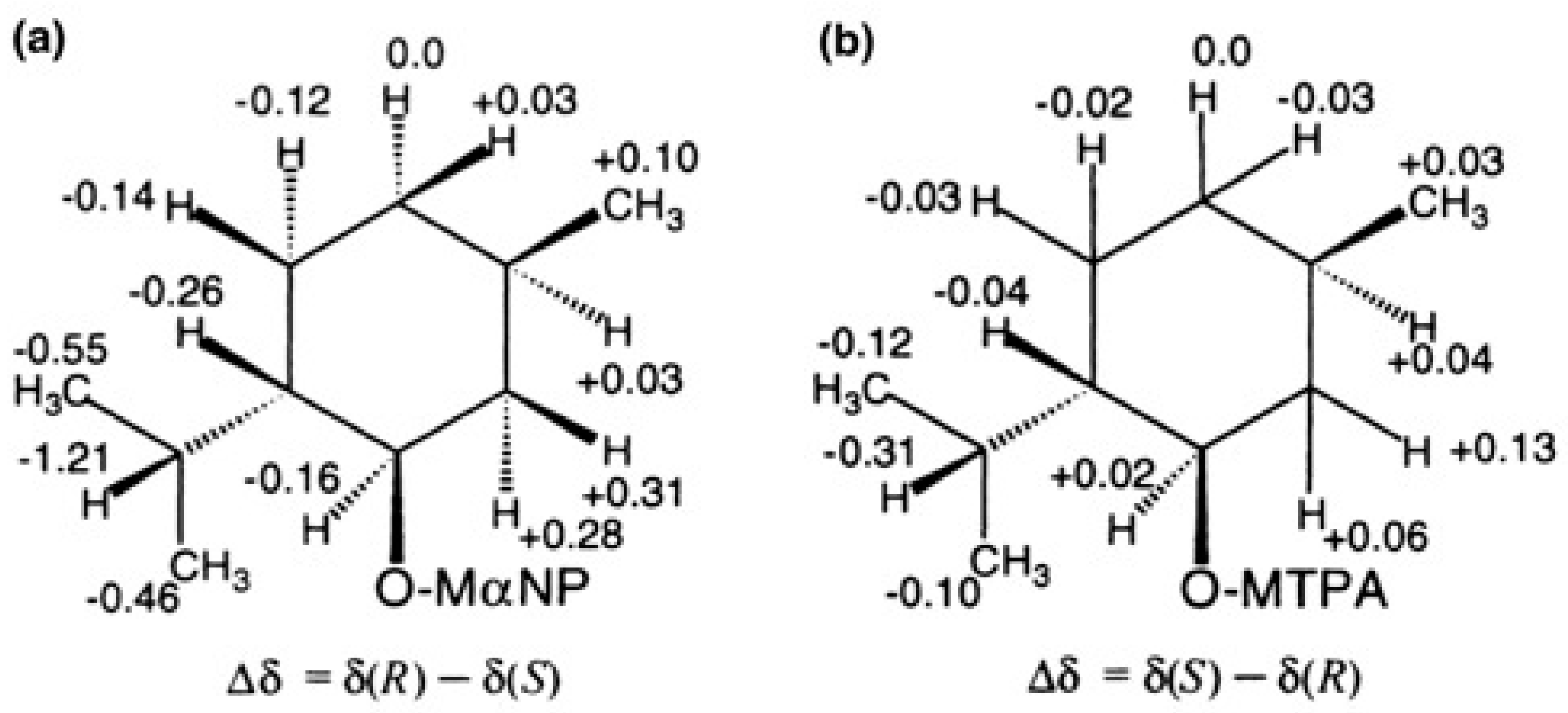
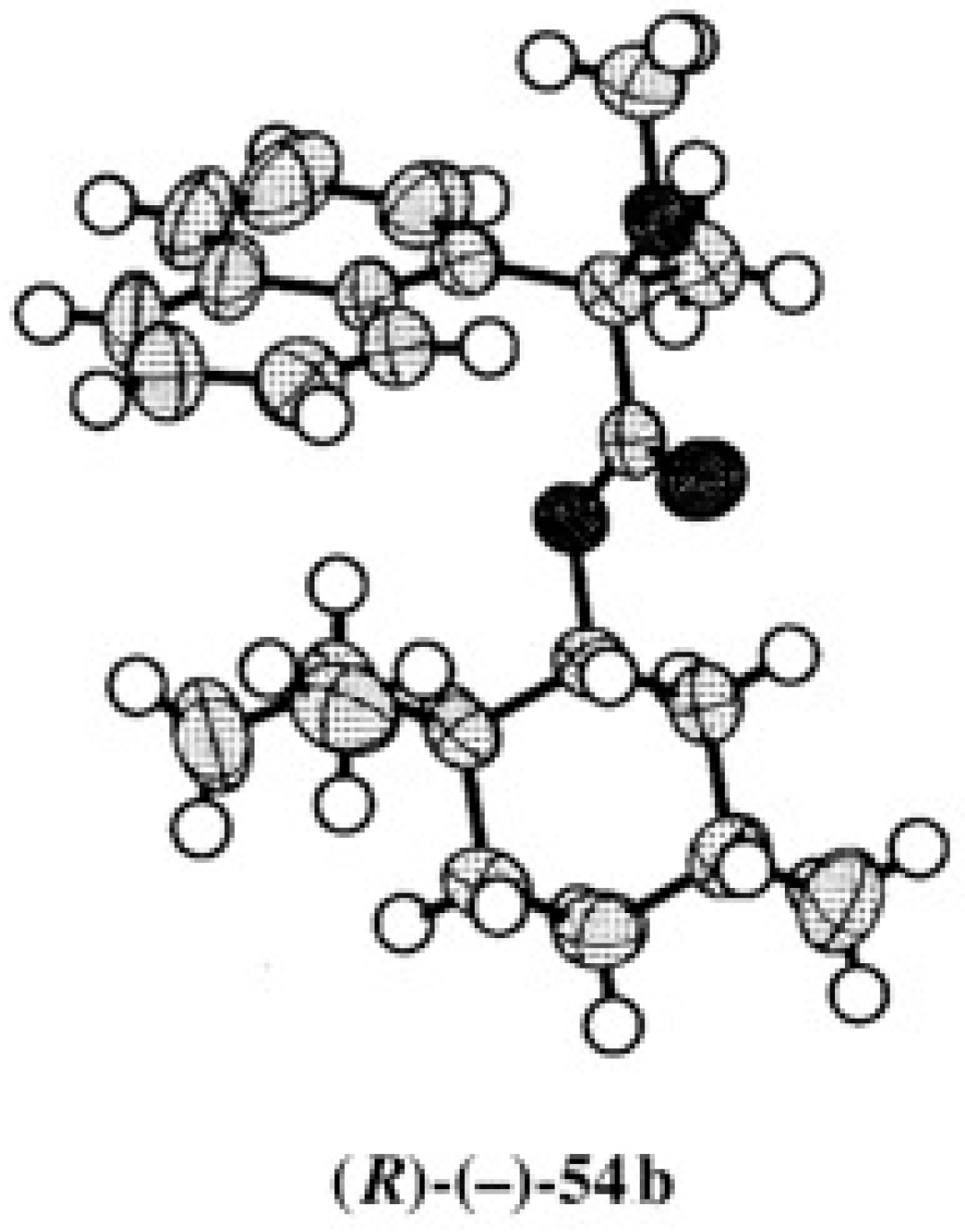
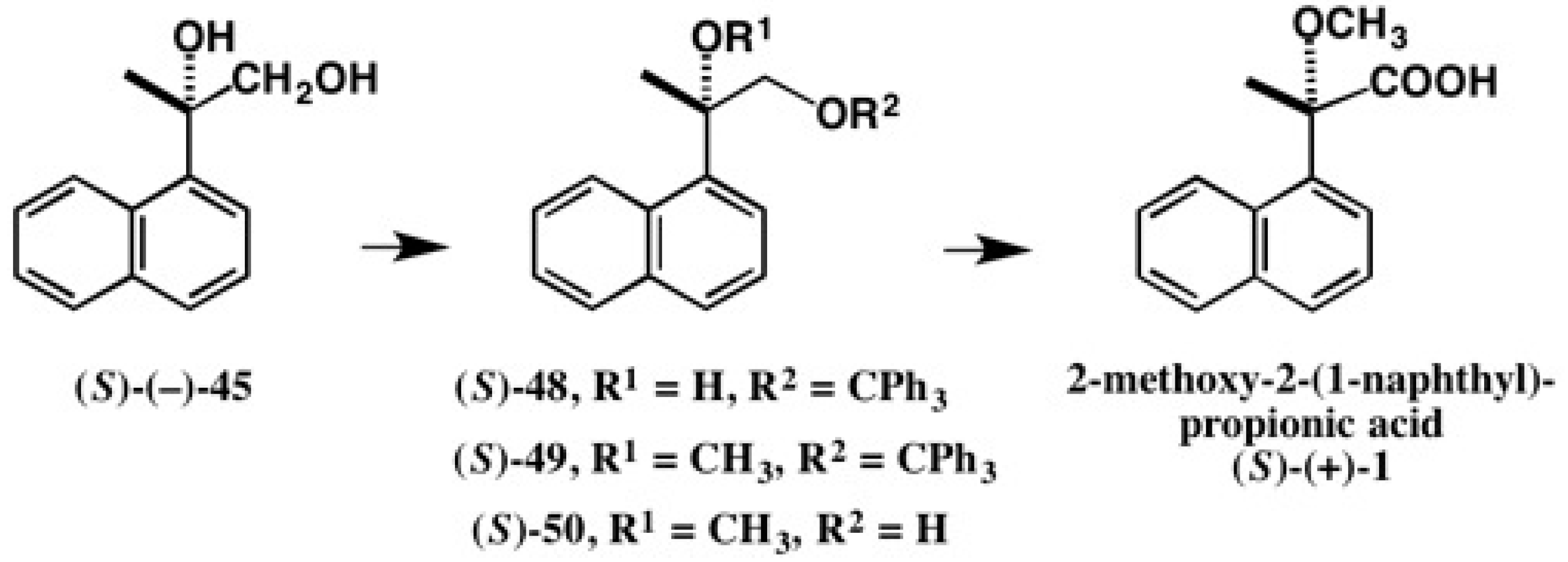
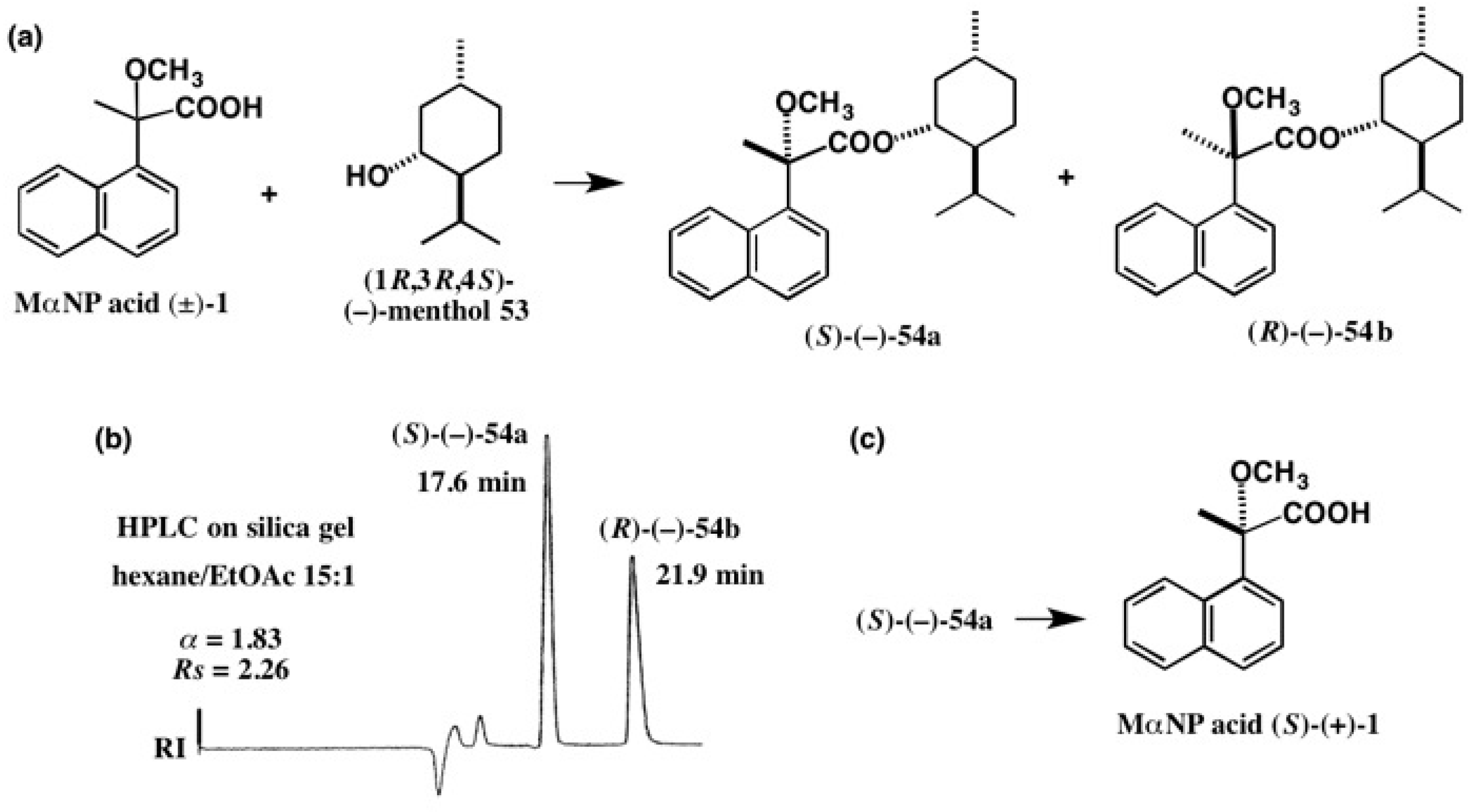
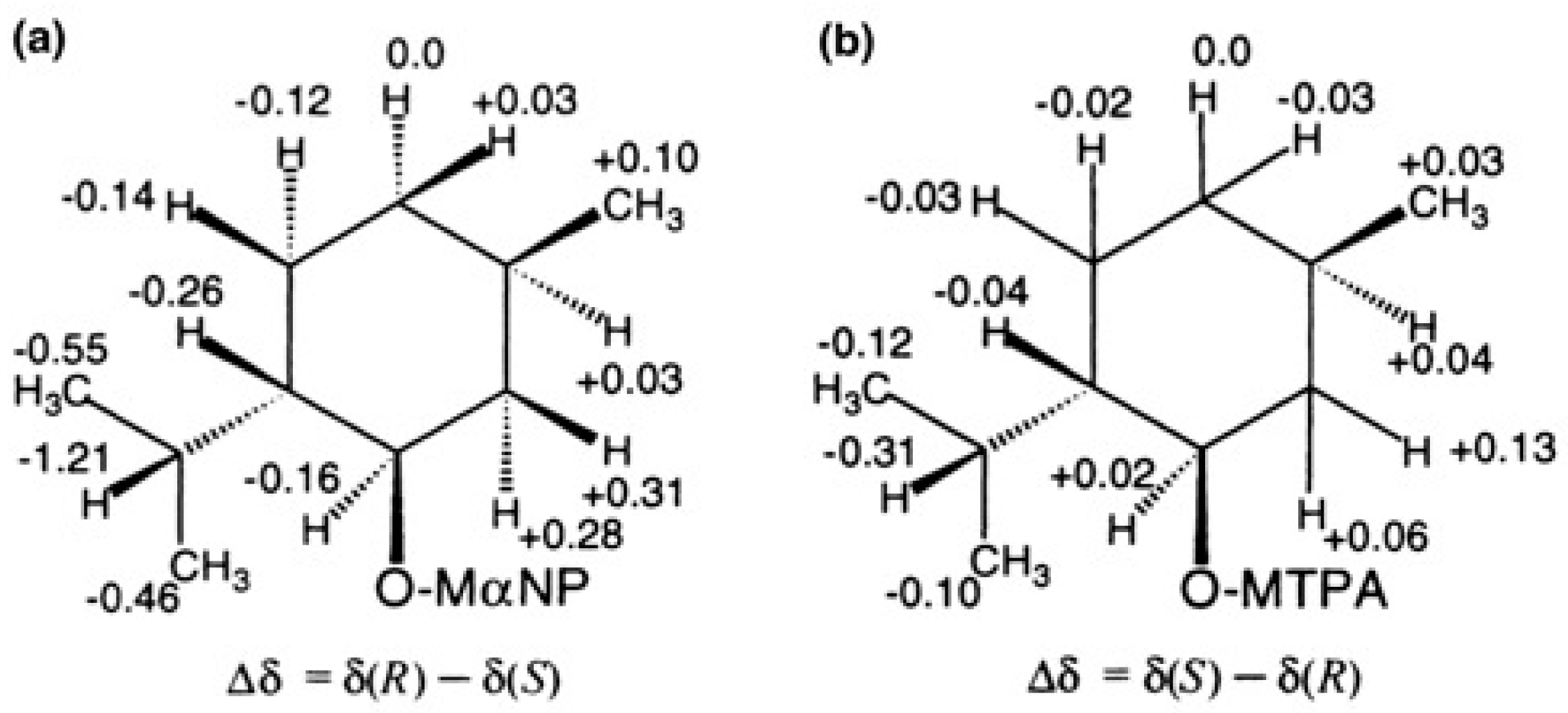
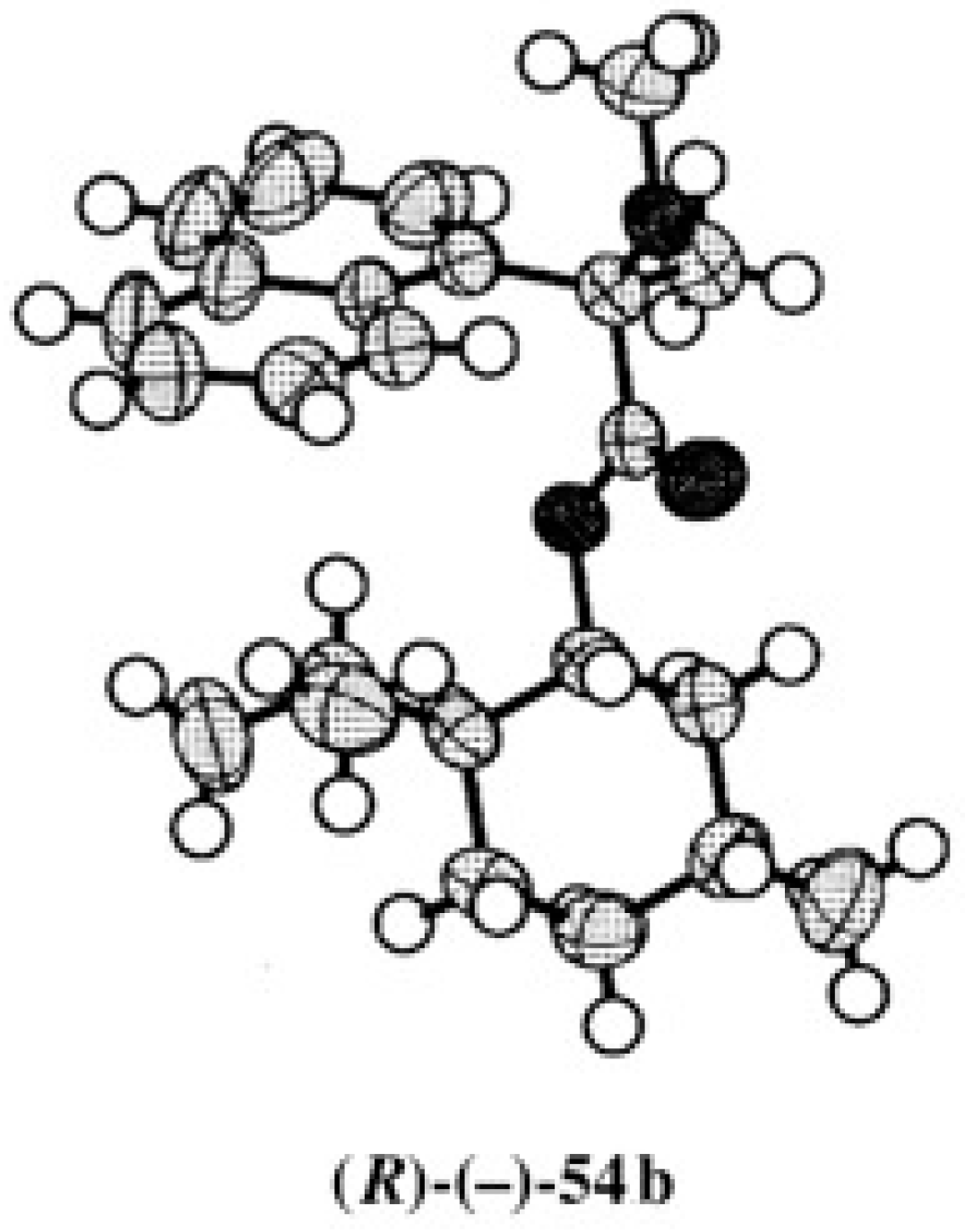
5.1. Synthesis of Enantiopure MαNP Acid, HPLC Separation, and AC Determination by 1H-NMR Diamagnetic Anisotropy
5.2. Application of the MαNP Acid Method to Aromatic Alcohol
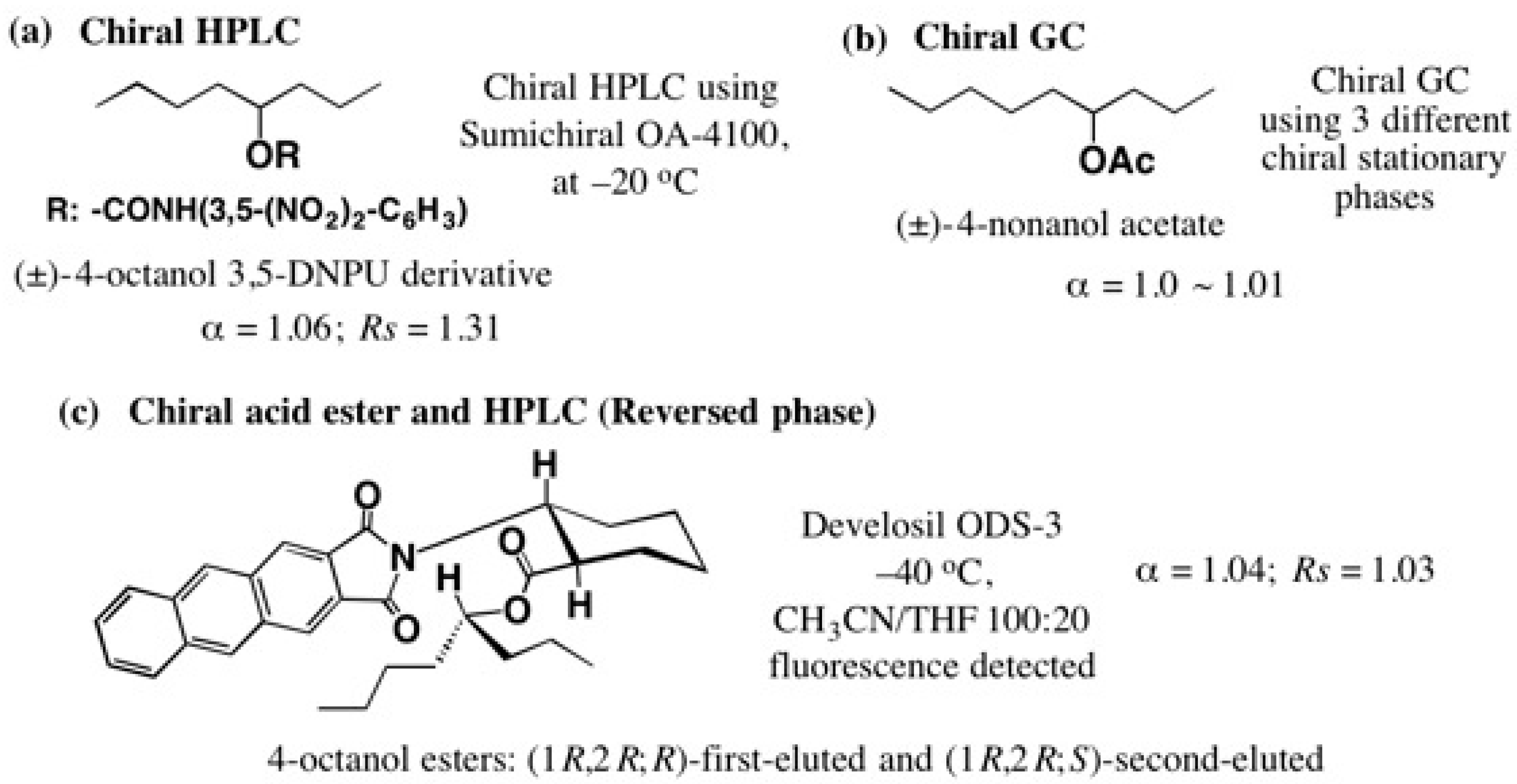
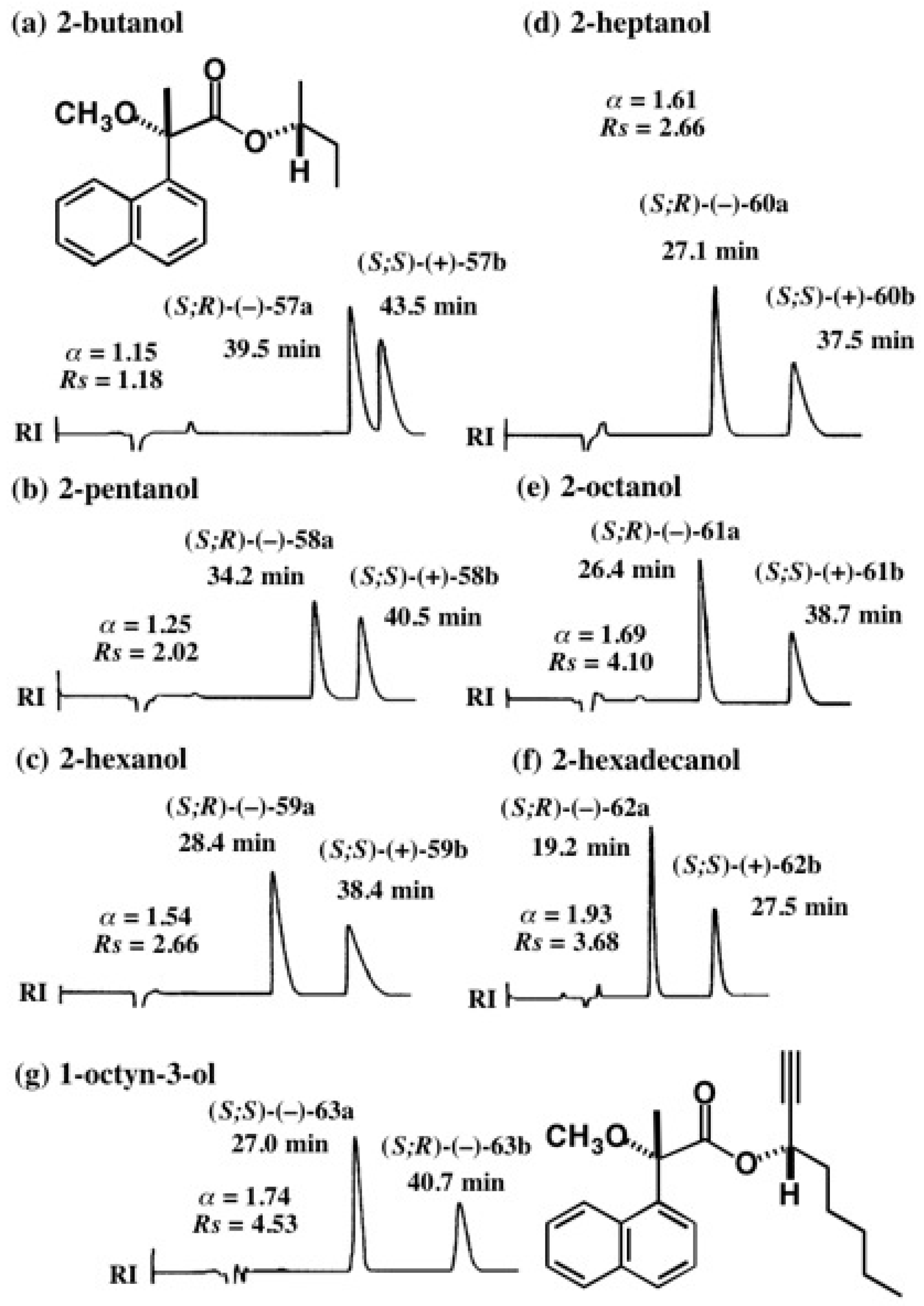
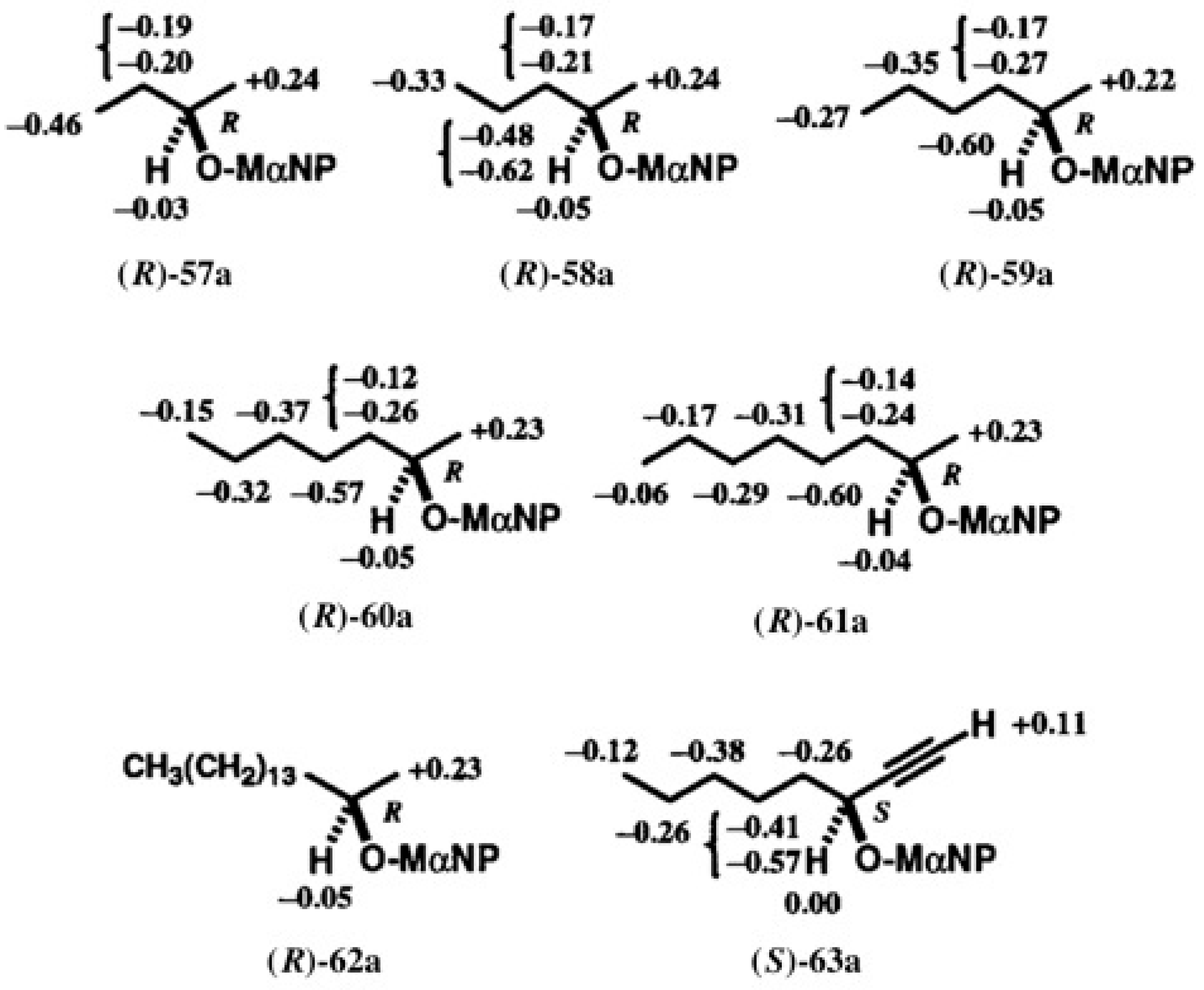
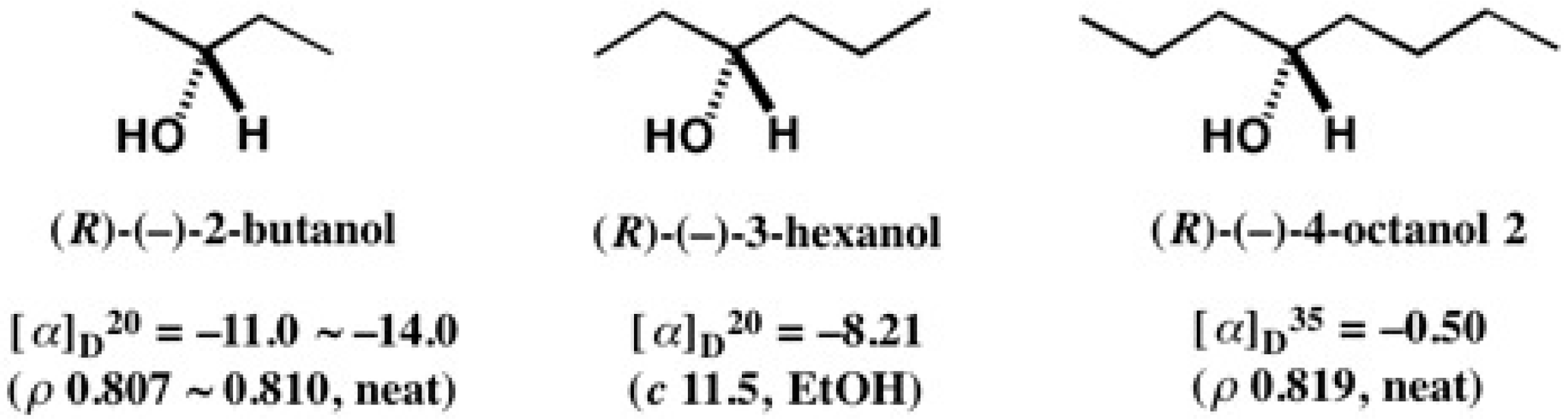
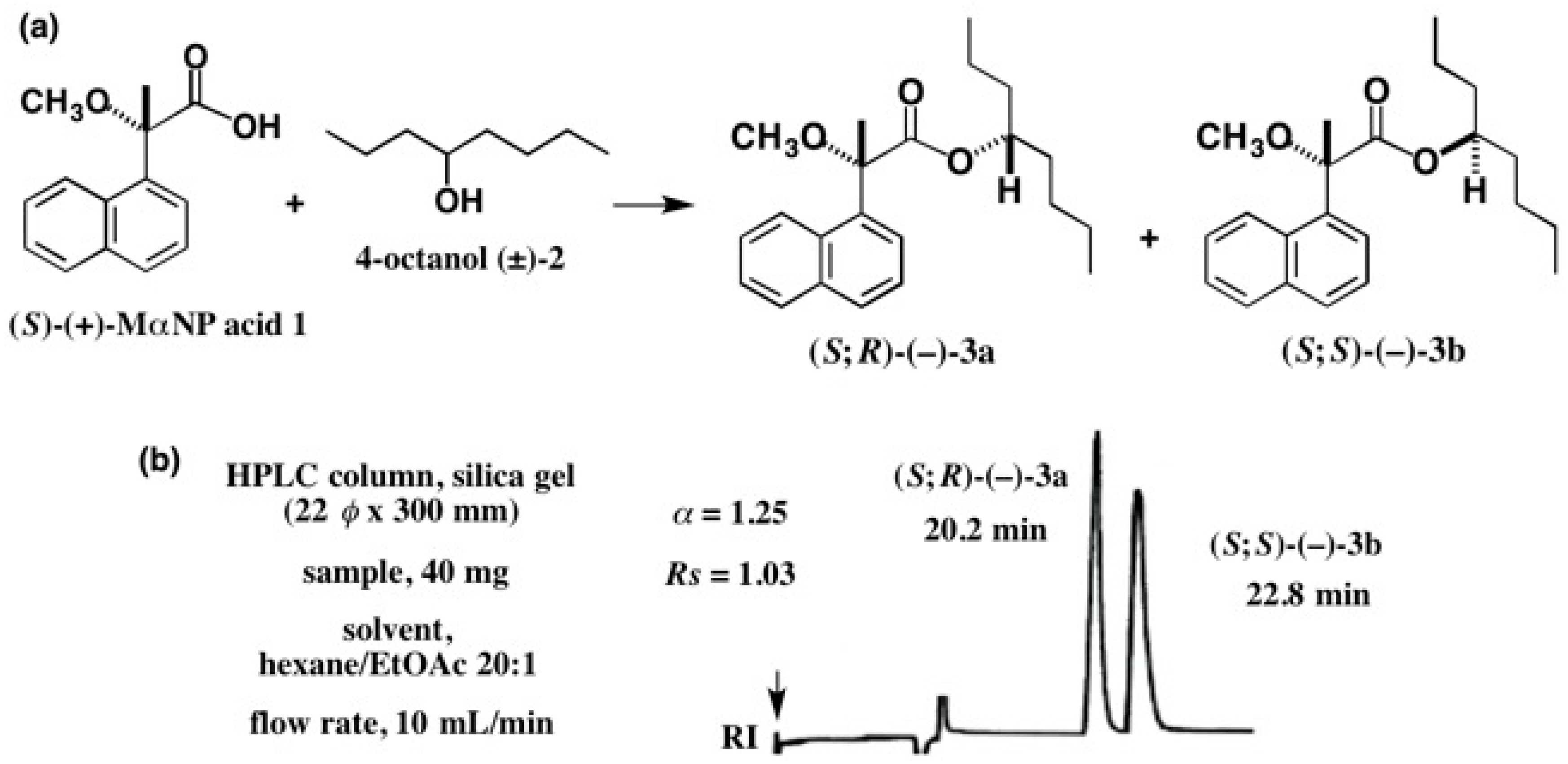
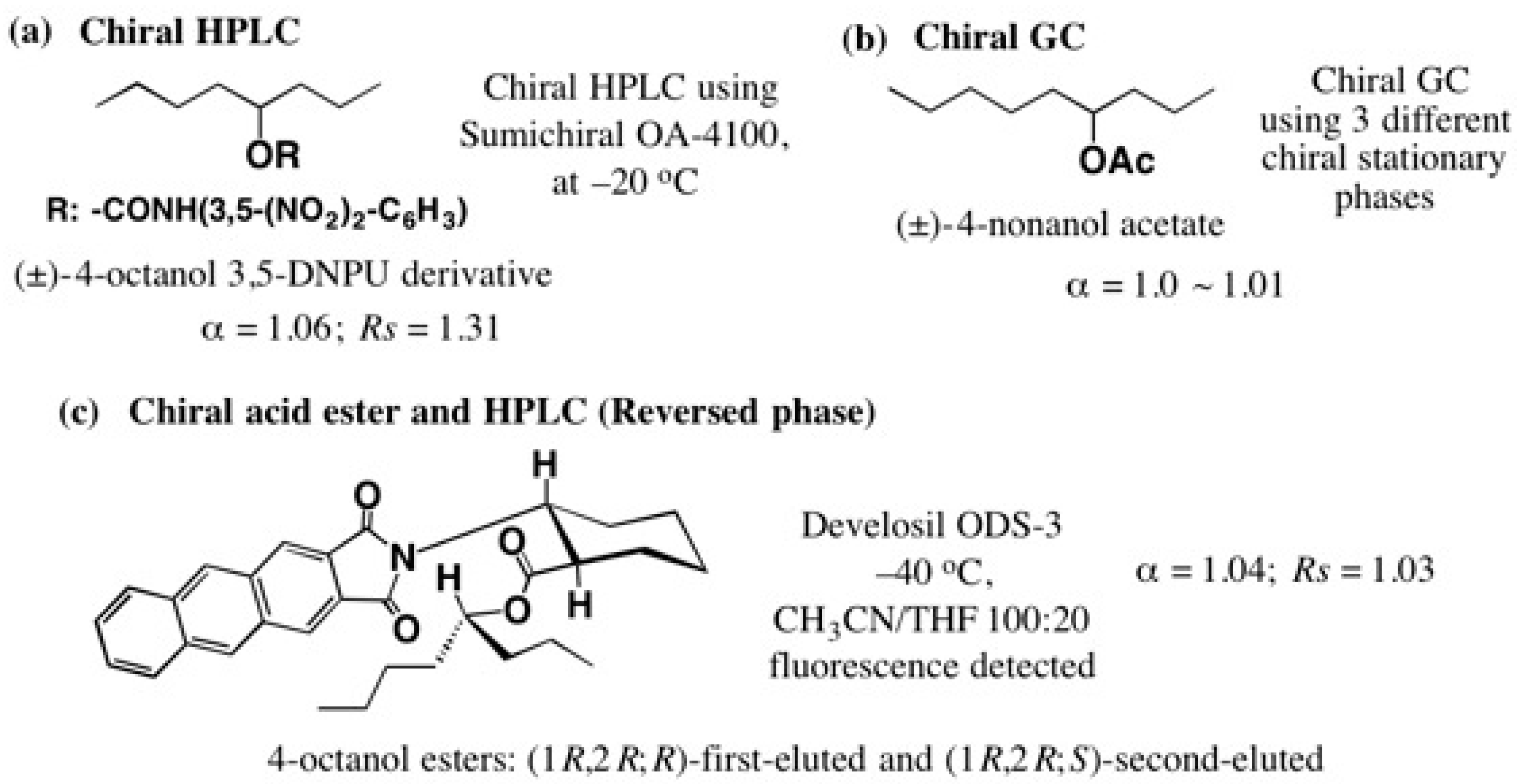
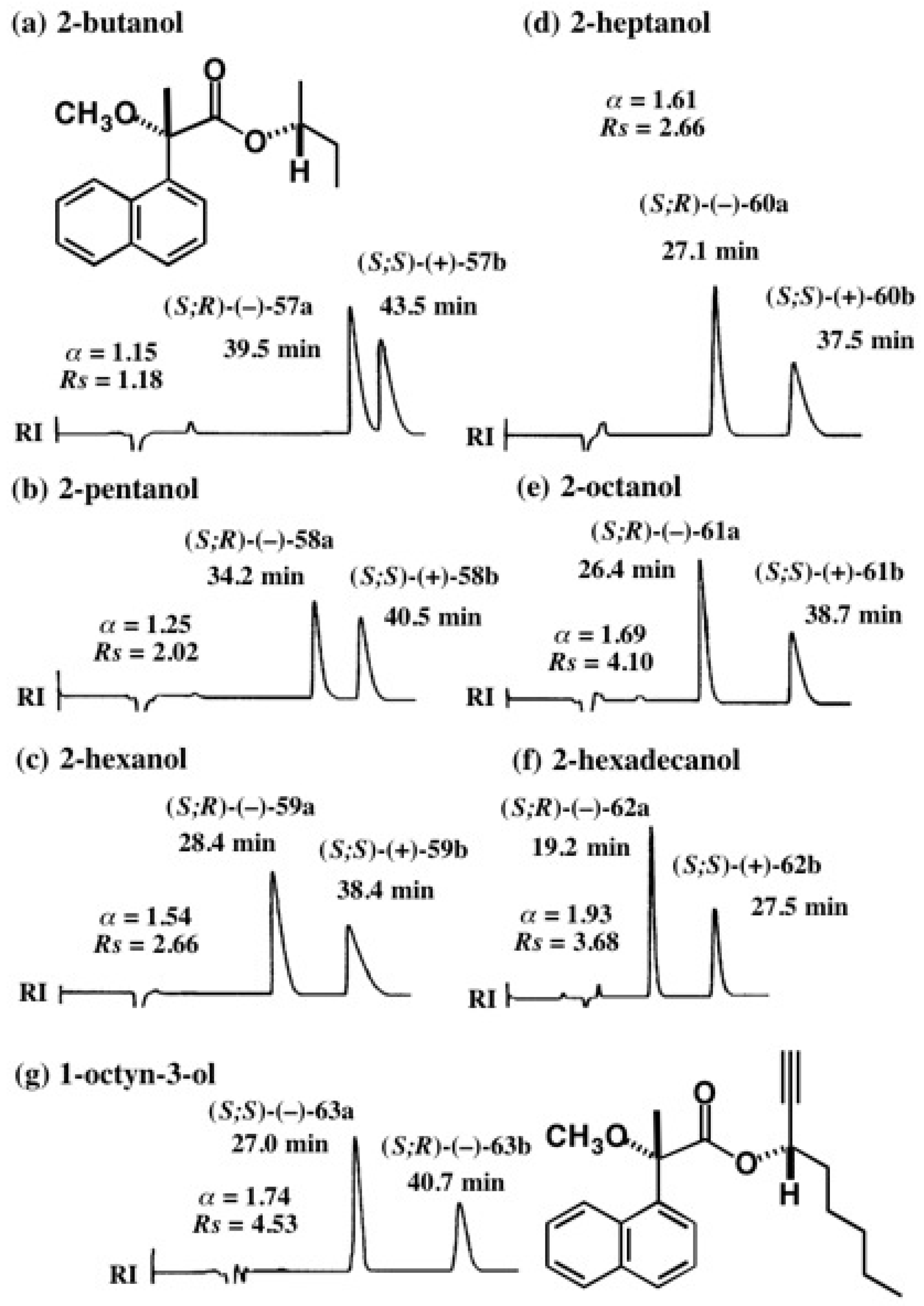
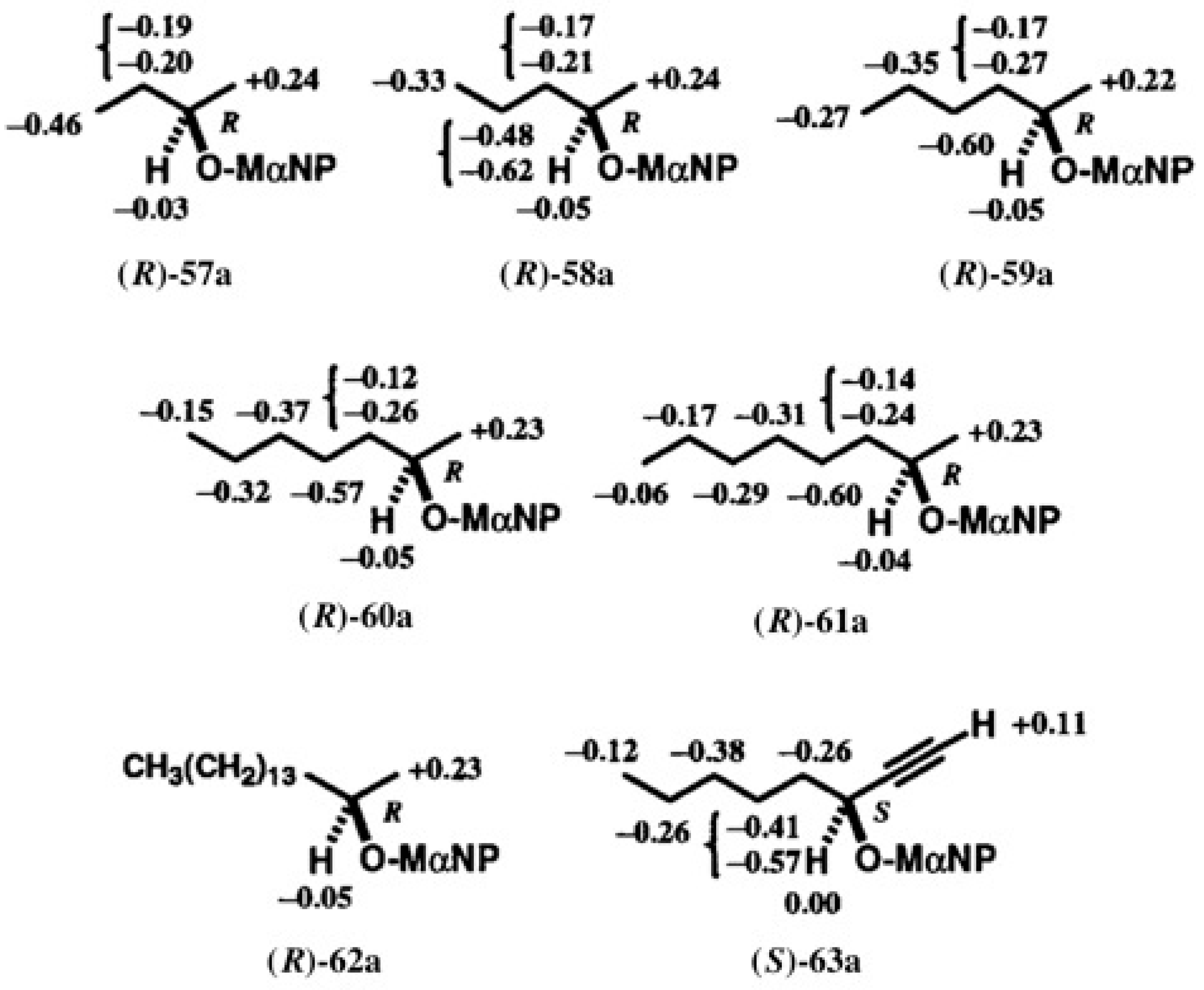
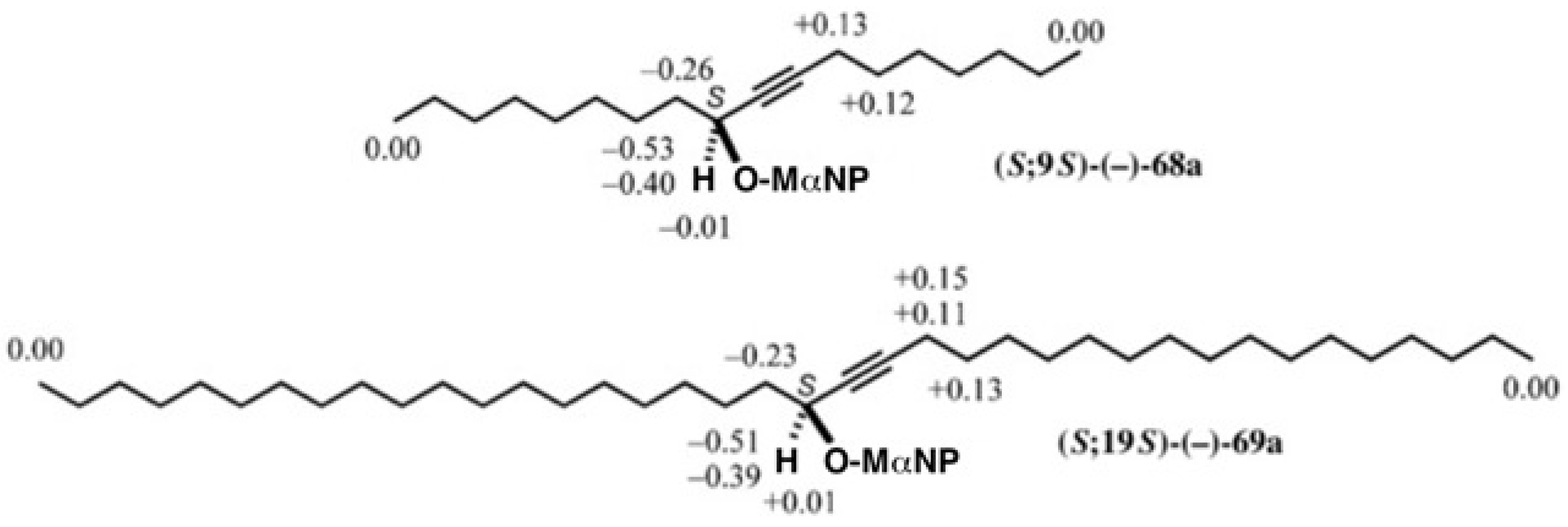
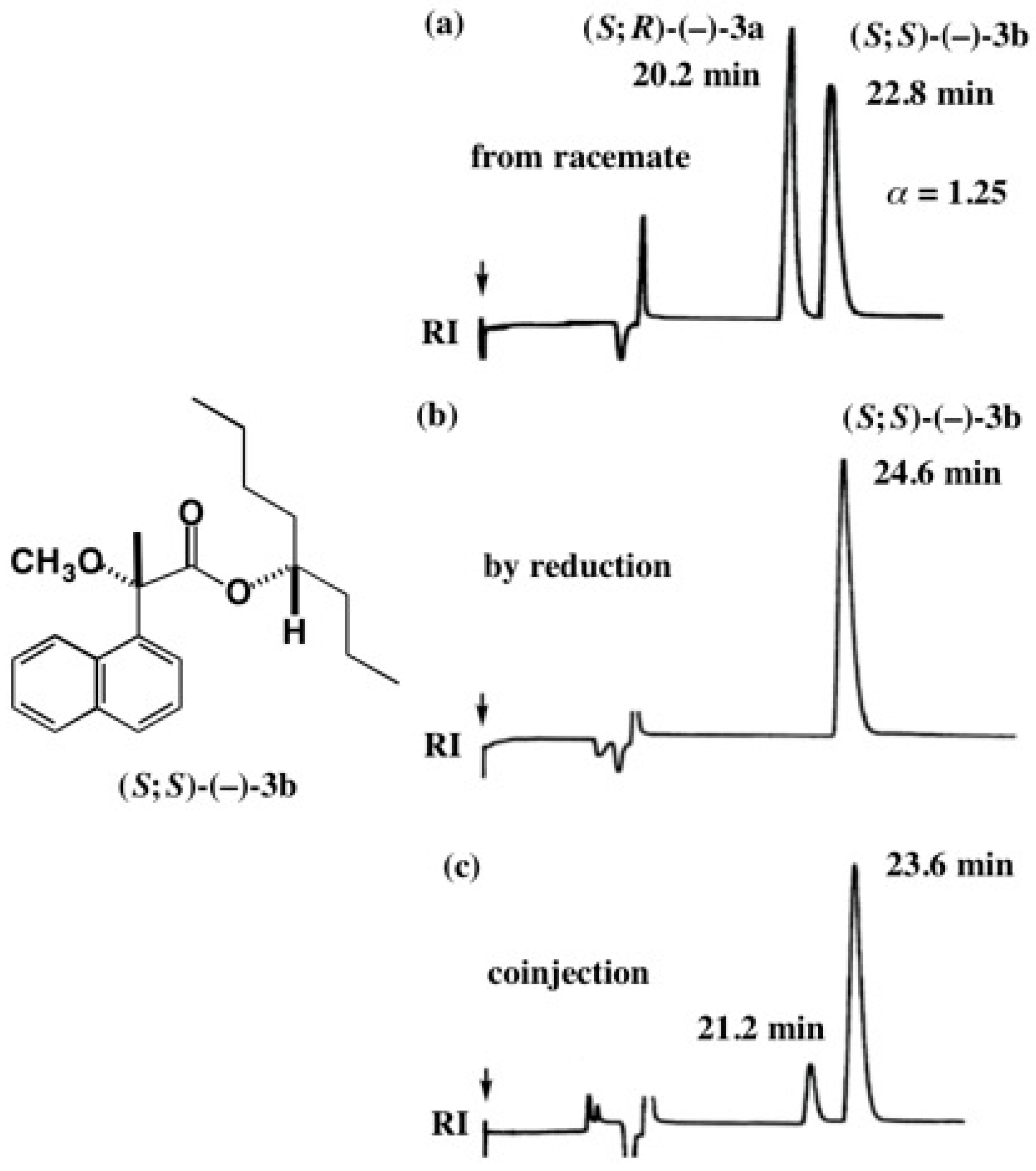
5.3. Application of Aliphatic Chain Alcohols
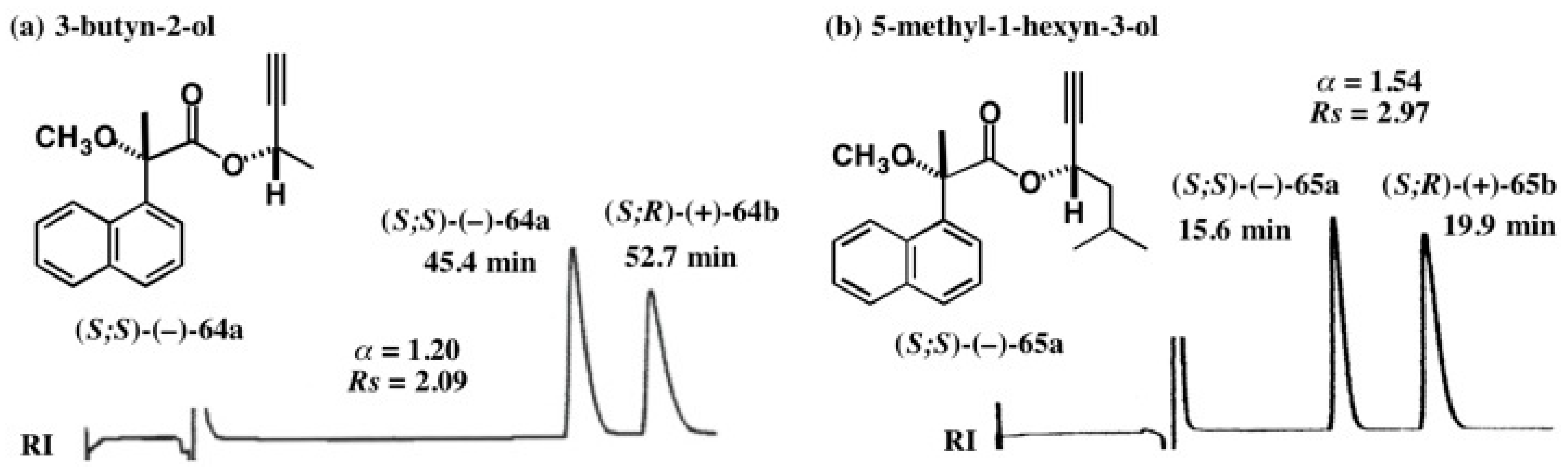
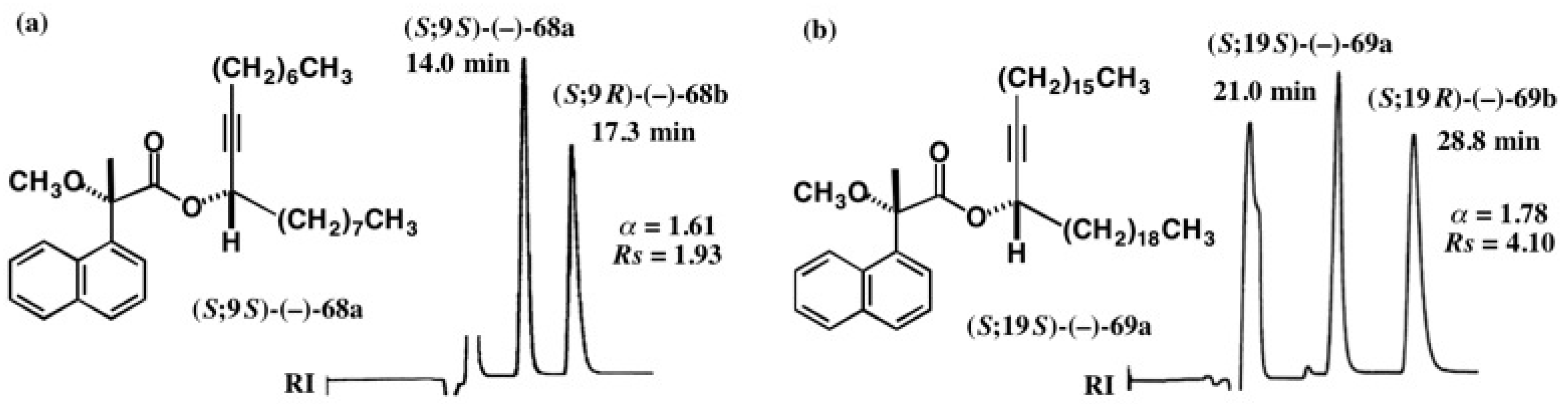
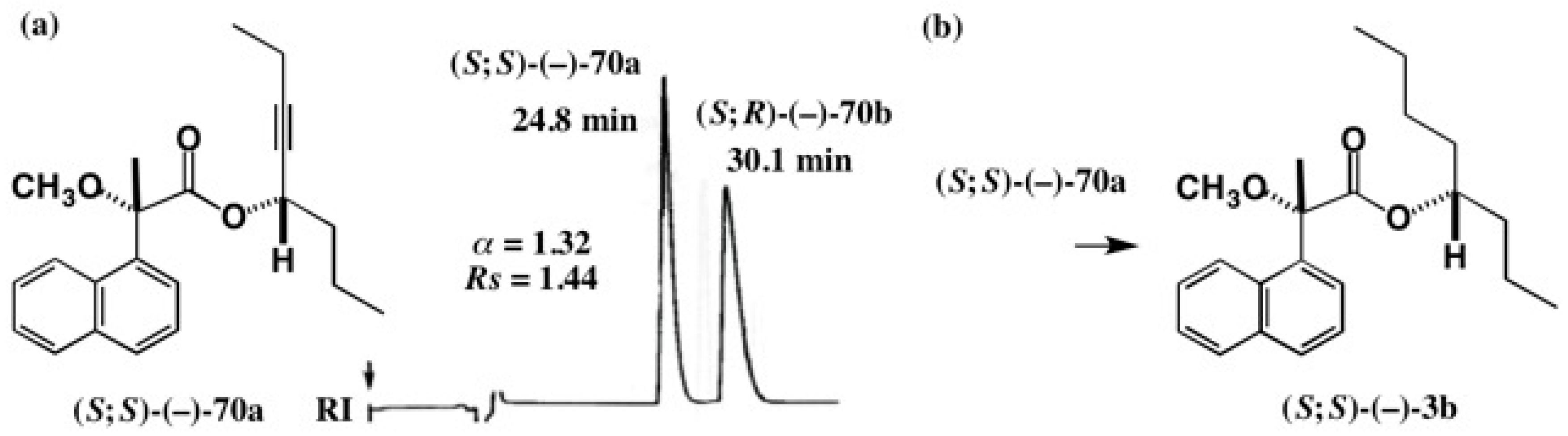
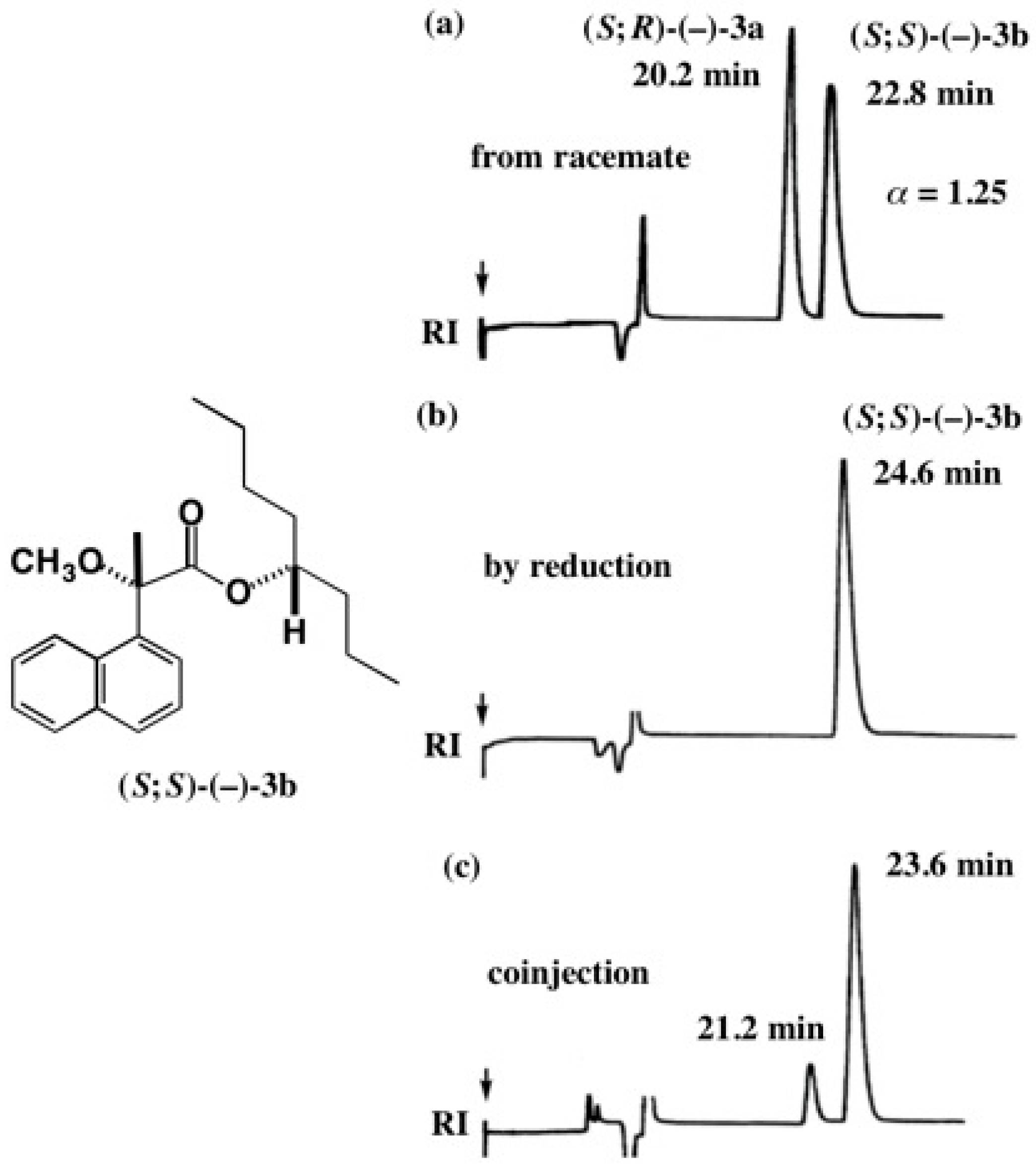
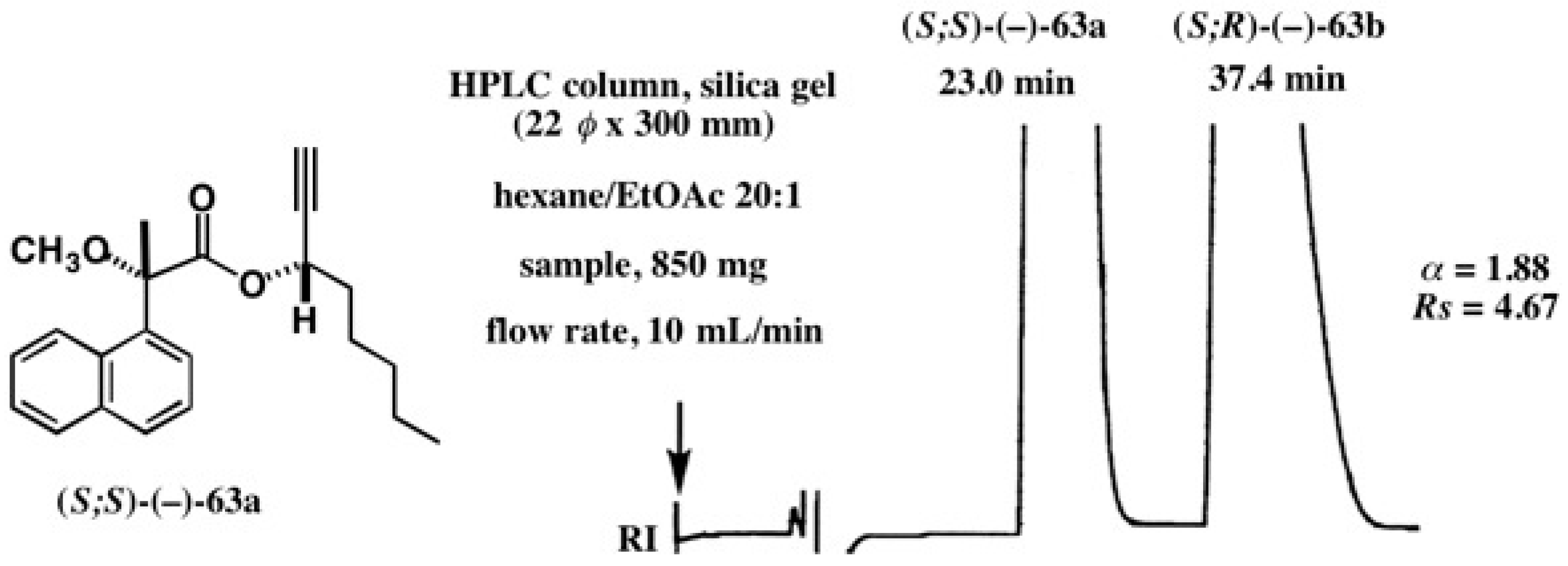
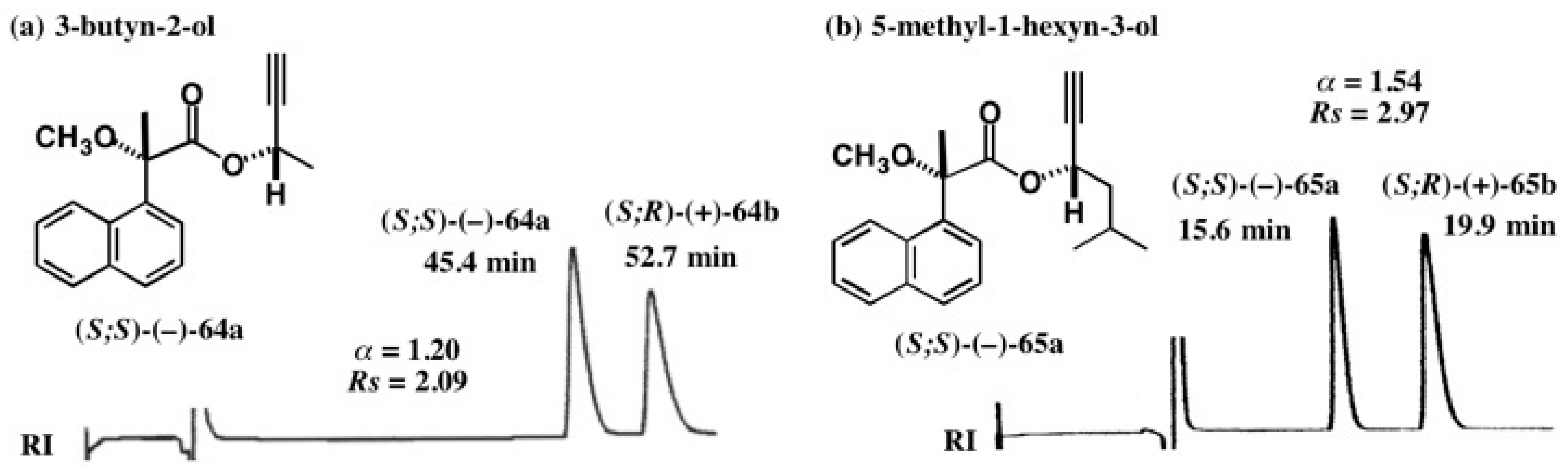
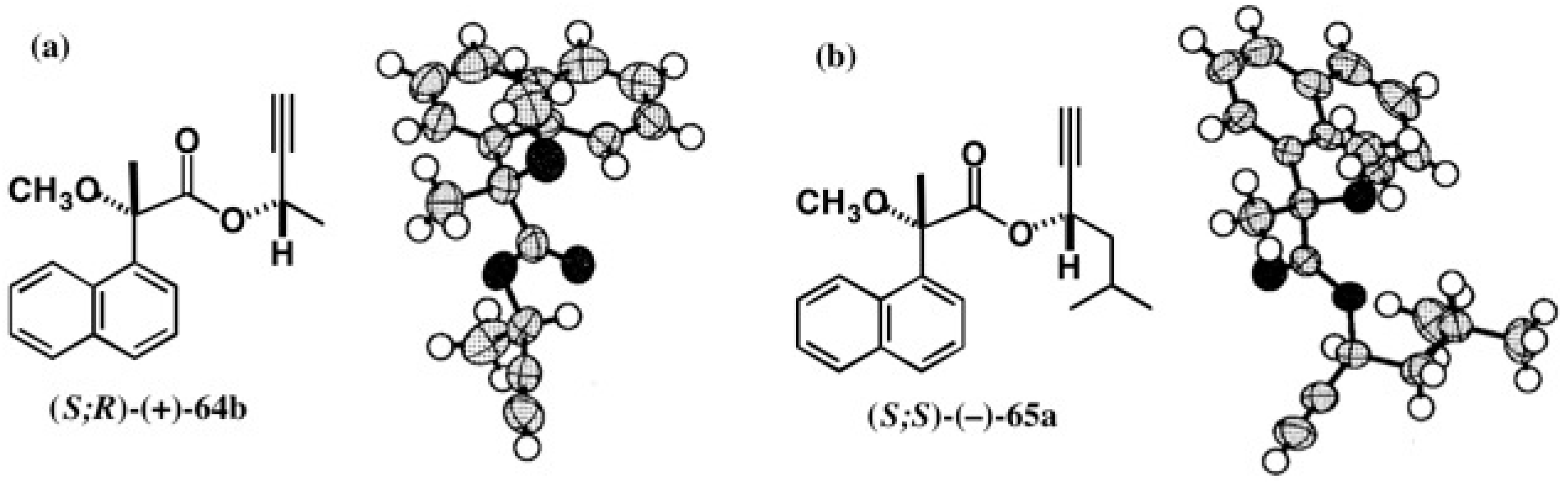
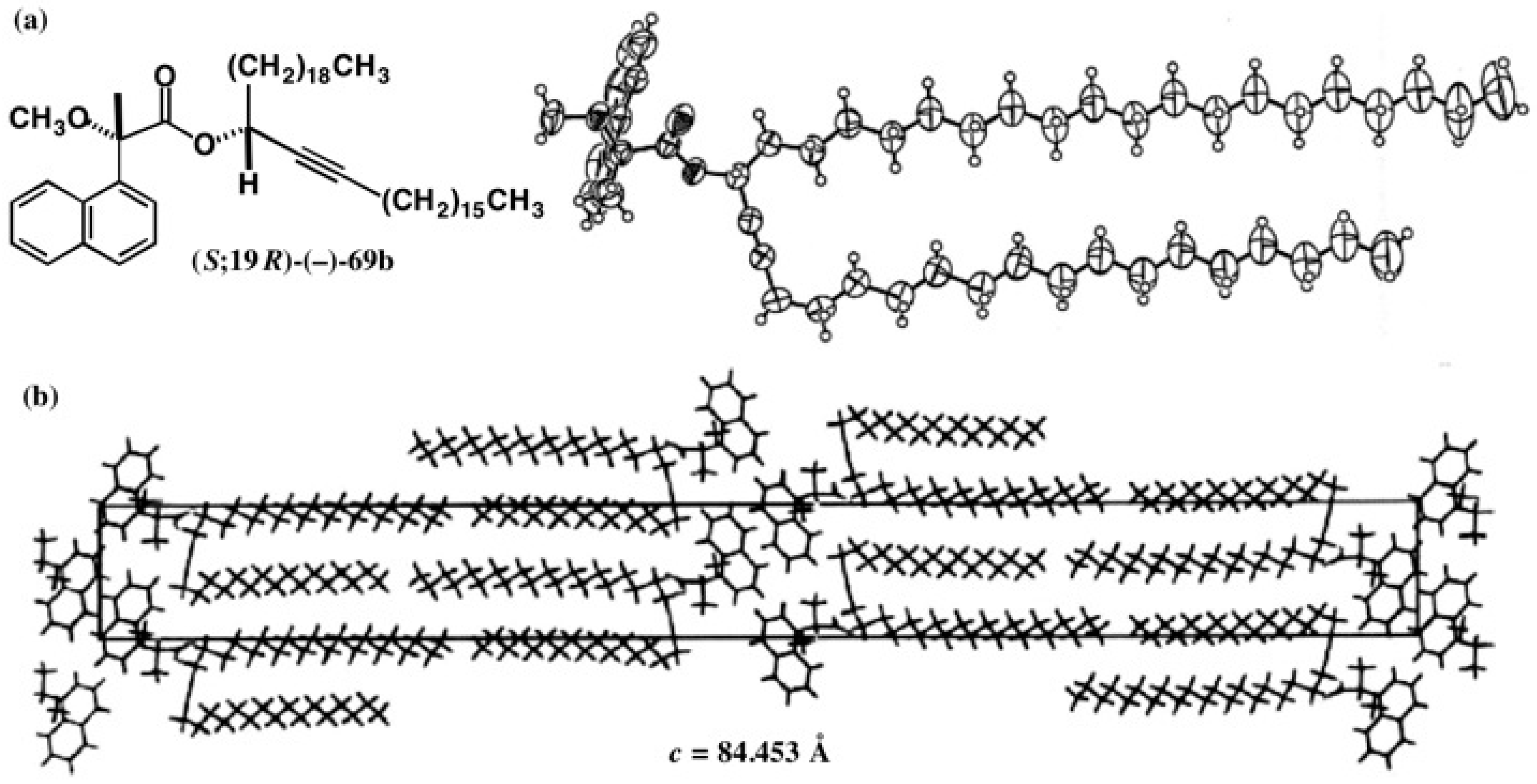
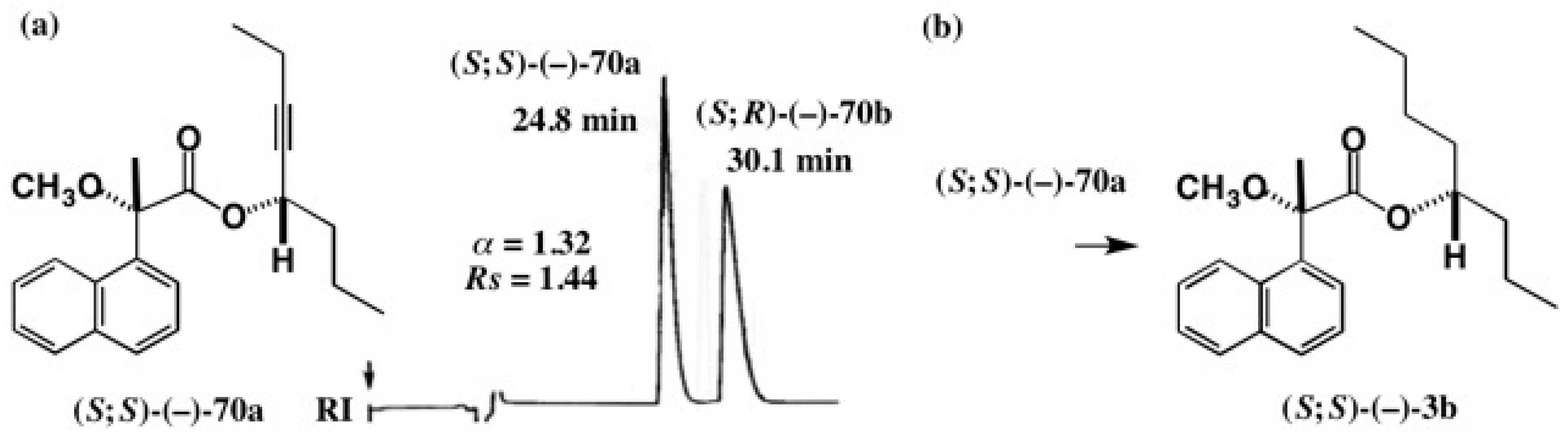
5.4. Application of Acetylene Chain Alcohols
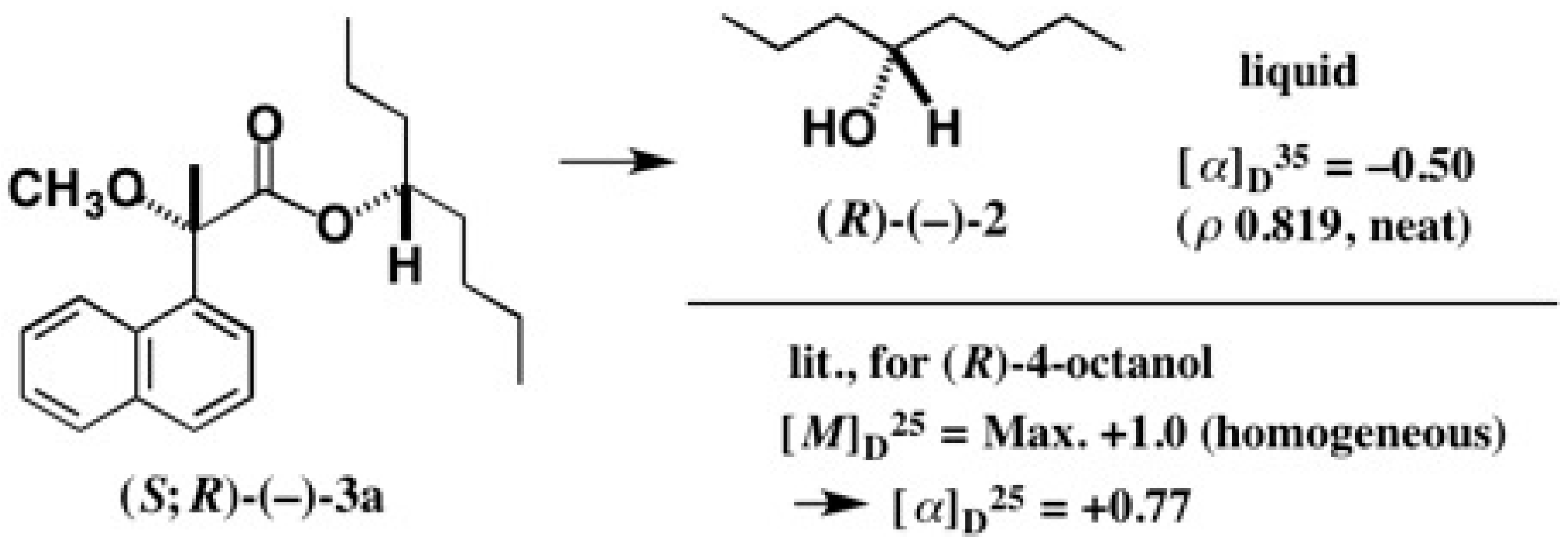
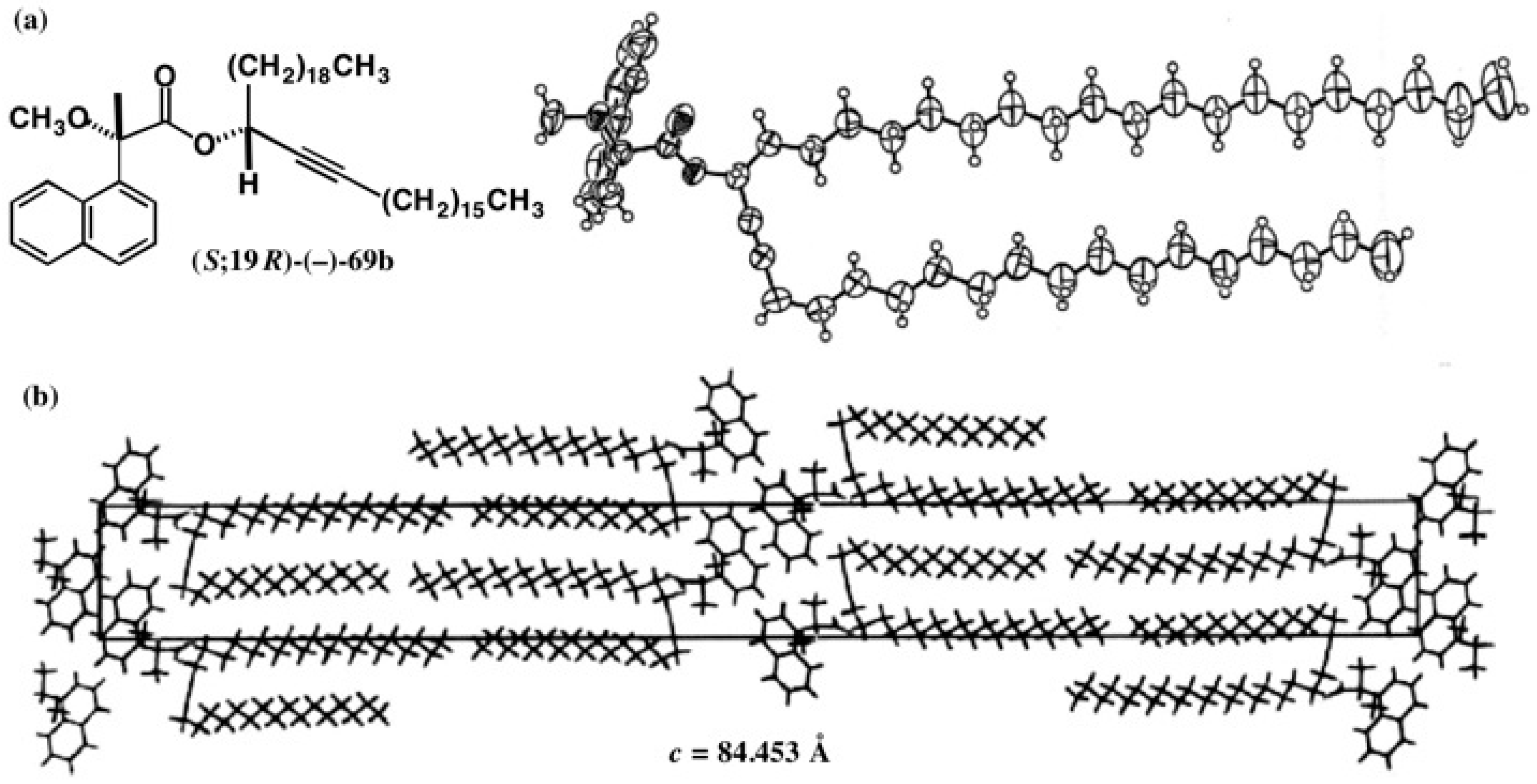
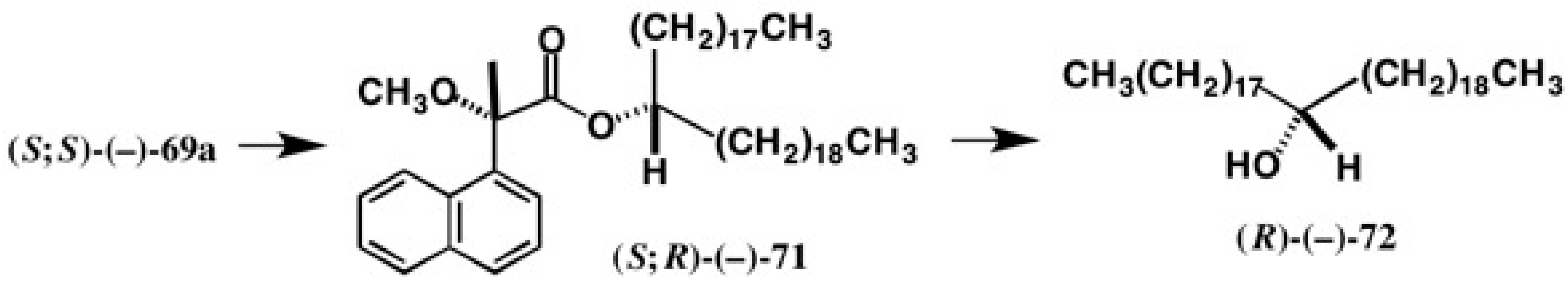
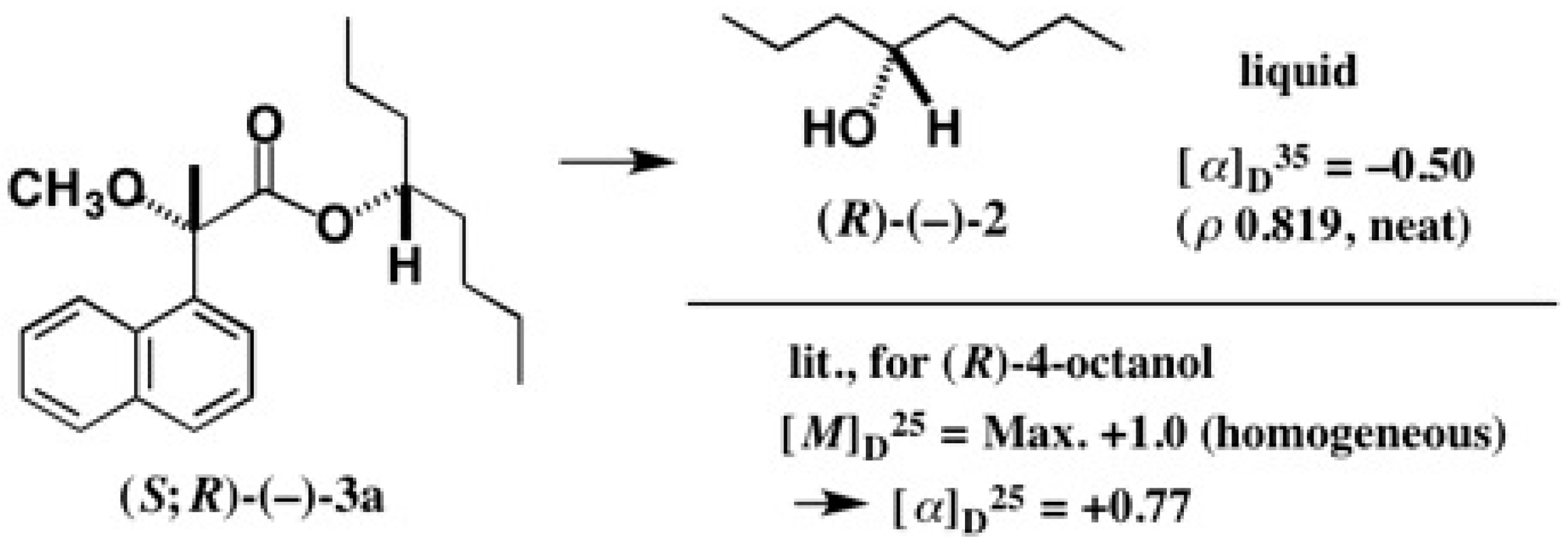
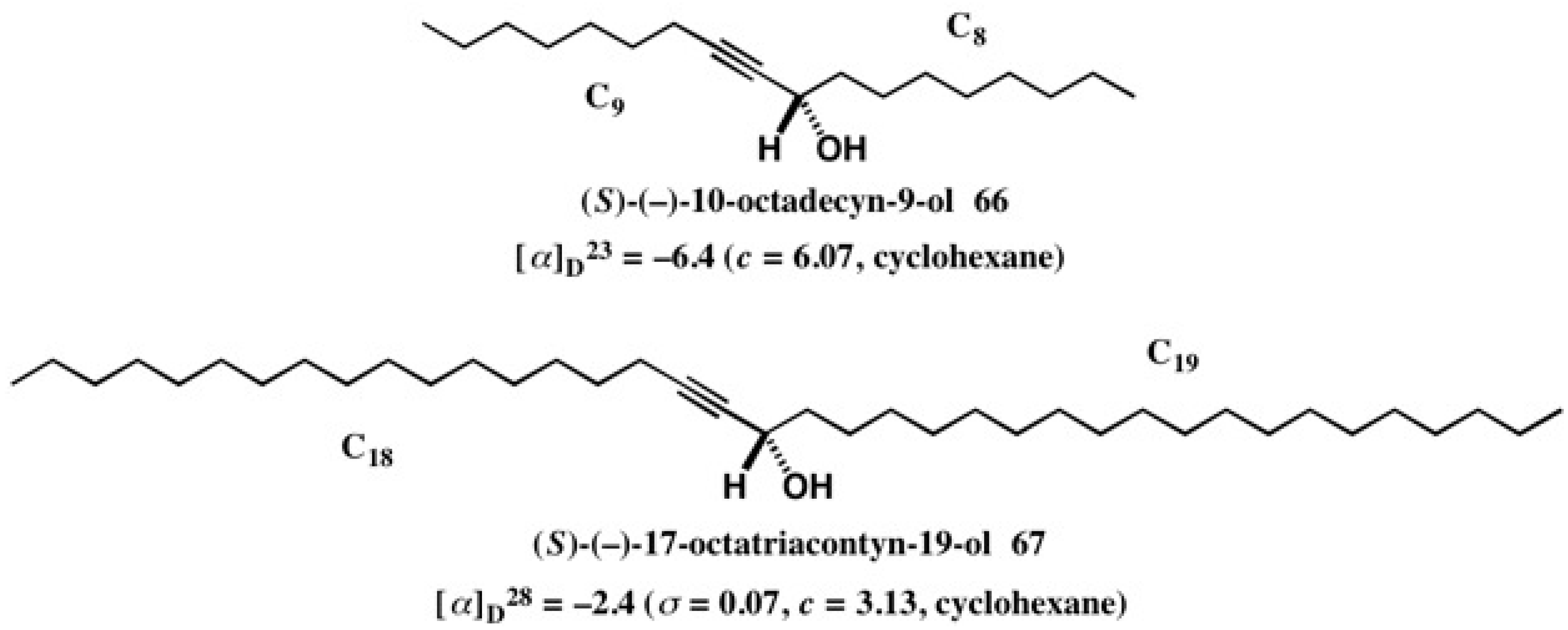
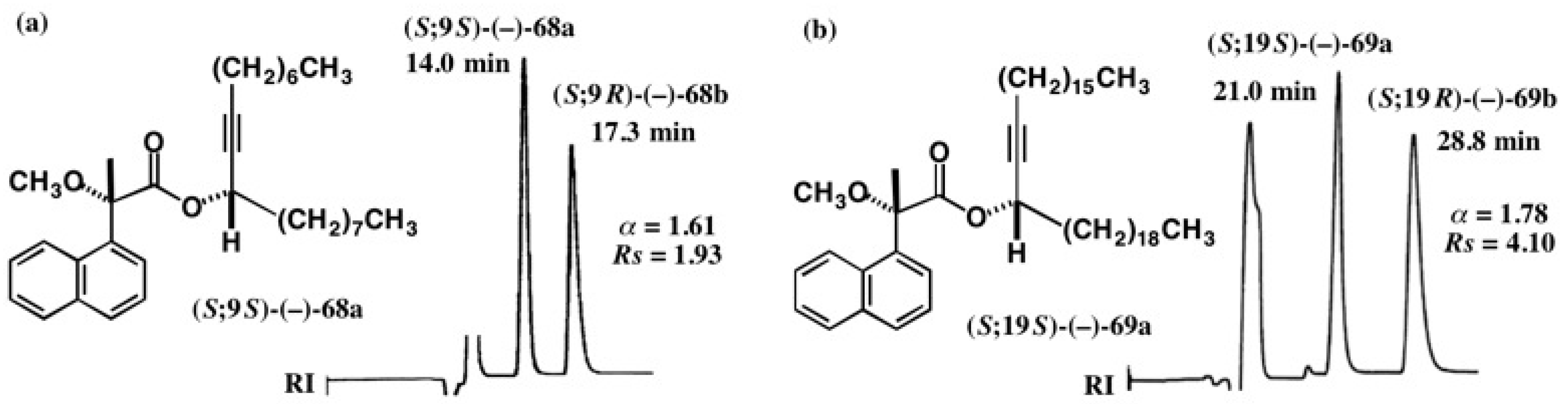
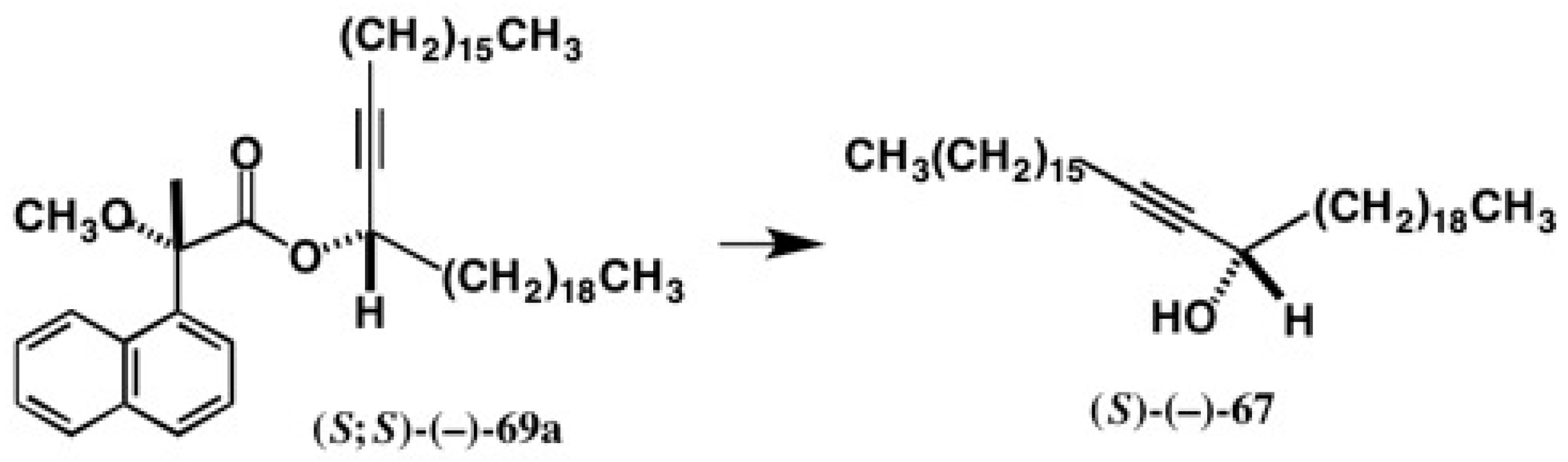
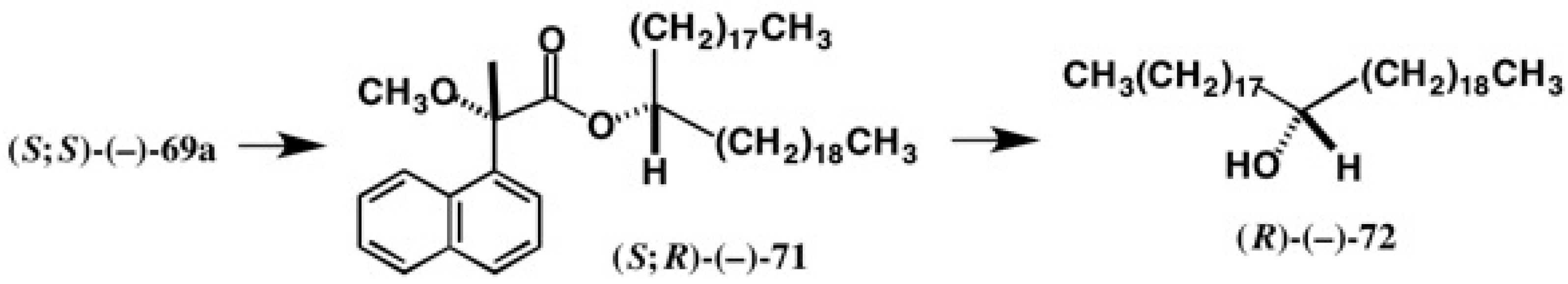
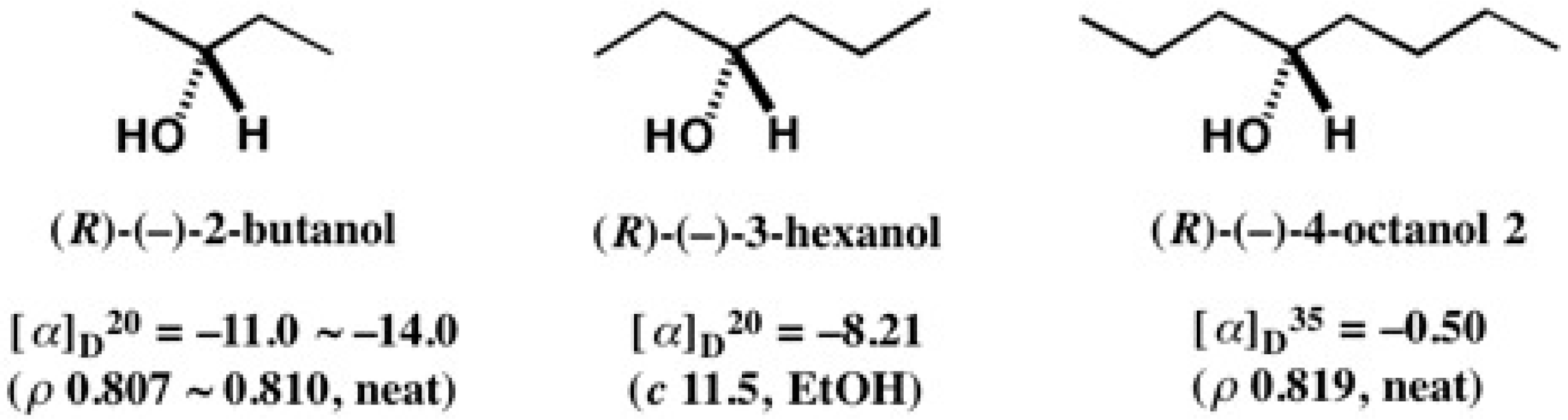
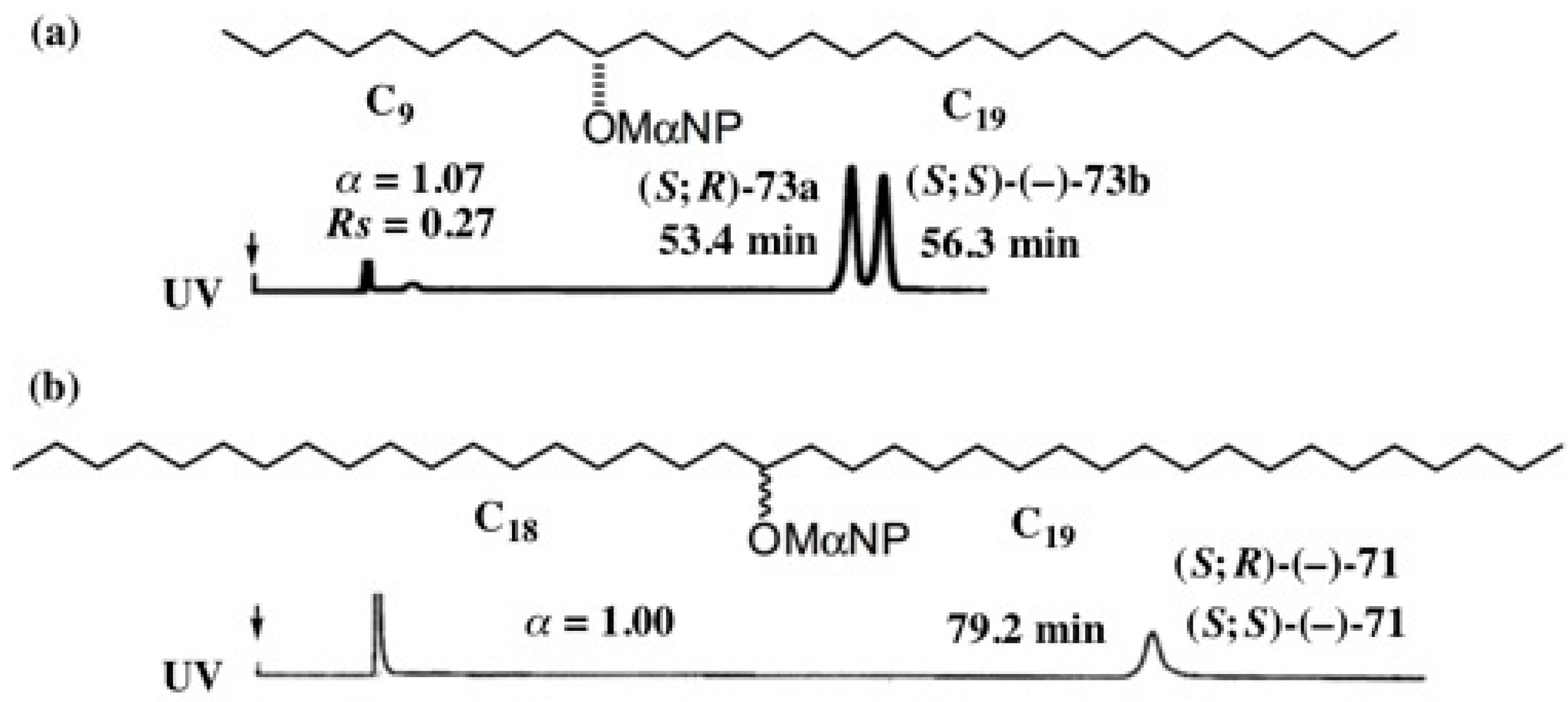
5.5. Synthesis of Enantiopure Saturated Long Chain Alcohol with Established AC
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Okamoto, Y.; Ikai, T. Chiral HPLC for Efficient Resolution of Enantiomers. Chem. Soc. Rev. 2008, 37, 2593–2608. [Google Scholar] [CrossRef] [PubMed]
- Wolf, C.; Pirkle, W.H. Conformational Effects on the Enantioselective Recognition of 4-(3,5-Dinitro-benzamido)-1,2,3,4-tetrahydrophenanthrene Derivatives by a Naproxen-derived Chiral Stationary Phase. Tetrahedron 2002, 58, 3597–3603. [Google Scholar] [CrossRef]
- Davankov, V.A. Separation of Enantiomeric Compounds Using Chiral HPLC Systems. A Brief Review of General Principles, Advances, and Development Trends. Chromatographia 1989, 27, 475–482. [Google Scholar] [CrossRef]
- Allenmark, S.; Andersson, S. Optical Resolution of Some Biologically Active Compounds by Chiral Liquid Chromatography on BSA-silica (Resolvosil) Columns. Chirality 1989, 1, 154–160. [Google Scholar] [CrossRef]
- Breitbach, A.S.; Lim, Y.; Xu, Q.-L.; Kurti, L.; Armstrong, D.W.; Breitbach, A.Z. Enantiomeric Separations of α-Aryl Ketones with Cyclofructan Chiral Stationary Phases via High Performance Liquid Chromatography and Supercritical Fluid Chromatography. J. Chromatogr. A 2016, 1427, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Harada, N. Powerful Novel Chiral Acids for Enantioresolution, Determination of Absolute Configuration, and MS Spectral Determination of Enantiomeric Excess. TCI MAIL 2003, 117, 2–27. [Google Scholar]
- Harada, N. Chiral Auxiliaries Powerful for Both Enantiomer Resolution and Determination of Absolute Configuration by X-Ray Crystallography. In Topics in Stereochemistry; Denmark, S.E., Siegel, J.S., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006; Volume 25, Chapter 6; pp. 177–203. [Google Scholar]
- Harada, N. Powerful Chiral Molecular Tools for Preparation of Enantiopure Alcohols and Simultaneous Determination of Their Absolute Configurations by X-Ray Crystallography and/or 1H-NMR Anisotropy Methods. In Chirality in Drug Research; Francotte, E., Lindner, W., Eds.; Wiley-VCH: Weinheim, Germany, 2006; Chapter 9; pp. 283–321. [Google Scholar]
- Harada, N. Determination of Absolute Configurations by X-Ray Crystallography and 1H-NMR Anisotropy. Chirality 2008, 20, 691–723. [Google Scholar] [CrossRef] [PubMed]
- Harada, N. X-Ray Crystallography and 1H-NMR Anisotropy Methods for Determination of Absolute Configurations. In Stereoselective Synthesis of Drugs and Natural Products; Andrushko, V., Andrushko, N., Eds.; Wiley-Blackwell: New York, NY, USA, 2013; Chapter 46; pp. 1629–1662. [Google Scholar]
- Harada, N. Determination of Absolute Configurations by Electronic CD Exciton Chirality, Vibrational CD, 1H-NMR Anisotropy, and X-Ray Crystallography Methods—Principles, Practices, and Reliability. In Structure Elucidation in Organic Chemistry; Cid, M.M., Bravo, J., Eds.; Wiley-VCH: Weinheim, Germany, 2015; Chapter 11; pp. 393–443. [Google Scholar]
- Ohrui, H.; Terashima, H.; Imaizumi, K.; Akasaka, K. A Solution of the “Intrinsic Problem” of Diastereomer Method in Chiral Discrimination. Development of a Method for Highly Efficient and Sensitive Discrimination of Chiral Alcohols. Proc. Jpn. Acad. Ser. B 2002, 78, 69–72. [Google Scholar] [CrossRef]
- Akagi, M.; Sekiguchi, S.; Taji, H.; Kasai, Y.; Kuwahara, S.; Watanabe, M.; Harada, N. A General Method for the Synthesis of Enantiopure Aliphatic Chain Alcohols with Established Absolute Configurations. Part 2, via Catalytic Reduction of Acetylene Alcohol MαNP Esters. Tetrahedron Asymmetry 2014, 25, 1466–1477. [Google Scholar] [CrossRef]
- Takagi, T.; Aoyanagi, N.; Nishimura, K.; Ando, Y.; Ota, T. Enantiomer Separations of Secondary Alkanols with Little Asymmetry by High-performance Liquid Chromatography on Chiral Columns. J. Chromatogr. 1993, 629, 385–388. [Google Scholar] [CrossRef]
- Nie, M.-Y.; Zhou, L.-M.; Liu, X.-L.; Wang, Q.-H.; Zhu, D.-Q. Gas Chromatographic Enantiomer Separation on Long-chain Alkylated β-Cyclodextrin Chiral Stationary Phases. Anal. Chim. Acta 2000, 408, 279–284. [Google Scholar] [CrossRef]
- Ohtaki, T.; Akasaka, K.; Kabuto, C.; Ohrui, H. Chiral Discrimination of Secondary Alcohols by Both 1H-NMR and HPLC After Labeling with a Chiral Derivatization Reagent, 2-(2,3-Anthracenedicarbox-imide)cyclohexane Carboxylic Acid. Chirality 2005, 17, S171–S176. [Google Scholar] [CrossRef] [PubMed]
- Bijvoet, J.M.; Peerdeman, A.F.; van Bommel, A.J. Determination of the Absolute Configuration of Optically Active Compounds by Means of X-rays. Nature 1951, 168, 271–272. [Google Scholar] [CrossRef]
- Harada, N.; Nakanishi, K. Circular Dichroic Spectroscopy—Exciton Coupling in Organic Stereochemistry; University Science Books: Mill Valley, CA, USA, 1983. [Google Scholar]
- Berova, N.; Polavarapu, P.; Nakanishi, K.; Woody, R.W. Comprehensive Chiroptical Spectroscopy; John Wiley & Sons: Hoboken, NJ, USA, 2012; Volumes 1 and 2. [Google Scholar]
- Levene, P.A.; Rothen, A. Optical Activity and Chemical Structure. J. Org. Chem. 1936, 1, 76–133. [Google Scholar] [CrossRef]
- Murai, S.; Soutome, T.; Yoshida, N.; Osawa, S.; Harada, N. Synthesis, Circular Dichroism, and Absolute Stereochemistry of a Fecht Acid Analog and Related Compounds. Enantiomer 2000, 5, 197–202. [Google Scholar] [PubMed]
- Murai, S. Synthesis of Chiral Spiro[3.3]heptane Compounds and Determination of their Absolute Configurations. Master’s Thesis, Tohoku University, Sendai, Japan, 1991. [Google Scholar]
- Harada, N.; Soutome, T.; Murai, S.; Uda, H. A Chiral Probe Useful for Optical Resolution and X-Ray Crystallographic Determination of the Absolute Stereochemistry of Carboxylic Acids. Tetrahedron Asymmetry 1993, 4, 1755–1758. [Google Scholar] [CrossRef]
- Soutome, T. Synthesis of Chiral Bridged Aromatic Compounds and Determination of their Absolute Configurations. Master’s Thesis, Tohoku University, Sendai, Japan, 1993. [Google Scholar]
- Falk, H.; Reich-Rohrwig, P.; Schloegl, K. Absolute Configuration and Circular Dichroism of Optically Active [2.2]Paracyclophane Derivatives. Tetrahedron 1970, 26, 511–527. [Google Scholar] [CrossRef]
- Eberhardt, H.; Schloegl, K. Stereochemistry of Planar-chiral Compounds. II. Preparation, Chiroptical Properties, and Absolute Configuration of [10]Paracyclophane Derivatives. Eur. J. Org. Chem. 1972, 760, 157–170. [Google Scholar]
- Nakazaki, M.; Yamamoto, K.; Ito, M.; Tanaka, S. Preparations of Optically Active [8][8]- and [8][10]-Paracyclophanes with Known Absolute Configurations. J. Org. Chem. 1977, 42, 3468–3473. [Google Scholar] [CrossRef]
- Schwartz, L.H.; Bathija, B.L. Absolute Configuration of an Ansa Compound: Gentisic Acid Nona-methylene Ether. J. Am. Chem. Soc. 1976, 98, 5344–5347. [Google Scholar] [CrossRef]
- Schloegl adopted the configurations of (R)-(–)-12, (R)-(+)-14, and (R)-(–)-15: Schloegl, K. Planar Chiral Molecular Structures. Top. Curr. Chem. 1984, 125, 27–62. [Google Scholar]
- Harada, N.; Soutome, T.; Nehira, T.; Uda, H.; Oi, S.; Okamura, A.; Miyano, S. Revision of the Absolute Configurations of [8]Paracyclophane-10-carboxylic and 15-Methyl[10]paracyclophane-12-carboxylic Acids. J. Am. Chem. Soc. 1993, 115, 7547–7548. [Google Scholar] [CrossRef]
- Harada, N.; Nehira, T.; Soutome, T.; Hiyoshi, N.; Kido, F. Chiral Phthalic Acid Amide, a Chiral Auxiliary Useful for Enantiomer Resolution and X-Ray Crystallographic Determination of the Absolute Stereochemistry of Alcohols. Enantiomer 1996, 1, 35–39. [Google Scholar]
- Nehira, T. Development of Novel Chiral Resolution Methods and Determination of the Absolute Configurations of Twisted π-Electron Compounds. Ph.D. Thesis, Tohoku University, Sendai, Japan, 1996. [Google Scholar]
- Harada, N.; Koumura, N.; Feringa, B.L. Chemistry of Unique Chiral Olefins. 3. Synthesis and Absolute Stereochemistry of trans- and cis-1,1′,2,2′,3,3′,4,4′-Octahydro-3,3′-dimethyl-4,4′-biphenanthrylidenes. J. Am. Chem. Soc. 1997, 119, 7256–7264. [Google Scholar] [CrossRef]
- Koumura, N.; Harada, N. X-Ray Crystallographic Determination of the Stereochemistry of a Unique Chiral Olefin, [CD(–)238.0]-(3R,3′R)-(P,P)-(Z)-(+)-1,1′,2,2′,3,3′,4,4′-Octahydro-3,3′-dimethyl-4,4′-biphenan-thrylidene. Enantiomer 1998, 3, 251–253. [Google Scholar]
- Koumura, N.; Harada, N. Photochemistry and Absolute Stereochemistry of Unique Chiral Olefins, trans- and cis-1,1′,2,2′,3,3′,4,4′-Octahydro-3,3′-dimethyl-4,4′-biphenanthrylidenes. Chem. Lett. 1998, 1998, 1151–1152. [Google Scholar] [CrossRef]
- Koumura, N.; Zijlstra, R.W.J.; van Delden, R.A.; Harada, N.; Feringa, B.L. Light-driven Monodirectional Molecular Rotor. Nature 1999, 401, 152–155. [Google Scholar] [PubMed]
- Fujita, T.; Kuwahara, S.; Harada, N. A New Model of Light Powered Chiral Molecular Motor with Higher Speed of Rotation (1). Synthesis and Absolute Stereostructure. Eur. J. Org. Chem. 2005, 2005, 4533–4543. [Google Scholar] [CrossRef]
- Harada, N.; Koumura, N.; Robillard, M. Chiral Dichlorophthalic Acid Amide, an Improved Chiral Auxiliary Useful for Enantioresolution and X-Ray Crystallographic Determination of Absolute Stereo-chemistry. Enantiomer 1997, 2, 303–309. [Google Scholar]
- Cervinka, O.; Belovsky, O.; Fabryova, A.; Dudek, V.; Grohman, K. Asymmetric Reactions. XII. A Limited Use of Asymmetric Meerwein-Ponndorf-Verley Type Reduction for Determination of Absolute Configuration of Alcohols. Collect. Czech. Chem. Commun. 1967, 32, 2618–2624. [Google Scholar] [CrossRef]
- Seebach, D.; Beck, A.K.; Roggo, S.; Wonnacott, A. Enantioselective Addition of Aryl Groups to Aromatic Aldehydes using Chiral Aryltitanium binaphthol Derivatives. Eur. J. Inorg. Chem. 1985, 118, 3673–3682. [Google Scholar] [CrossRef]
- Watanabe, M.; Kuwahara, S.; Harada, N.; Koizumi, M.; Ohkuma, T. Enantioresolution by the Chiral Phthalic Acid Method: Absolute Configurations of (2-Methylphenyl)phenylmethanol and Related Compounds. Tetrahedron Asymmetry 1999, 10, 2075–2078. [Google Scholar] [CrossRef]
- Kuwahara, S. Chiral Resolution of Alcohols by the Chiral Phthalic Acid Method and Determination of their Absolute Configurations: New Developments. Master’s Thesis, Tohoku University, Sendai, Japan, 1999. [Google Scholar]
- Kosaka, M.; Kuwahara, S.; Watanabe, M.; Harada, N.; Job, G.E.; Pirkle, W.H. Comparison of CD Spectra of (2-Methylphenyl)- and (2,6-Dimethylphenyl)-phenylmethanols Leads to Erroneous Absolute Configurations. Enantiomer 2002, 7, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, M. Chiral Resolution of Aromatic Alcohols by the Chiral Phthalic Acid Method and Determination of their Absolute Configurations: Applications in the Synthetic Studies of Chloromonilicin. Master’s Thesis, Tohoku University, Sendai, Japan, 2000. [Google Scholar]
- Fujita, T.; Obata, K.; Kuwahara, S.; Nakahashi, A.; Monde, K.; Decatur, J.; Harada, N. (R)-(+)- [VCD(−)984]-4-Ethyl-4-methyloctane, a Cryptochiral Hydrocarbon with a Quaternary Chirality Center. (1) Synthesis of Enantiopure Compound and Unambiguous Determination of Absolute Configuration. Eur. J. Org. Chem. 2010, 2010, 6372–6384. [Google Scholar] [CrossRef]
- Hoeve, W.T.; Wynberg, H. Chiral Tetraalkylmethanes. Two Syntheses of Optically Active Butylethyl-methylpropylmethane of Known and High Optical Purity. J. Org. Chem. 1980, 45, 2754–2763. [Google Scholar] [CrossRef]
- Caporusso, A.M.; Consoloni, C.; Lardicci, L. Stereoselective Construction of Quaternary Carbon Centers via Reaction of Heterocuprates with Chiral Allenic Bromides: A Facile Synthesis of Butylethyl-methylpropylmethane in High Enantiomeric Purity. Gazz. Chim. Ital. 1988, 118, 857–859. [Google Scholar]
- Kuwahara, S.; Obata, K.; Fujita, T.; Miura, N.; Nakahashi, A.; Monde, K.; Harada, N. (R)-(+)- [VCD(–)984]-4-Ethyl-4-methyloctane: A Cryptochiral Hydrocarbon with a Quaternary Chiral Center. (2) Vibrational CD Spectra of Both Enantiomers and Absolute Configurational Assignment. Eur. J. Org. Chem. 2010, 2010, 6385–6392. [Google Scholar] [CrossRef]
- Matsumoto, T.; Ishida, T.; Takeda, Y.; Soh, K.; Kubo, I.; Sakamoto, M. Enantioselective Biotransformation of 1-Isopropylnaphthalene in Rabbits. Chem. Pharm. Bull. 1995, 43, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, S.; Fujita, K.; Watanabe, M.; Harada, N.; Ishida, T. Enantioresolution and Absolute Stereochemistry of 2-(1-Naphthyl)propane-1,2-diol and Related Compounds. Enantiomer 1999, 4, 141–145. [Google Scholar]
- Harada, N.; Watanabe, M.; Kuwahara, S.; Sugio, A.; Kasai, Y.; Ichikawa, A. 2-Methoxy-2-(1-naphthyl)-propionic Acid, a Powerful Chiral Auxiliary for Enantioresolution of Alcohols and Determination of Their Absolute Configurations by the 1H-NMR Anisotropy Method. Tetrahedron: Asymmetry 2000, 11, 1249–1253. [Google Scholar] [CrossRef]
- Dale, J.A.; Mosher, H.S. Nuclear Magnetic Resonance Enantiomer Reagents. Configurational Correlations via Nuclear Magnetic Resonance Chemical Shifts of Diastereomeric Mandelate, O-Methylmandelate, and α-Methoxy-α-trifluoromethyl-phenylacetate (MTPA) Esters. J. Am. Chem. Soc. 1973, 95, 512–519. [Google Scholar] [CrossRef]
- Ohtani, I.; Kusumi, T.; Kashman, Y.; Kakisawa, H. High-field FT NMR Application of Mosher’s Method. The Absolute Configurations of Marine Terpenoids. J. Am. Chem. Soc. 1991, 113, 4092–4096. [Google Scholar] [CrossRef]
- Trost, B.M.; Belletire, J.L.; Godleski, S.; McDougal, P.G.; Balkovec, J.M.; Baldwin, J.J.; Christy, M.E.; Ponticello, G.S.; Varga, S.L.; Springer, J.P. On the Use of the O-Methylmandelate Ester for Establishment of Absolute Configuration of Secondary Alcohols. J. Org. Chem. 1986, 51, 2370–2374. [Google Scholar] [CrossRef]
- Kasai, Y.; Sugio, A.; Sekiguchi, S.; Kuwahara, S.; Matsumoto, T.; Watanabe, M.; Ichikawa, A.; Harada, N. Conformational Analysis of MαNP Esters, Powerful Chiral Resolution and 1H-NMR Anisotropy Tools—Aromatic Geometry and Solvent Effects on ∆δ Values. Eur. J. Org. Chem. 2007, 2007, 1811–1826. [Google Scholar] [CrossRef]
- Kuwahara, S.; Naito, J.; Yamamoto, Y.; Kasai, Y.; Fujita, T.; Noro, K.; Shimanuki, K.; Akagi, M.; Watanabe, M.; Matsumoto, T.; et al. Crystalline State Conformational Analysis of MαNP Esters, Powerful Resolution and Chiral 1H-NMR Anisotropy Tools. Eur. J. Org. Chem. 2007, 2007, 1827–1840. [Google Scholar] [CrossRef]
- Fujita, T.; Obata, K.; Kuwahara, S.; Miura, N.; Nakahashi, A.; Monde, K.; Decatur, J.; Harada, N. (R)-(+)-[VCD(+)945]-4-Ethyl-4-methyloctane, the Simplest Chiral Saturated Hydrocarbon with a Quaternary Stereogenic Center. Tetrahedron Lett. 2007, 48, 4219–4222. [Google Scholar] [CrossRef]
- Taji, H.; Kasai, Y.; Sugio, A.; Kuwahara, S.; Watanabe, M.; Harada, N.; Ichikawa, A. Practical Enantioresolution of Alcohols with 2-Methoxy-2-(1-naphthyl)propionic Acid and Determination of Their Absolute Configurations by the 1H-NMR Anisotropy Method. Chirality 2002, 14, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Kasai, Y.; Taji, H.; Fujita, T.; Yamamoto, Y.; Akagi, M.; Sugio, A.; Kuwahara, S.; Watanabe, M.; Harada, N.; Ichikawa, A.; et al. MαNP Acid, a Powerful Chiral Molecular Tool for Preparation of Enantio- pure Alcohols by Resolution and Determination of Their Absolute Configurations by the 1H-NMR Anisotropy Method. Chirality 2004, 16, 569–585. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Akagi, M.; Shimanuki, K.; Kuwahara, S.; Watanabe, M.; Harada, N. A General Method for the Synthesis of Enantiopure Aliphatic Chain Alcohols with Established Absolute Configurations. Part 1, Application of the MαNP Acid Method to Acetylene Alcohols. Tetrahedron Asymmetry 2014, 25, 1456–1465. [Google Scholar] [CrossRef]
- Akagi, M. Synthesis of Ultimate Cryptochiral Alcohols with Aliphatic Long Chains and Development of the Novel Methods for Determining their Absolute Configurations. Master’s Thesis, Tohoku University, Sendai, Japan, 2004. [Google Scholar]
- Sekiguchi, S.; Akagi, M.; Naito, J.; Yamamoto, Y.; Taji, H.; Kuwahara, S.; Watanabe, M.; Ozawa, Y.; Toriumi, K.; Harada, N. Synthesis of Enantiopure Aliphatic Acetylene Alcohols and Determination of Their Absolute Configurations by 1H-NMR Anisotropy and/or X-Ray Crystallography. Eur. J. Org. Chem. 2008, 2008, 2313–2324. [Google Scholar] [CrossRef]
- Mori, K.; Ohtaki, T.; Ohrui, H.; Berkebile, D.R.; Carlson, D.A. Synthesis of the Four Stereoisomers of 6-Acetoxy-19-methylnonacosane, the Most Potent Component of the Female Sex Pheromone of the New World Screwworm Fly, with Special Emphasis on Partial Racemization in the Course of Catalytic Hydrogenation. Eur. J. Org. Chem. 2004, 2004, 1089–1096. [Google Scholar] [CrossRef]
- TCI Catalog. Research Chemicals, 2016. Available online: http://www.tcichemicals.com/ (accessed on 3 October 2016).
- Coke, J.L.; Shue, R.S. Nucleophilic Ring Opening of Optically Pure (R)-(+)-1,2-Epoxybutane. Synthesis of New (R)-2-Butanol Derivatives. J. Org. Chem. 1973, 38, 2210–2211. [Google Scholar] [CrossRef]



























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Preparation of Diastereomers | HPLC | AC Determination |
|---|---|---|
 | α, not determined Rs = 0.7~2.87 av. 1.74 | X-ray, 6 examples |
 | α = 1.05~1.1 av. 1.09 Rs = 1.3~1.6 av. 1.4 | X-ray, 3 examples |
 | α = 1.14~1.25 av. 1.20 Rs = 0.91~1.94 av. 1.31 | X-ray, 4 examples |
 | α = 1.15~1.93 av. 1.58 Rs = 1.18~5.97 av. 2.98 | 1H-NMR, 13 examples X-ray, 5 examples |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harada, N. HPLC Separation of Diastereomers: Chiral Molecular Tools Useful for the Preparation of Enantiopure Compounds and Simultaneous Determination of Their Absolute Configurations. Molecules 2016, 21, 1328. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21101328
Harada N. HPLC Separation of Diastereomers: Chiral Molecular Tools Useful for the Preparation of Enantiopure Compounds and Simultaneous Determination of Their Absolute Configurations. Molecules. 2016; 21(10):1328. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21101328
Chicago/Turabian StyleHarada, Nobuyuki. 2016. "HPLC Separation of Diastereomers: Chiral Molecular Tools Useful for the Preparation of Enantiopure Compounds and Simultaneous Determination of Their Absolute Configurations" Molecules 21, no. 10: 1328. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21101328