
Kinetic Spectrophotometric Method for the 1,4-Diionic Organophosphorus Formation in the Presence of Meldrum′s Acid: Stopped-Flow Approach

Abstract
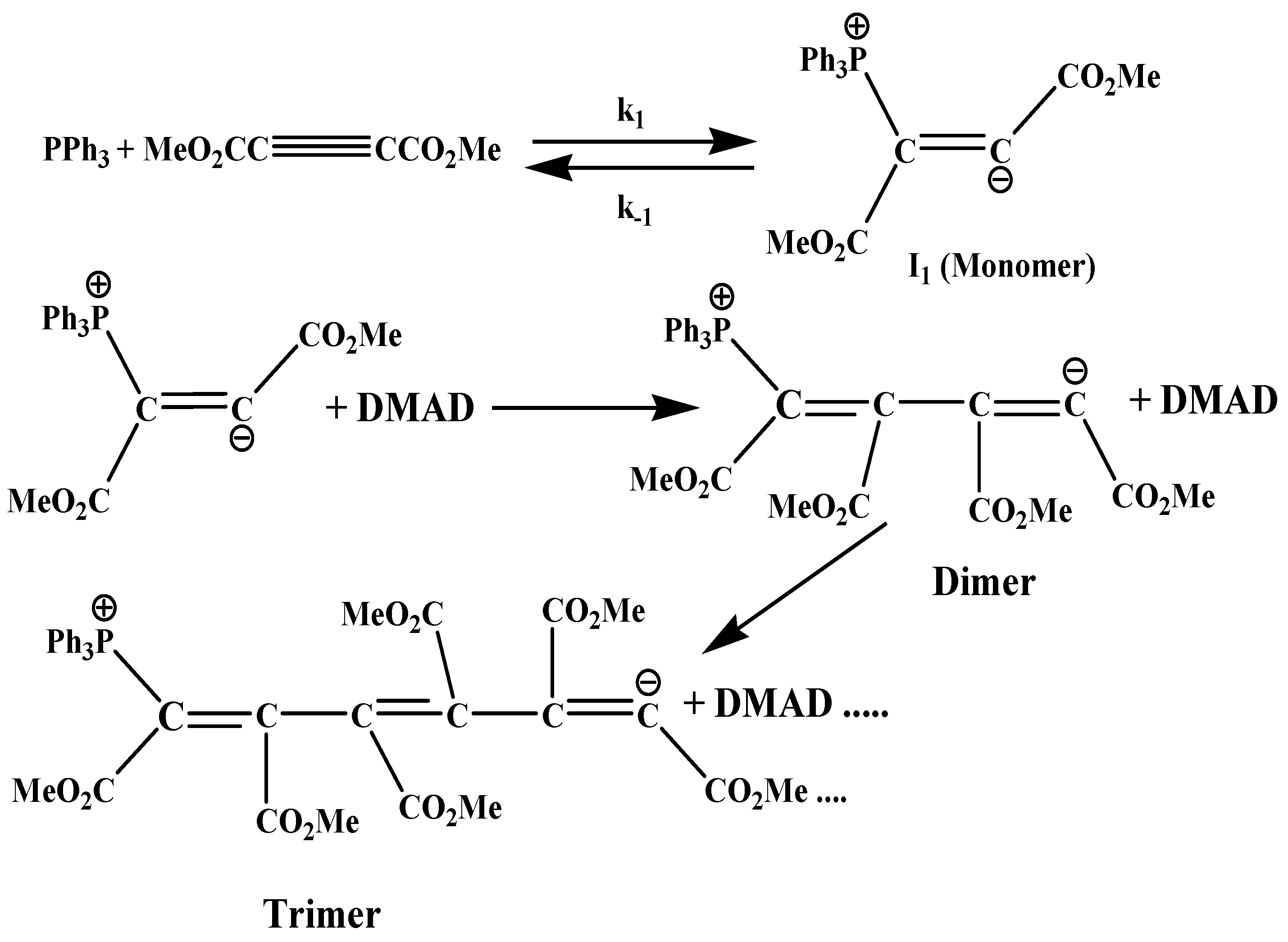
:1. Introduction
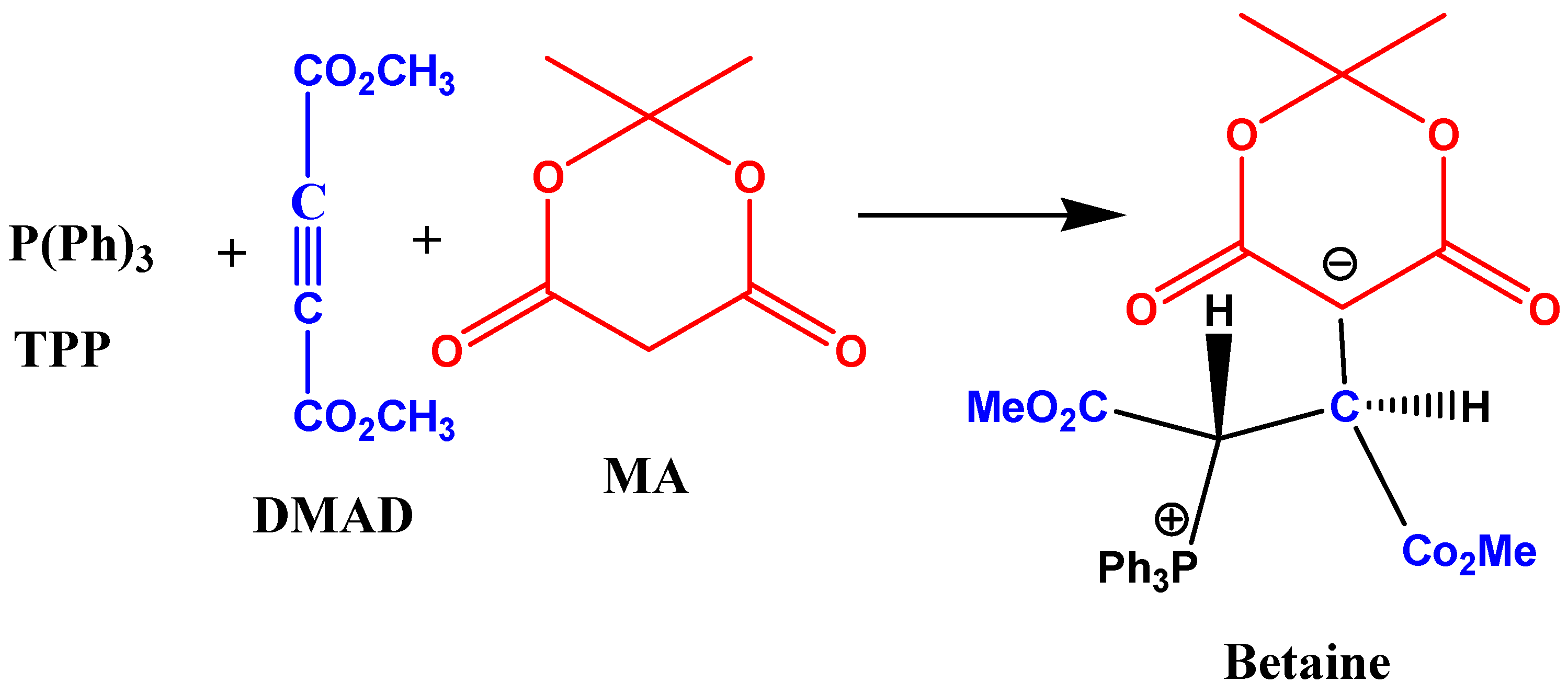
2. Results and Discussion
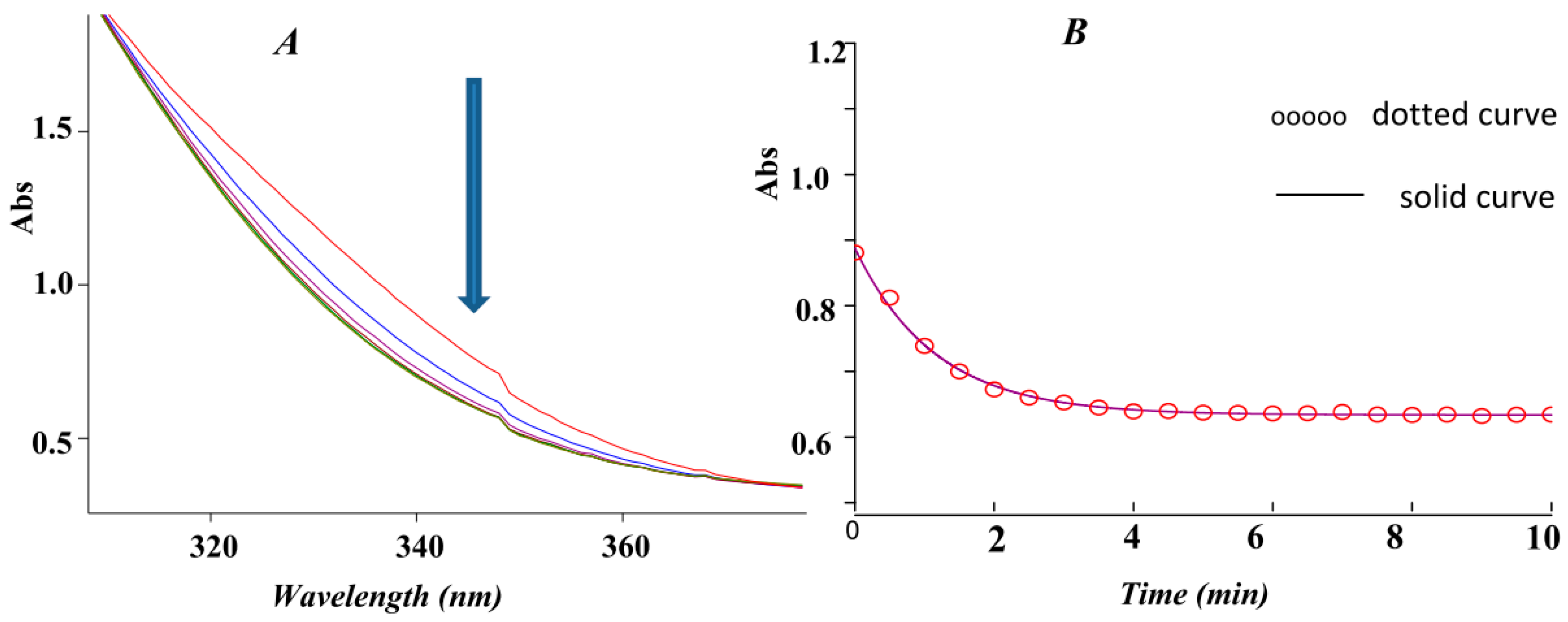
2.1. Spectral and Kinetic Studies of the Overall Three-Component Reaction by the UV-VIS Spectrophotometry Technique
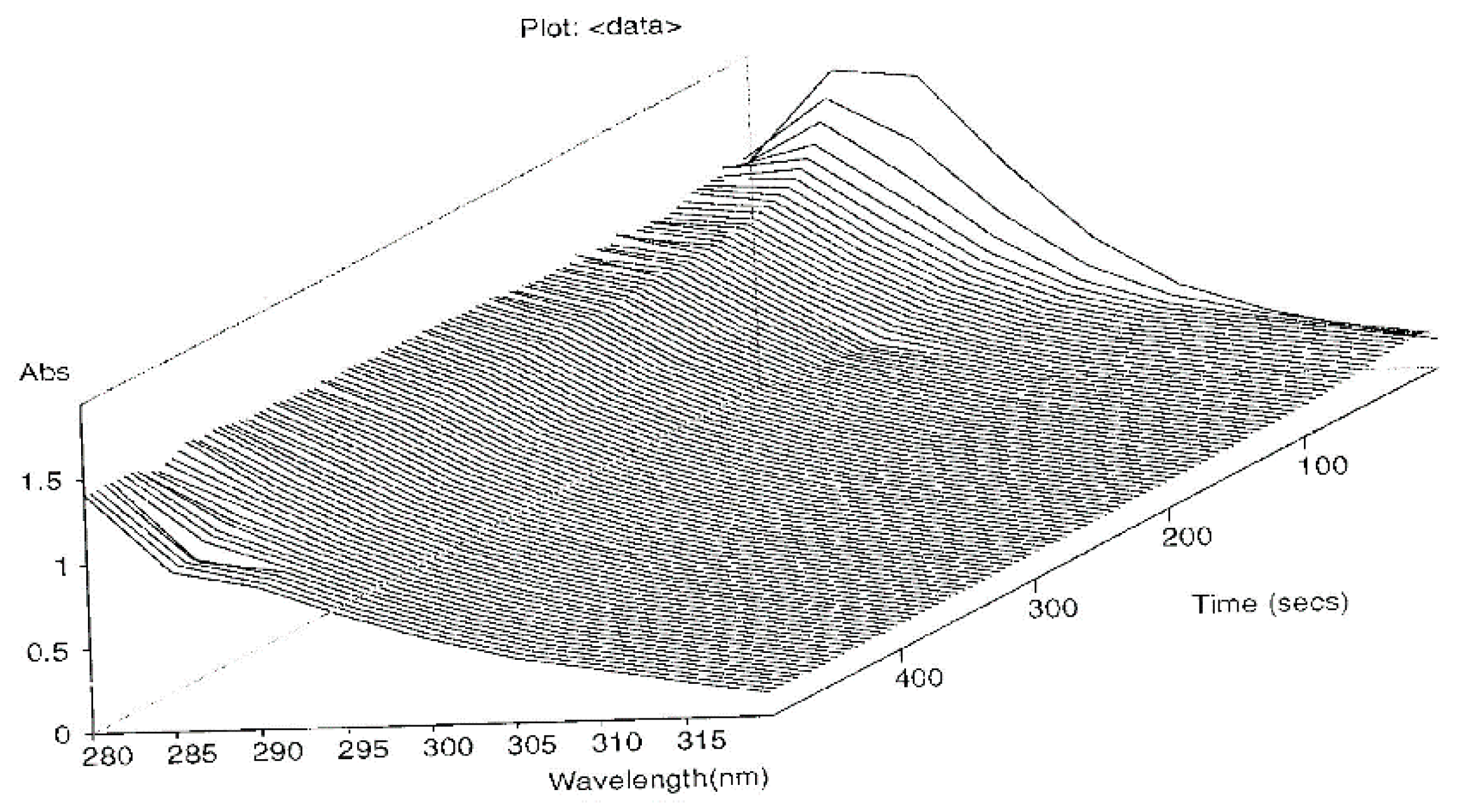
2.2. Stopped Flow Apparatus
Order of the Fast Step
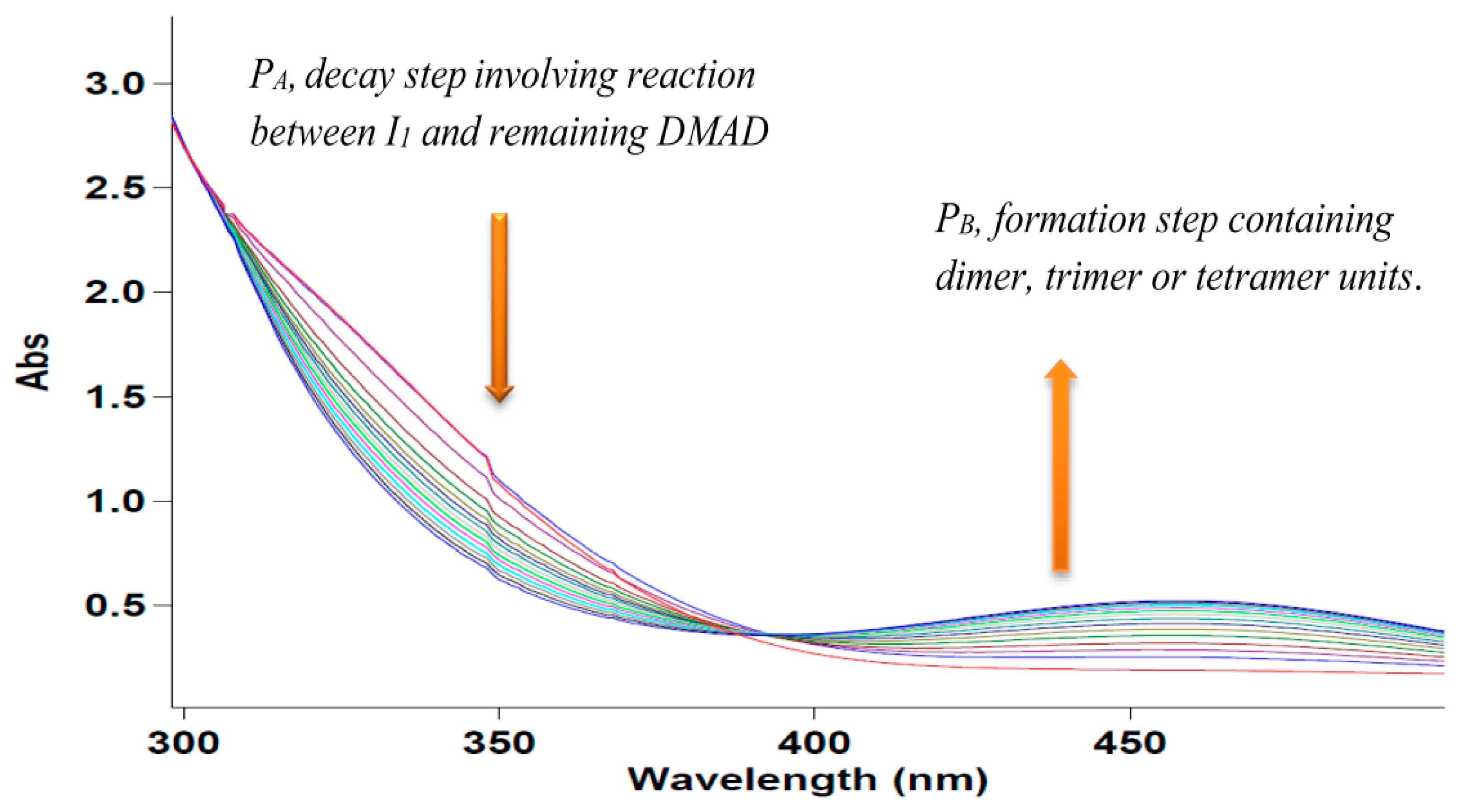
2.3. UV-VIS Experiments
Overall Order
2.4. Effect of Concentration
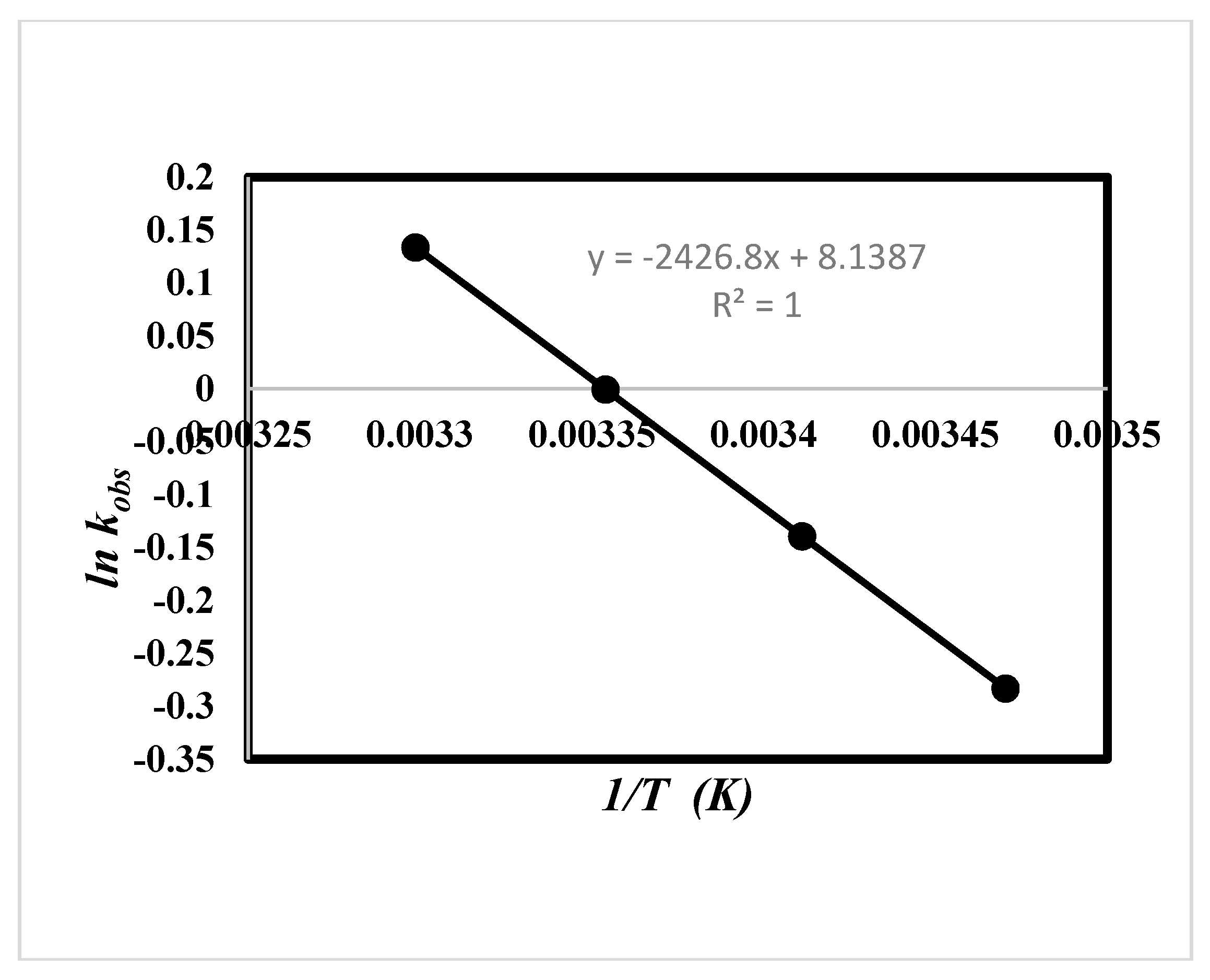
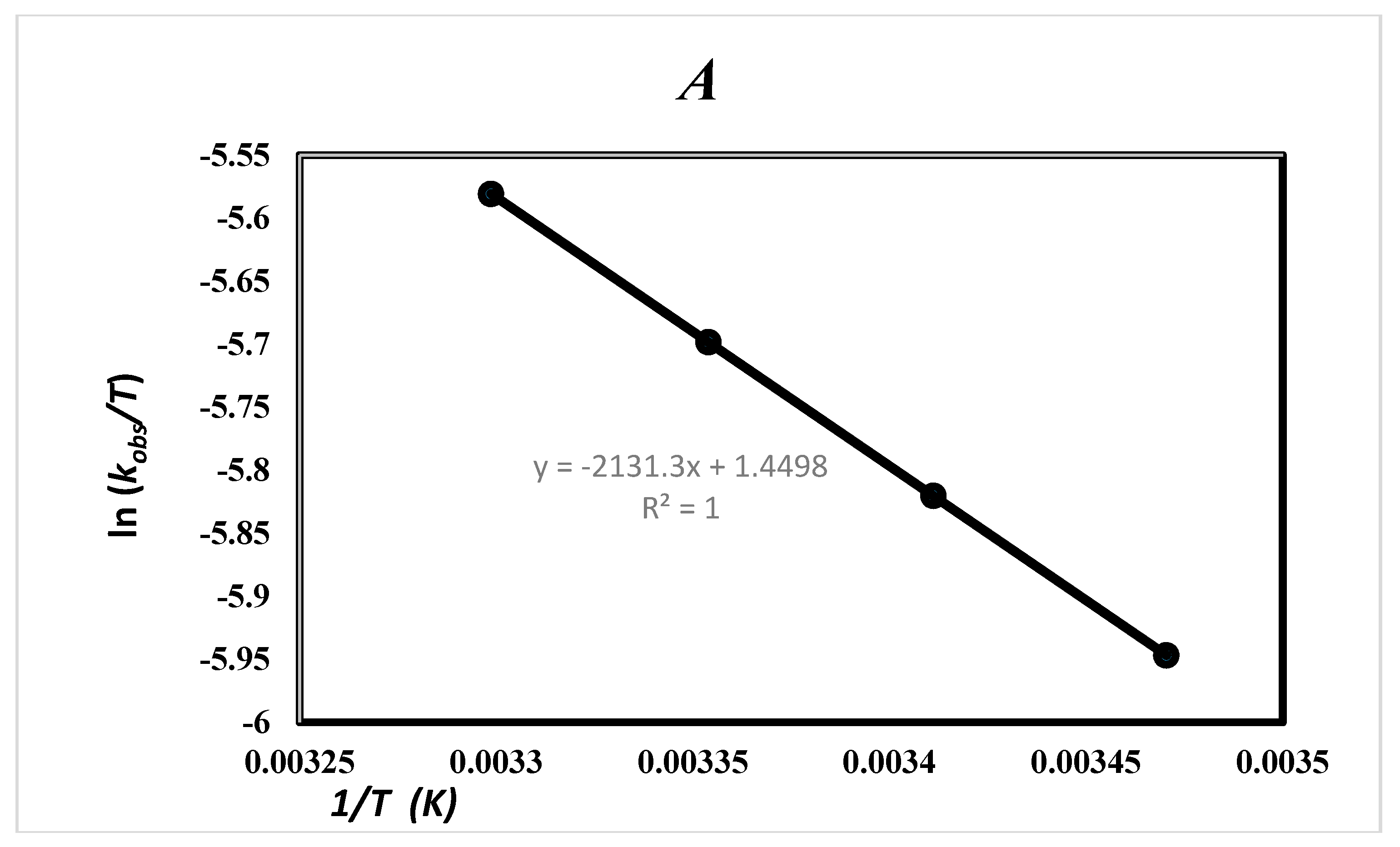
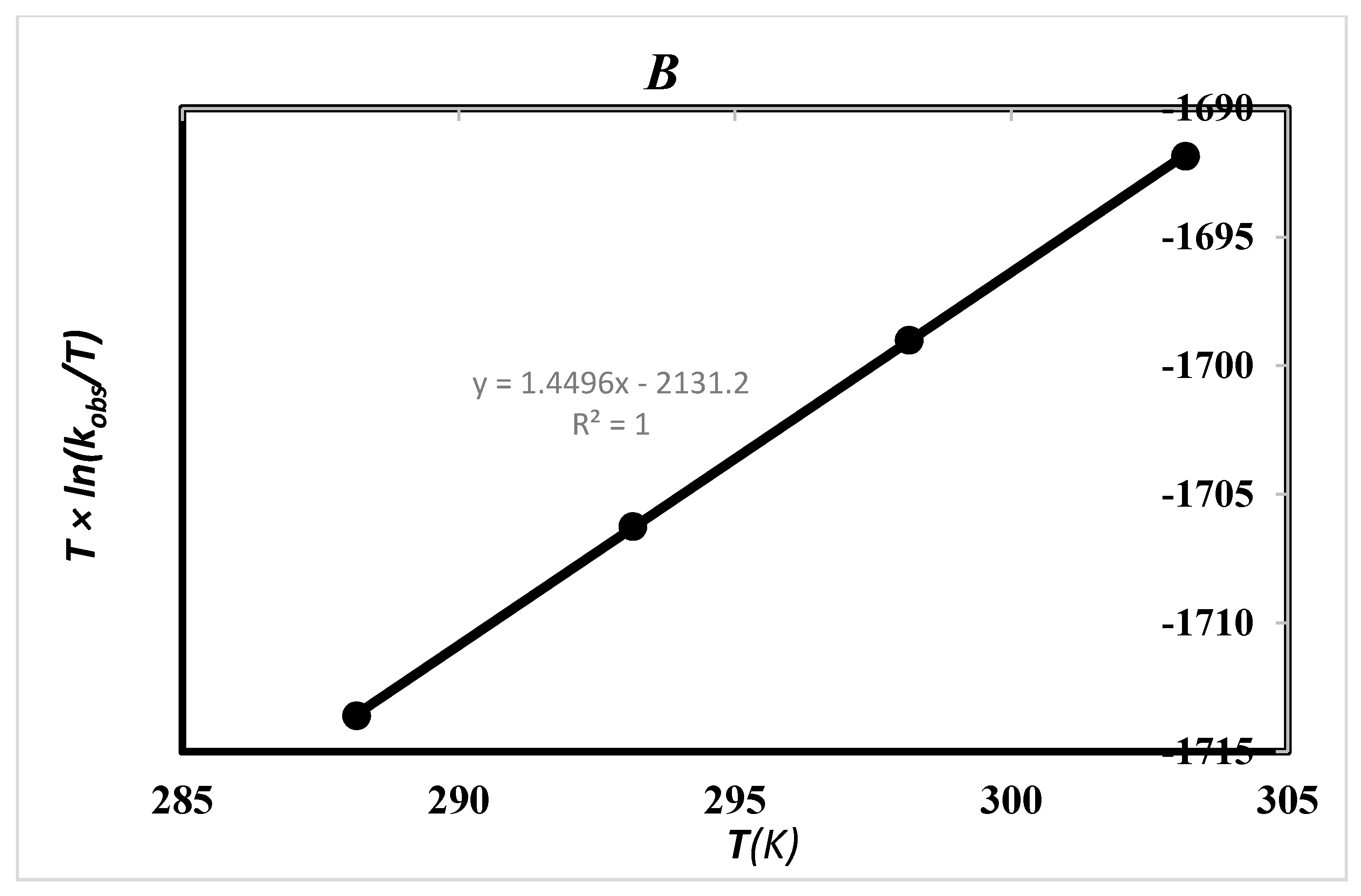
2.5. Effect of Solvent and Temperature
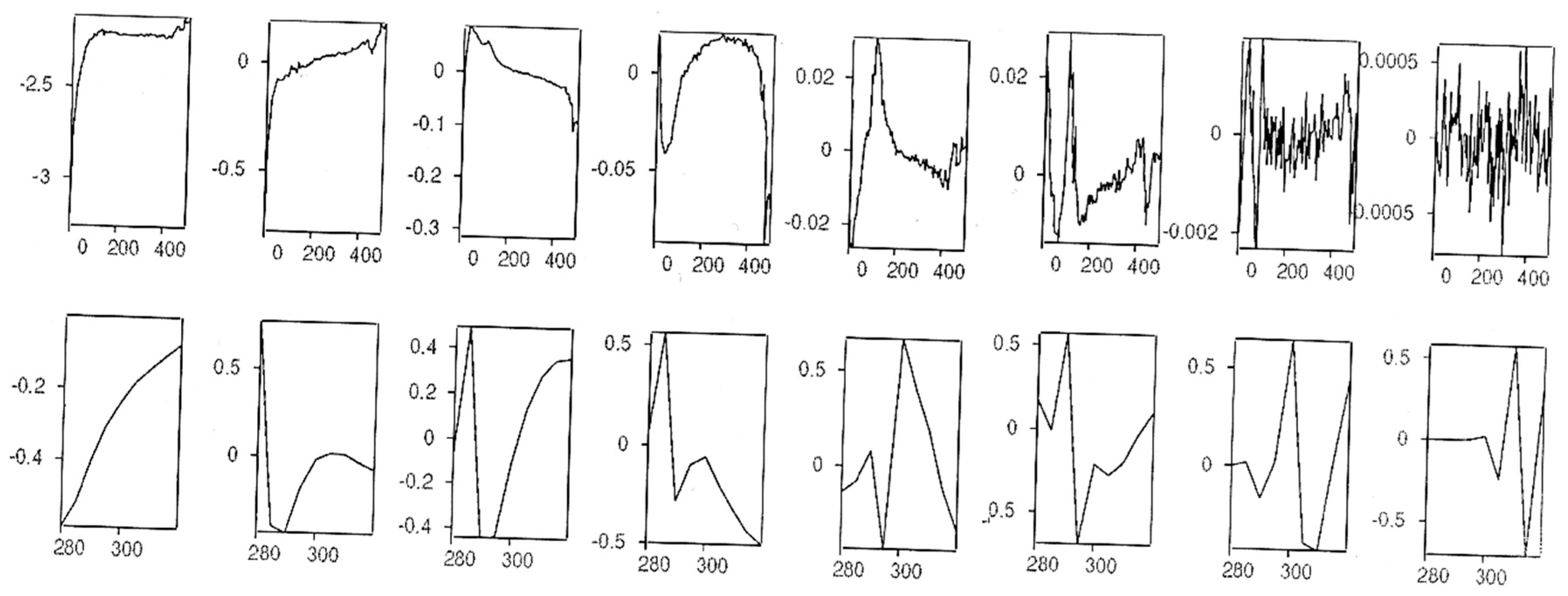
2.6. Global Kinetic Analysis of Data for the Two- and Three-Component Reactions Using the Pro-K Software
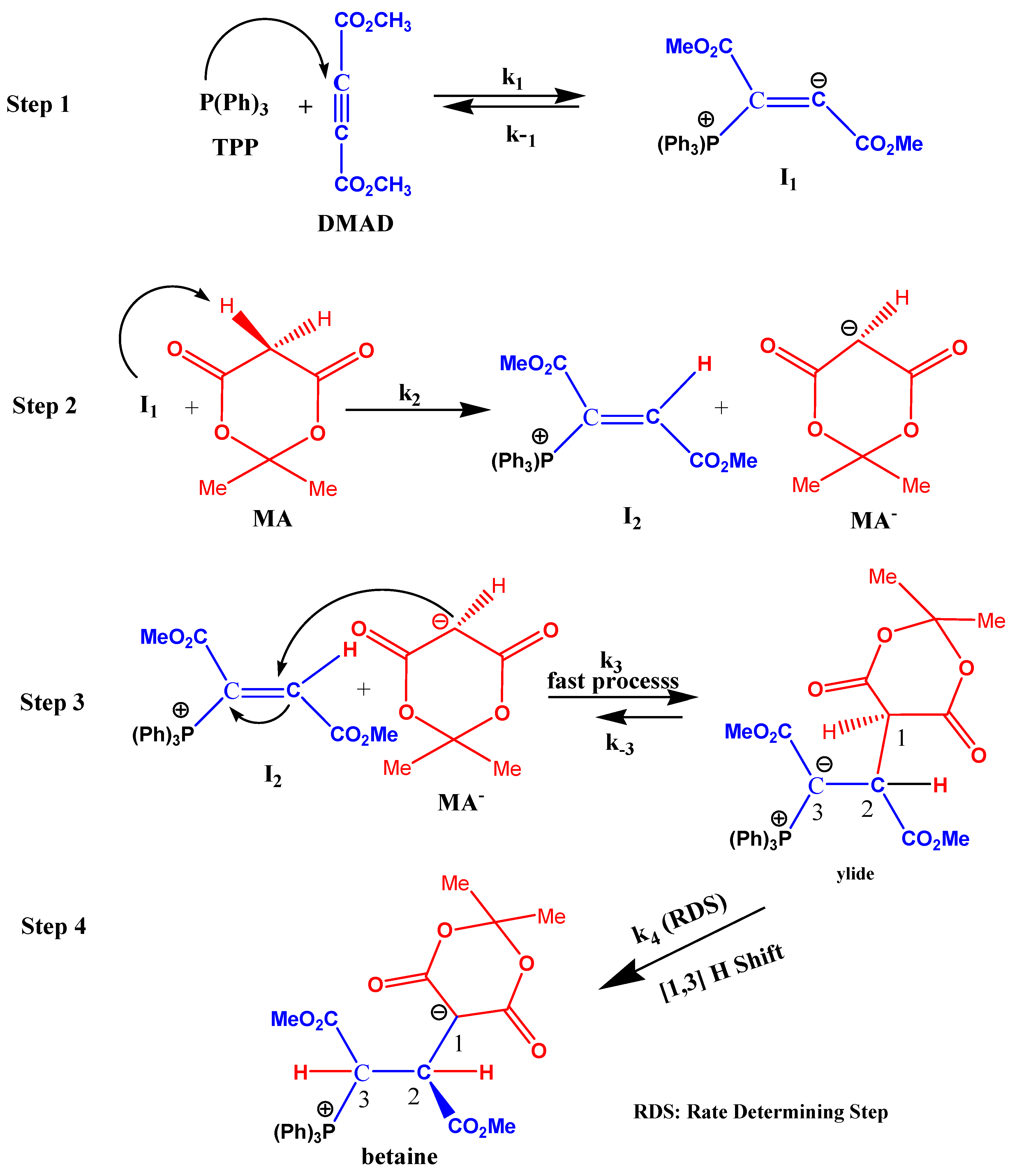
2.7. Mechanism Discussion
3. Chemicals and Apparatus
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nakamura, M.; Miki, M.; Majima, T.J. Effects of methylene chains on photoreactions of diphenylalkanediones and phenylalkenons. J. Chem. Soc. Perkin Trans. 1 2000, 3, 415–420. [Google Scholar] [CrossRef]
- Yasui, S.; Tojo, S.; Majima, T. Reaction of triarylphosphine radical cations generated from photoinduced electron transfer in the presence of oxygen. J. Org. Chem. 2005, 70, 1276–1280. [Google Scholar] [CrossRef] [PubMed]
- Corbridge, D.E.C. Phosphorus: An Outline of its Chemistry, Biochemistry and Technology, 4th ed.; Elsevier: Amsterdam, The Netherlands; New York, NY, USA, 1990; Chapter 14; pp. 994–1007. [Google Scholar]
- Engel, R.; Cohen, J.L. Synthesis of Carbon-Phosphorus Bond, 2nd ed.; CRC Press: Boca Raton, FL, USA, 1988. [Google Scholar]
- Cadogan, J.I.G. Organophosphorus Reagents in Organic Synthesis; Academic: New York, NY, USA, 1979. [Google Scholar]
- Johnson, A.W. Ylide Chemistry; Academic Press: New York, NY, USA, 1966. [Google Scholar]
- Shen, Y. New synthetic methodologies for carbon-carbon double bond formation. Acc. Chem. Res. 1998, 31, 584–592. [Google Scholar] [CrossRef]
- Bestmann, H.J.; Vostrowsky, O. Selected; topics of the witting reaction in the synthesis of natural products. Top. Curr. Chem. 2005, 109, 85–163. [Google Scholar]
- Bestmann, H.J.; Gross, A. Reaktionen von vinylketonen mit stabilisierten phosphoniumyliden. Tetrahedron Lett. 1997, 38, 4765–4768. [Google Scholar] [CrossRef]
- Nasiri, F.; Bidar, M. A facile synthesis of stable 1,4-diionic phosphorus compounds in aqueous media. Phosphorus Sulfur Silicon 2008, 183, 1344–1349. [Google Scholar] [CrossRef]
- Yavari, I.; Maghsoodlou, M.T. A facile synthesis of stable 1,4-diionic phosphorus compounds. Tetrahedron Lett. 1998, 39, 4579–4580. [Google Scholar] [CrossRef]
- Yavari, I.; Islami, M.R.; Bijanzadeh, H.R. A facile synthesis of diastereoisomeric 1,4-diionic organophosphorus compounds. Tetrahedron 1999, 55, 5547–5554. [Google Scholar] [CrossRef]
- Yavari, M.; Anary-Abbasinejad, A.; Alizadeh, A. Synthesis and dynamic NMR study of atropisomerism in stable 1,4-diionic phosphorus compounds. Phosphorus Sulfur Silicon 2002, 177, 93–103. [Google Scholar] [CrossRef]
- Shaabani, A.; Teimouri, M.B.; Yavari, I.; Arasi, H.N.; Bijanzadeh, H.R. 1,4-Diionic organophosphorus compounds: Stereoselective synthesis of dialkyl 2-(1,1,1,5,5,5-hexafluoro-2,4-dioxo-pentane-3-yl-3-yide)-3-triphenylphosphoniobutane-1,4-dioates. J. Fluor. Chem. 2000, 103, 155–157. [Google Scholar] [CrossRef]
- Yavari, I.; Hossaini, Z.; Alizadeh, A. Diastreoselective synthesis of meso-bisphosphonates from Trialkyl (aryl) phosphites and activated acetylenes in the presence of 4-nitrophenol. Mon. Chem. 2006, 137, 1083–1088. [Google Scholar] [CrossRef]
- Maghsoodlou, M.T.; Hazeri, N.; Habibi-Khorassani, S.M.; Nasiri, M.; Marandi, G.; Shahzadeh, A.G.; Bijanzadeh, H.R. Synthesis of hydroxybenzaldehyde stable phosphorus ylides from the reaction between acetylenic esters with triphenylphosphine in the presence of 2,3-dihydroxybenzaldehyde and 2-hydroxy-4-methoxybenzaldehyde. Phosphorus Sulfur Silicon 2006, 181, 1117–1122. [Google Scholar] [CrossRef]
- Maghsoodlou, M.T.; Hazeri, N.; Habibi-Khorassani, S.M.; Moeeni, Z.; Marandi, G.; Lashkari, M.; Ghasemzadeh, M. Water-acetone media enforced chemoselective synthesis of 2-substituted pyrrole stable phosphorus ylides from reaction between pyrrole and acetylenic esters in the presence of triphenylphosphine. J. Chem. Res. 2007, 10, 566–568. [Google Scholar] [CrossRef]
- Burton, D.J.; Greenwald, J.R. The reaction of triphenylphosphine with trihalo-acid derivatives-a convenient preparation of α-halo vinyl esters and nitriles. Tetrahedron Lett. 1967, 8, 1535–1539. [Google Scholar] [CrossRef]
- Charati, F.R.; Maghsoodlou, M.T.; Khorasani, S.M.H.; Makha, M. Green diastereoselective synthesis of highly functionalised trifluoromethylated γ-lactone phosphonate esters bearing a thioester or ketothiophene. Tetrahedron Lett. 2008, 49, 343–347. [Google Scholar] [CrossRef]
- Maghsoodlou, M.T.; Hazeri, N.; Habibi-Khorassani, S.M.; Mahmoodi Moghadam, H.; Nassiri, M.; Salehzadeh, A. Facile synthesis of stable phosphorus ylides containing chlorine and sulfur derived from 6-chloro-2-benzoxazolethiol and 2-chloro-phenothiazine. J. Phosphorus Sulfur Silicon Relat. Elem. 2009, 184, 1713–1721. [Google Scholar] [CrossRef]
- Heydari, R.; Maghsoodlou, M.T.; NejatYami, R. An efficient method for synthesis of organophosphorus compounds in aqueous media. Chin. Chem. Lett. 2009, 20, 1175–1178. [Google Scholar] [CrossRef]
- Maghsoodlou, M.T.; Hazeri, N.; Habibi-Khorassani, S.M.; Heydari, R.; Marandi, G.; Lashkari, M.; Bagherpour, K.; Gharechaei, Z. Synthesis of phosphonato esters involving heterocyclic biological bases in a highly diastereoselective and chemoselective route. Monatsh. Chem. 2010, 141, 351–356. [Google Scholar] [CrossRef]
- Habibi-Khorassani, S.M.; Maghsoodlou, M.T.; Ebrahimi, A.; Roohi, H.; Zakarianezhad, M. UV spectrophotometric study of the kinetics and mechanism of the reactions between triphenylphosphine, dialkyl acetylenedicarboxylates and NH-acid. J. Iran. Chem. Soc. 2006, 3, 223–232. [Google Scholar] [CrossRef]
- Habibi-Khorassani, S.M.; Maghsoodlou, M.T.; Nassiri, M.; Zakarianezhad, M.; Fattahi, M. Synthesis of stable phosphorus ylides from 3, 5-dimethylpyrazole and kinetic investigation of the reaction by UV spectrophotometry. Arkivoc 2006, 168–184. [Google Scholar]
- Shahraki, M.S.M.; Habibi-Khorassani, A.; Ebrahimi, M.; Maghsoodlou, Y. Ghalandarzehi Intramolecular hydrogen bonding in chemoselective synthesized 2-substituted pyrrole stable phosphorus ylide: GIAO, AIM, and NBO approaches. Struct. Chem. 2013, 24, 623–635. [Google Scholar] [CrossRef]
- Habibi-Khorassani, S.M.; Maghsoodlou, M.T.; Shahraki, M.; Poorshamsoddin, M.A.; Karima, M.; Abbasi, M. Sucrose catalyzes synthesis of 2-amino-4 H-chromene: Insight to the kinetics. Iran. J. Catal. 2015, 5, 79–87. [Google Scholar]
- Shaharaki, M.; Habibi-Khorassani, S.M.; Purpanah, S.S. Kinetic Aspects of Tetrahydrobenzo[b]pyran Formation in the Presence of Agar as a Green Catalyst: A Mechanistic Investigation. Biosci. Biotech. Res. Asia. 2016, 13, 715–723. [Google Scholar] [CrossRef]
- Pourpanah, S.S.H.; Habibi-Khorassani, S.M.; Shahraki, M. Fructose-catalyzed synthesis of tetrahydrobenzo[b]pyran derivatives: Investigation of kinetics and mechanism. Chin. J. Catal. 2015, 36, 757–763. [Google Scholar] [CrossRef]
- Ziyaadini, M.; Maghsoodlou, M.T.; Hazeri, N.; Habibi-Khorassani, S.M. Novel synthesis of stable 1,5-diionic organophosphorus compounds from the reaction between triphenylphosphine and acetylenedicarboxylic acid in the presence of N–H heterocyclic compounds. Monatsh. Chem. 2012, 143, 1681–1685. [Google Scholar] [CrossRef]
- Dehdab, M.; Habibi-Khorassani, S.M.; Shahraki, M. Kinetics and Mechanism Investigation of the Synthesized Highly Diasteroselective Substituted Tetrahydropyridines in the Presence of La(NO3)3·6H2O as a Catalyst. Catal. Lett. 2014, 144, 1790–1796. [Google Scholar] [CrossRef]
- Jacewicz, D.; Dąbrowska, A.; Chmurzyński, L. Stopped-Flow Spectrophotometric Study of the Kinetics and Mechanism of CO2 Uptake by cis-[Cr(C2O4)(BaraNH2)(OH2)2]+ Cation and the Acid-Catalyzed Decomposition of cis-[Cr(C2O4)(BaraNH2)OCO2]− Anion in Aqueous Solution. Molecules 2011, 16, 7746–7761. [Google Scholar] [CrossRef] [PubMed]
- Fisher, H.F. The Application of Transient-State Kinetic Isotope Effects to the Resolution of Mechanisms of Enzyme-Catalyzed Reactions. Molecules 2013, 18, 8230–8242. [Google Scholar] [CrossRef] [PubMed]
- Habibi-Khorassani, S.M.; Ebrahimi, A.; Maghsoodlou, M.; Asheri, O.; Shahraki, M.; Akbarzadeh, N.; Ghalandarzehi, Y. Kinetic and mechanistic insights into the pathway leading to cyclic crystalline phosphorus ylide formation in the presence of 3-chloropentane-2,4-dione: Theoretical and stopped-flow approaches. Int. J. Chem. Kinet. 2013, 45, 596–612. [Google Scholar] [CrossRef]
- Shahraki, M.; Habibi-Khorassani, S.M. Kinetic spectrophotometric approach to the reaction mechanism of pyrrole phosphorus ylide formation based on monitoring the zwitterionic intermediate by using the stopped-flow technique. J. Phys. Org. Chem. 2015, 28, 396–402. [Google Scholar] [CrossRef]
- Arnett, E.M.; Anderson, J.A. Ion pairing and reactivity of enolate anions. 7. A spectacular example of the importance of rotational barriers: The ionization of Meldrum’s acid. J. Am. Chem. Soc. 1987, 109, 809–812. [Google Scholar] [CrossRef]
- Wang, X.; Houk, K.N. Theoretical elucidation of the origin of the anomalously high acidity of Meldrum’s acid. J. Am. Chem. Soc. 1988, 110, 1870–1872. [Google Scholar] [CrossRef]
- Lee, I.; Han, I.S.; Kim, C.H.; Lee, H.W. Theoretical Studies on the Structure and Acidity of Meldrum’s Acid and Related Compounds. Bull. Korean Chem. Soc. 2003, 24, 1141–1149. [Google Scholar]
- Evansek, J.D.; Houk, K.N.; Biggs, J.M.; Jorgensen, W.L. Quantification of Solvent Effects on the Acidities of Z and E Esters from Fluid Simulations. J. Am. Chem. Soc. 1994, 110, 10630–10634. [Google Scholar] [CrossRef]
- Schwartz, L.M.; Gelb, R.I. Alternative method of analyzing first-order kinetic data. Anal. Chem. 1978, 50, 1592–1594. [Google Scholar] [CrossRef]
- Lente, G.; Fabian, I.; Poe, A.J. A common misconception about the Eyring equation. New J. Chem. 2005, 29, 759–760. [Google Scholar] [CrossRef]
- Wang, R.Y.; Lu, Y.T. Kinetic spectrophotometric method for determination of perphenazine based on monitoring the oxidation intermediate by applying a stopped-flow technique. Spectrochim. Acta Part A 2005, 61, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.; Jorgenson, J.W. Automated instrumentation for comprehensive two-dimensional high-performance liquid chromatography of proteins. Anal. Chem. 1990, 62, 161–167. [Google Scholar]
- Henry, E.R.; Hofrichter, J. Singular value decomposition: Application to analysis of experimental data. J. Methods Enzymol. 1992, 210, 129–192. [Google Scholar]
- Ramazani, A.; Kazemizadeh, A.R.; Ahmadi, E.; Noshiranzadeh, N.; Souldozi, A. synthesis and reactions of stabilized phosphorus ylides. Curr. Organ. Chem. 2008, 12, 59–82. [Google Scholar] [CrossRef]
- Habibi-Khorassani, S.M.; Ebrahimi, A.; Maghsoodlou, M.T.; Shahraki, M.; Price, D. Establishing a new conductance stopped-flow apparatus to investigate the initial fast step of reaction between 1,1,1-trichloro-3-methyl-3-phospholene and methanol under a dry inert. Analyst 2011, 136, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- Shahraki, M.; Habibi-Khorassani, S.M.; Dehdab, M. Effect of different substituents on the one-pot formation of 3,4,5-substituted furan-2(5H)-ones: A kinetics and mechanism study. RSC Adv. 2015, 5, 52508–52515. [Google Scholar] [CrossRef]
- Darijani, M.; Habibi-Khorassani, S.M.; Shahraki, M. A Thermodynamic and Kinetic Insight into the Pathways Leading to a Highly Functionalized Ketenimine: A Computational Study. Int. J. Chem. Kinet. 2015, 47, 751–763. [Google Scholar] [CrossRef]
- Caesar, J.C.; Griffiths, D.V.; Griffiths, P.A.; Tebby, J.C. Studies of the reaction of trivalent phosphorus compounds with dialkyl acetylenedicarboxylates in the presence of carbon dioxide. J. Chem. Soc. Perkin Trans. 1 1989, 2425–2430. [Google Scholar] [CrossRef]
- Caesar, J.C.; Griffiths, D.V.; Griffiths, P.A.; Tebby, J.C. Reactions of ylides formed from trialkyl phosphites with dialkyl acetylenedicarboxylates in the presence of carbon dioxide. J. Chem. Soc. Perkin Trans. 1 1990, 2329–2334. [Google Scholar] [CrossRef]
- Dillon, R.L.; Pearson, R.G. Rates of Ionization of Pseudo Acids.1 IV. Relation between Rates and Equilibria. J. Am. Chem. Soc. 1953, 75, 2439–2443. [Google Scholar]
- Ireland, R.E.; Wipf, P.; Armstrong, J.D. Stereochemical control in the ester enolate Claisen rearrangement. 1. Stereoselectivity in silyl ketene acetal formation. J. Org. Chem. 1991, 56, 650–657. [Google Scholar] [CrossRef]
- Buncel, E.; Dust, J.M. Carbanion Chemistry Structure and Mechanisms; Oxford University Press: Washington, WA, USA, 2003; pp. 268–269, 282–285. [Google Scholar]
- Cram, D.J.; Uyeda, R.T. Electrophilic substitution at saturated carbon. L. Isoinversion without an available conducted tour pathway for proton transfer. J. Am. Chem. Soc. 1972, 94, 3521–3531. [Google Scholar]
- Sample Availability: Not Available.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | Ethanol (24.7 a) |
|---|---|
| k1 (M−1·S−1) | 1.02 × 102 |
| Completion time (s) | 20 |
| Temperature | Solvent: Ethanol (24.7) a | Temperature | Temperature | |
| T | T = 288.15 K | T = 293.15 K | T = 298.15 K | T = 303.15 K |
| kobs | 0.750 (0.002) b | 0.870 (0.002) | 0.990 (0.002) | 1.14 (0.002) |
| Solvent: 1,2-Dicholoroethan (10.37) a | ||||
| T | T = 293.15 K | |||
| kobs | 3.65 (0.004) | |||
| Solvent | ΔH‡ (kJ·mol−1) | ΔS‡ (J·mol−1) | TΔS‡ (kJ·mol−1) | ΔG‡ (kJ·mol−1) | Ln A | Ea (kJ/mol) |
|---|---|---|---|---|---|---|
| Ethanol (24.7) a | 17.72 ± 0.07 b1 | −185.49 ± 0.03 b1 | −54.38 b1 | 71.17 ± 0.02 b1 | 8.14 c | 20.16 c 20.17 d |
| 17.72 ± 0.05 b2 | −185.49± 0.02 b2 | −54.38 b2 | 72.09 ± 0.01 b2 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghodsi, F.; Habibi-Khorassani, S.M.; Shahraki, M. Kinetic Spectrophotometric Method for the 1,4-Diionic Organophosphorus Formation in the Presence of Meldrum′s Acid: Stopped-Flow Approach. Molecules 2016, 21, 1514. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21111514
Ghodsi F, Habibi-Khorassani SM, Shahraki M. Kinetic Spectrophotometric Method for the 1,4-Diionic Organophosphorus Formation in the Presence of Meldrum′s Acid: Stopped-Flow Approach. Molecules. 2016; 21(11):1514. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21111514
Chicago/Turabian StyleGhodsi, Fatemeh, Sayyed Mostafa Habibi-Khorassani, and Mehdi Shahraki. 2016. "Kinetic Spectrophotometric Method for the 1,4-Diionic Organophosphorus Formation in the Presence of Meldrum′s Acid: Stopped-Flow Approach" Molecules 21, no. 11: 1514. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21111514