
Synthesis and Pharmacological Evaluation of Novel 1-(1,4-Alkylaryldisubstituted-4,5-dihydro-1H-imidazo)-3-substituted Urea Derivatives

Abstract
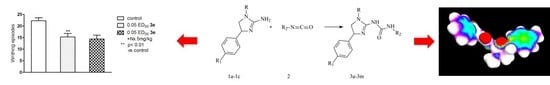
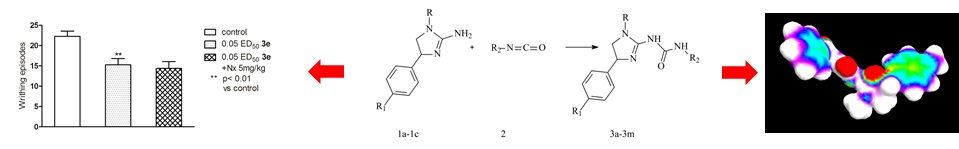
:
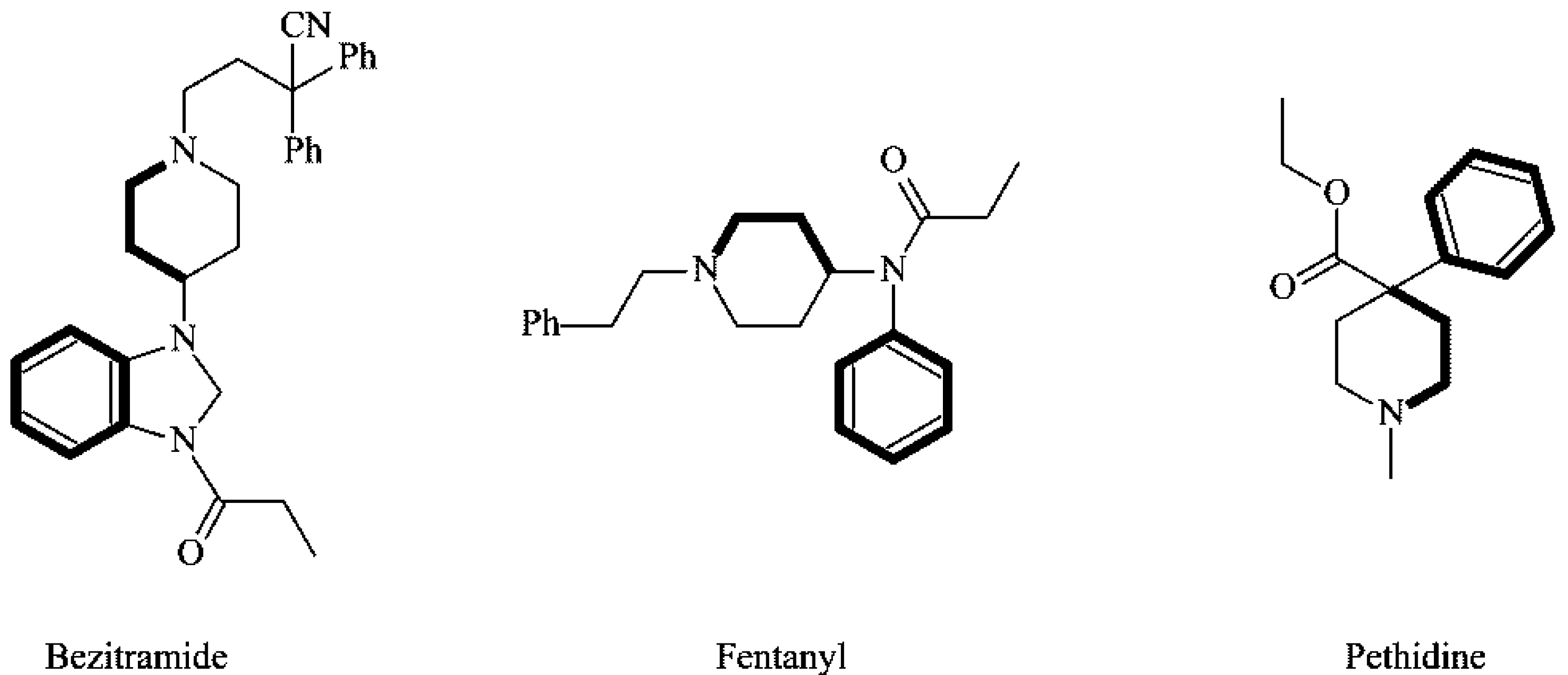
1. Introduction
2. Results and Discussion
2.1. Chemistry
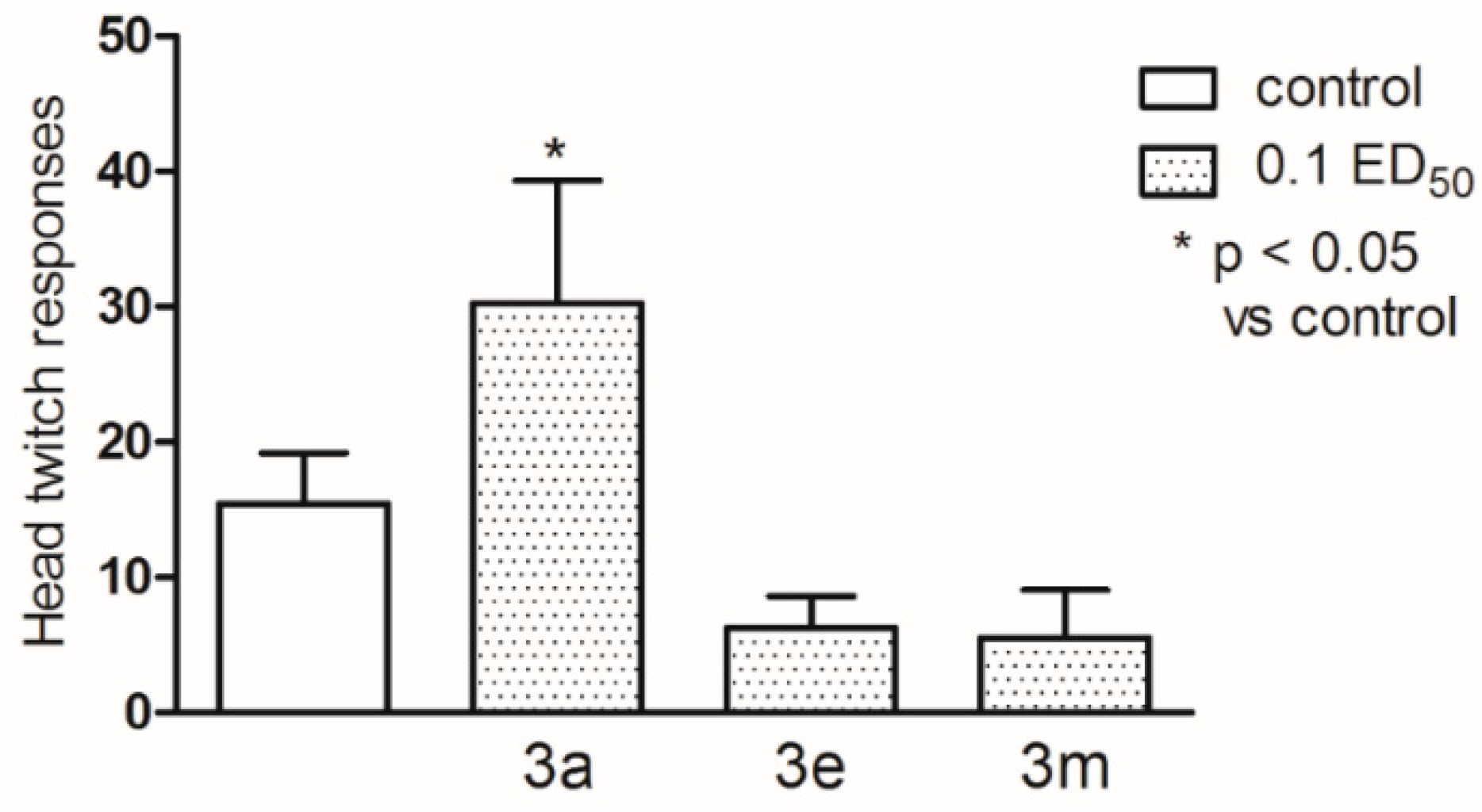
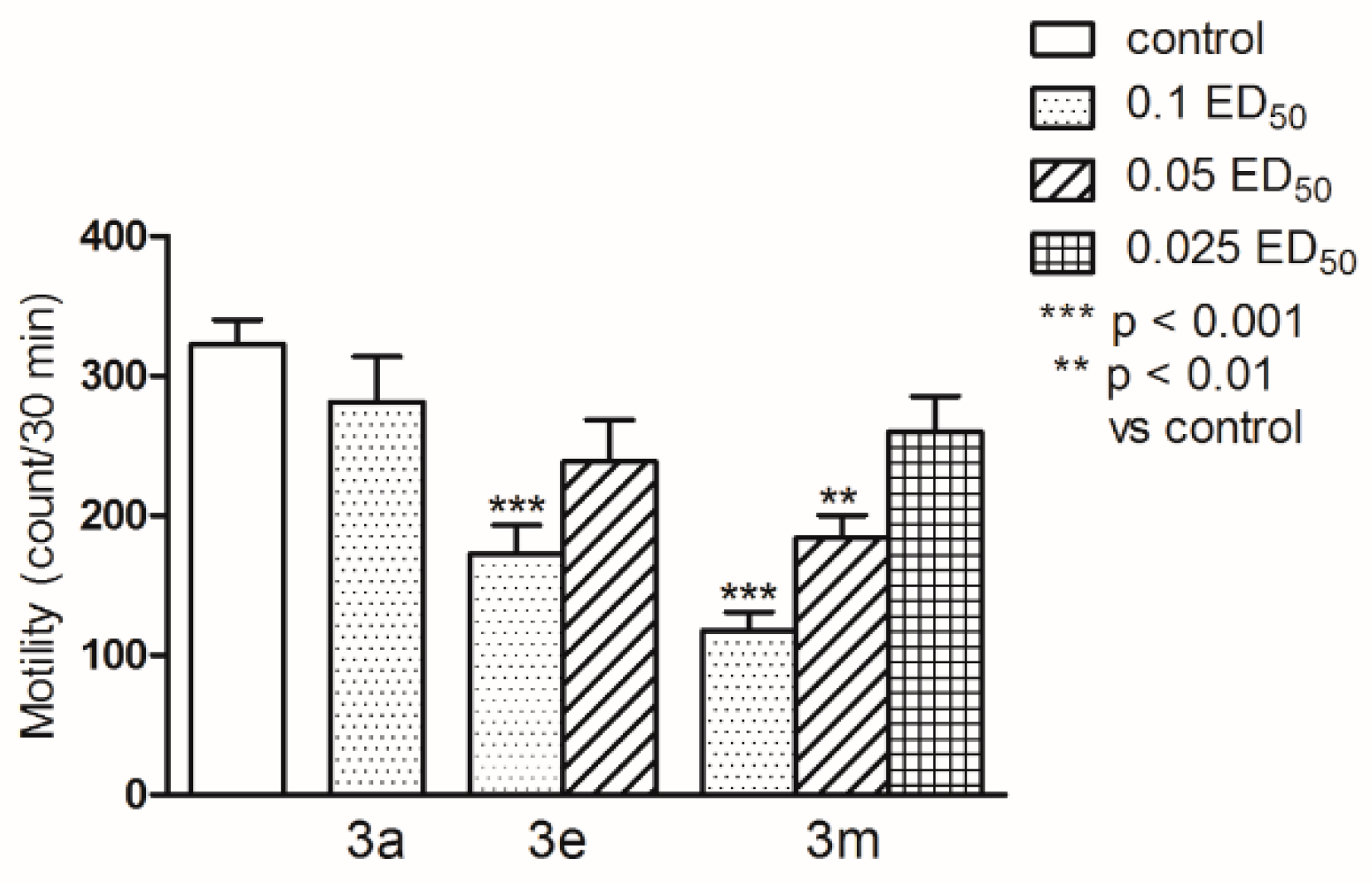
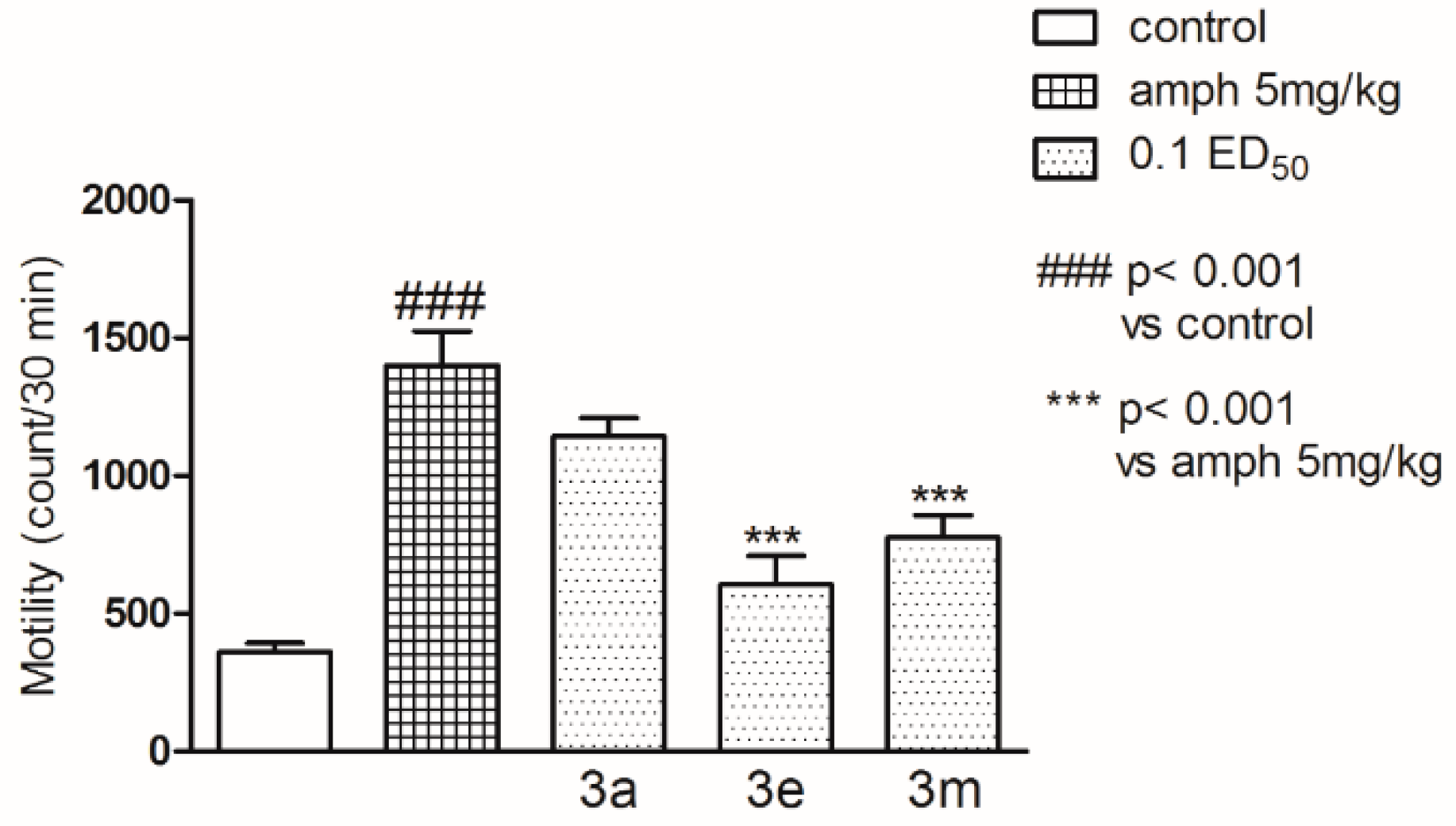
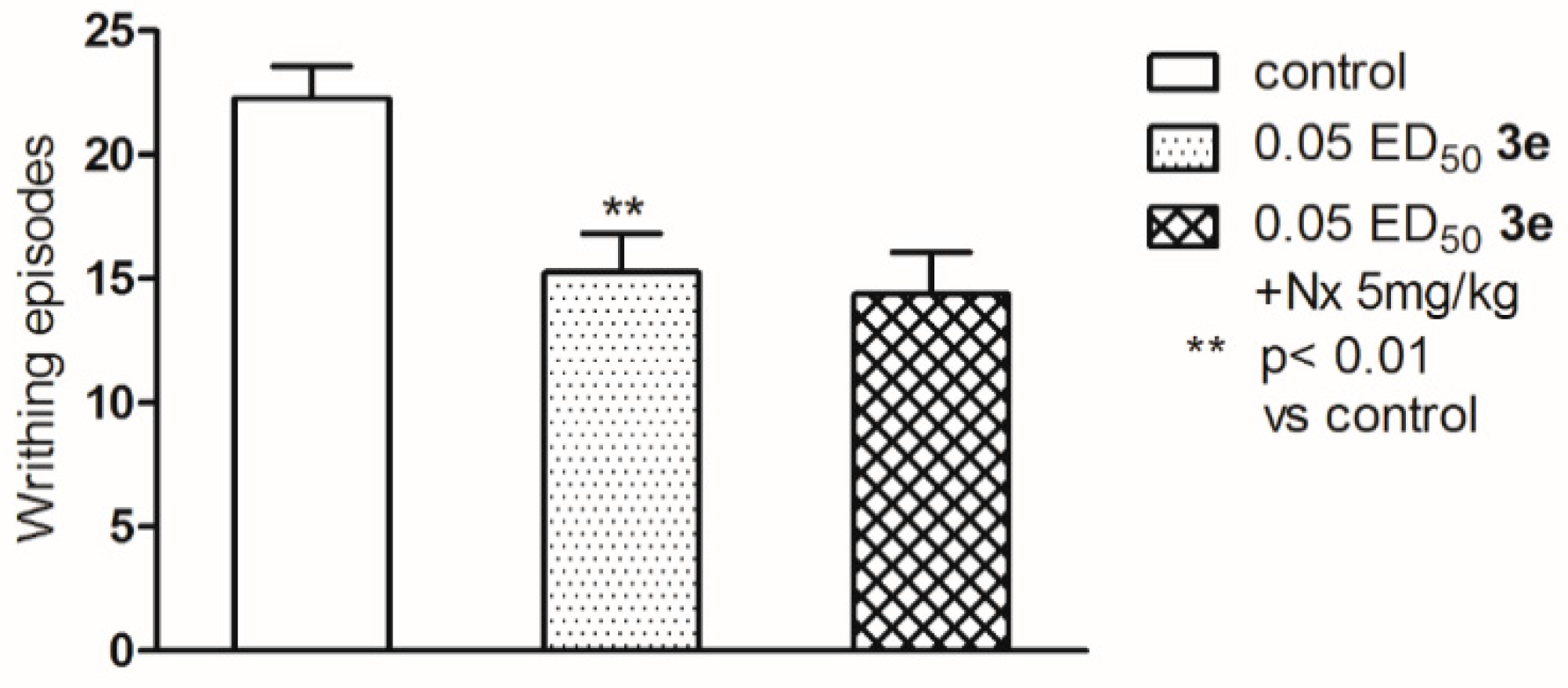
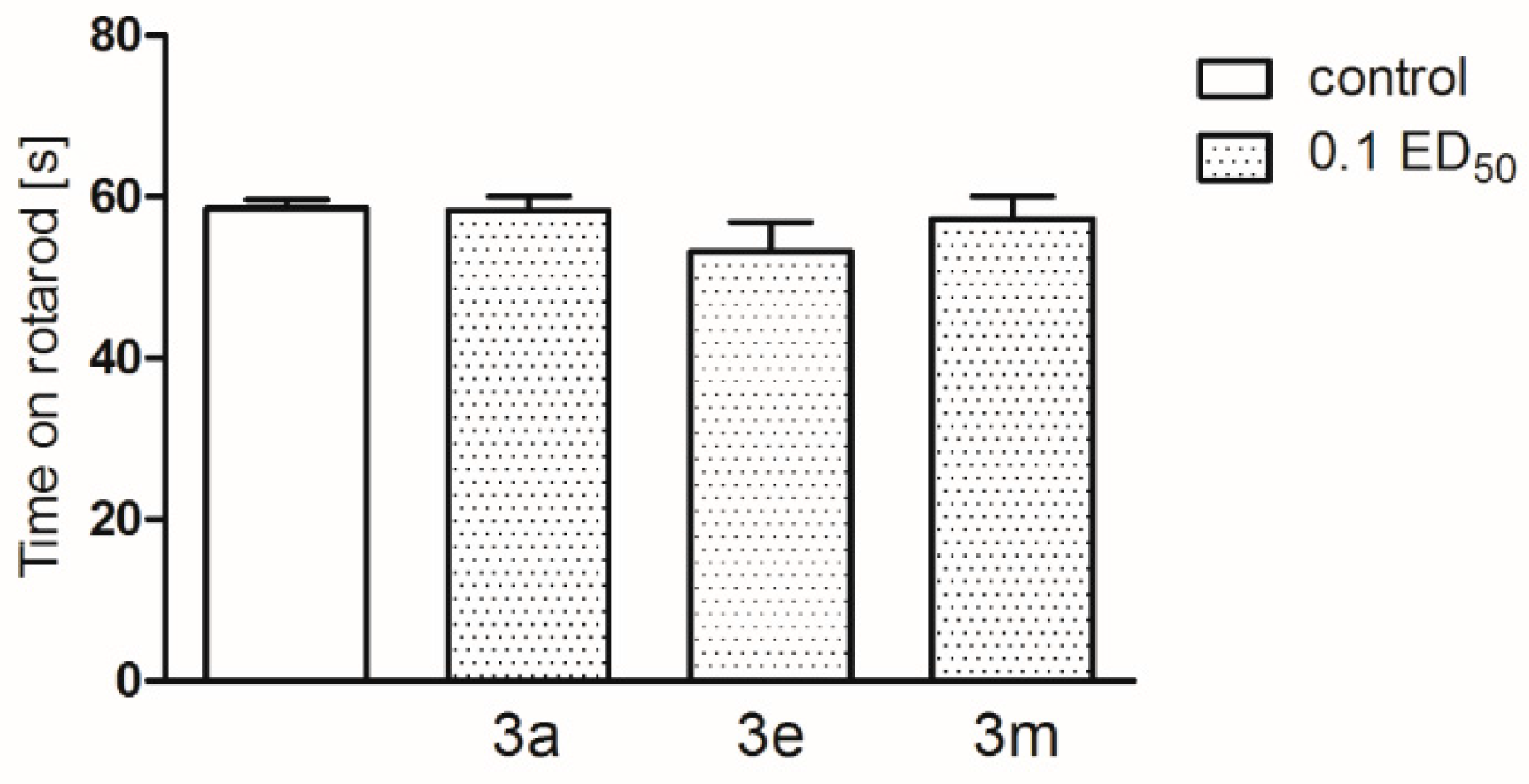
2.2. Pharmacological Activity
3. Materials and Methods
3.1. General Information
3.2. General Procedure for the Synthesis of Compounds 3a–m
3.2.1. 1-(1-Methyl-4-phenyl-4,5-dihydro-1H-imidazo)-3-benzoilourea (3a)
3.2.2. 1-(1-Methyl-4-phenyl-4,5-dihydro-1H-imidazo)-3-(4-acetylophenyl)urea (3b)
3.2.3. 1-(1-Methyl-4-phenyl-4,5-dihydro-1H-imidazo)-3-cyclopenthylurea (3c)
3.2.4. 1-(1-Methyl-4-phenyl-4,5-dihydro-1H-imidazo)-3-(2-chloroethyl)urea (3d)
3.2.5. 1-[1-Methyl-4-(4methylphenyl)-4,5-dihydro-1H-imidazo]-3-benzoilourea (3e)
3.2.6. 1-[1-Methyl-4-(4methylphenyl)-4,5-dihydro-1H-imidazo]-3-(4-acetylophenyl)urea (3f)
3.2.7. 1-[1-Methyl-4(4-methylphenyl)-4,5-dihydro-1H-imidazo]-3-cyclopenthylurea (3g)
3.2.8. 1-[1-Methyl-4(4-methylphenyl)-4,5-dihydro-1H-imidazo]-3-(2-chloroethyl)urea (3h)
3.2.9. 1-(1-Ethyl-4-phenyl-4,5-dihydro-1H-imidazo)-3-benzoilourea (3i)
3.2.10. 1-(1-Ethyl-4-phenyl-4,5-dihydro-1H-imidazo)-3-(4-acetylophenyl)urea (3j)
3.2.11. 1-(1-Ethyl-4phenyl-4,5-dihydro-1H-imidazo)-3-cyclopenthylurea (3k)
3.2.12. 1-(1-Ethyl-4-phenyl-4,5-dihydro-1H-imidazo)-3-(2-chloroethyl)urea (3l)
3.2.13. 1-(1-Ethyl-4-phenyl-4,5-dihydro-1H-imidazo)-3-(4-etoxycarbonylphenyl)urea (3m)
3.3. Molecular Modeling
3.4. Pharmacology
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Eguchi, M. Recent advances in selective opioid receptor agonists and antagonists. Med. Res. Rev. 2004, 24, 182–212. [Google Scholar] [PubMed]
- Rządkowska, M.; Szacoń, E.; Kaczor, A.A.; Fidecka, S.; Kędzierska, E.; Matosiuk, D. Synthesis, pharmacological activity and molecular modeling of 1-aryl-7-hydroxy-2,3-dihydroimidazo(1,2-a)pyrimidine-5(1H)-ones and their 6-substituted derivatives. Med. Chem. 2014, 10, 460–475. [Google Scholar] [CrossRef] [PubMed]
- Kaczor, A.A.; Matosiuk, D. Non-peptide opioid receptor ligands—Recent advances. Part I—Agonists. Curr. Med. Chem. 2002, 9, 1567–1589. [Google Scholar] [CrossRef] [PubMed]
- Kaczor, A.; Matosiuk, D. Non-peptide opioid receptor ligands—Recent advances. Part II-Antagonists. Curr. Med. Chem. 2002, 9, 1591–1603. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Kim, S.; Loew, G. Development of a common 3D pharmacophore for delta-opioid recognition from peptides and non-peptides using a novel computer program. J. Comput. Aided. Mol. Des. 1997, 11, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Rządkowska, M.; Szacoń, E.; Kaczor, A.A.; Fidecka, S.; Kędzierska, E.; Matosiuk, D. Synthesis, central nervous system activity, and structure–activity relationship of 1-aryl-6-benzyl-7-hydroxy-2,3-dihydroimidazo(1,2-a)pyrimidine-5(1H)-ones. Med. Chem. Res. 2014, 23, 4221–4237. [Google Scholar] [CrossRef] [PubMed]
- Szacoń, E.; Rządkowska, M.; Kaczor, A.A.; Kędzierska, E.; Mazur, A.; Fidecka, S.; Matosiuk, D. Synthesis, central nervous system activity and structure-activity relationship of N-substituted derivatives of 1-arylimidazolidyn-2-ylideneurea and products of their cyclization. J. Enzyme Inhib. Med. Chem. 2015, 30, 746–760. [Google Scholar] [CrossRef] [PubMed]
- Szacoń, E.; Rządkowska, M.; Kaczor, A.A.; Kędzierska, E.; Mazur, A.; Fidecka, S.; Matosiuk, D. Synthesis, central nervous system activity and structure-activity relationships of novel 1-(1-alkyl-4-aryl-4,5-dihydro-1H-imidazo)-3-substituted urea derivatives. Molecules 2015, 20, 3821–3840. [Google Scholar] [CrossRef] [PubMed]
- Matosiuk, D.; Fidecka, S.; Antkiewicz-Michaluk, L.; Dybała, I.; Kozioł, A.E. Synthesis and pharmacological activity of new carbonyl derivatives of 1-aryl-2-iminoimidazolidine. Part 1. Synthesis and pharmacological activity of chain derivatives of 1-aryl-2-iminoimidazolidine containing urea moiety. Eur. J. Med. Chem. 2001, 36, 783–797. [Google Scholar] [CrossRef]
- Matosiuk, D.; Fidecka, S.; Antkiewicz-Michaluk, L.; Dybała, I.; Kozioł, A.E. Synthesis and pharmacological activity of new carbonyl derivatives of 1-aryl-2-iminoimidazolidine. Part 3. Synthesis and pharmacological activity of 1-aryl-5,6(1H)dioxo-2,3-dihydroimidazo[1,2-a]imidazoles. Eur. J. Med. Chem. 2002, 37, 845–853. [Google Scholar] [CrossRef]
- Matosiuk, D.; Fidecka, S.; Antkiewicz-Michaluk, L.; Lipkowski, J.; Dybała, I.; Kozioł, A.E. Synthesis and pharmacological activity of new carbonyl derivatives of 1-aryl-2-iminoimidazolidine: Part 2. Synthesis and pharmacological activity of 1,6-diaryl-5,7(1H)dioxo-2,3-dihydroimidazo[1,2-a][1,3,5]triazines. Eur. J. Med. Chem. 2002, 37, 761–772. [Google Scholar] [CrossRef]
- Sztanke, K.; Fidecka, S.; Kędzierska, E.; Karczmarzyk, Z.; Pihlaja, K.; Matosiuk, D. Antinociceptive activity of new imidazolidine carbonyl derivatives. Part 4. Synthesis and pharmacological activity of 8-aryl-3,4-dioxo-2H,8H-6,7-dihydroimidazo(2,1-c)(1,2,4)triazines. Eur. J. Med. Chem. 2005, 40, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, L.T.; Wilcoxon, F. Simplified method of evaluating dose effect experiments. J. Pharmacol. Exp. Ther. 1949, 96, 99–113. [Google Scholar] [PubMed]
- Hermersdörfer, H. Handbook of Toxicology; Derelanko, M.J., Hollinger, M.A., Eds.; CRC Press: Boca Raton, FL, 1996. [Google Scholar]
- Tadanoa, M.; Hozumib, N.; Satoha, R.; Okaa, T.; Hishinumab, M.; Mizugakib, Y.; Araic, H.; Yasuharac, H.; Kinemuchid, F.; Niijimaa, O.; et al. Central Serotonergic Mechanisms on Head Twitch Response Induced by Benzodiazepine Receptor Agonists. Pharmacology 2001, 62, 157–162. [Google Scholar] [CrossRef]
- Peroutka, S.J.; Lebovitz, R.M.; Snyder, S.H. Two distinct central serotonin receptors with different physiological functions. Science 1981, 212, 827–829. [Google Scholar] [CrossRef] [PubMed]
- Colpaert, F.C.; Janssen, P.A. The head-twitch response to intraperitoneal injection of 5-hydroxytryptophan in the rat: Antagonist effects of purported 5-hydroxytryptamine antagonists and of pirenperone, an LSD antagonist. Neuropharmacology 1983, 22, 993–1000. [Google Scholar] [CrossRef]
- Green, A.R.; O’Shaughnessy, K.; Hammond, M.; Schachter, M.; Grahame-Smith, D.G. Inhibition of 5-hydroxytryptamine-mediated behaviour by the putative 5-HT2 antagonist pirenperone. Neuropharmacology 1983, 22, 573–578. [Google Scholar] [CrossRef]
- Goodwin, G.M.; Green, A.R. A behavioural and biochemical study in mice and rats of putative selective agonists and antagonists for 5-HT1 and 5-HT2 receptors. Br. J. Pharmacol. 1985, 84, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Darmani, N.A.; Martin, B.R.; Glennon, R.A. Withdrawal from chronic treatment with (+/−)-DOI causes super-sensitivity to 5-HT2 receptor-induced head-twitch behaviour in mice. Eur. J. Pharmacol. 1990, 186, 115–118. [Google Scholar] [PubMed]
- Darmani, N.A.; Martin, B.R.; Pandy, U.; Glennon, R.A. Do functional relationships exist between 5-HT1A and 5-HT2 receptors? Pharmacol. Biochem. Behav. 1990, 36, 901–906. [Google Scholar] [CrossRef]
- Darmani, N.A.; Martin, B.R.; Glennon, R.A. Behavioral evidence for differential adaptation of the serotonergic system after acute and chronic treatment with (±)-1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane (DOI) or ketanserin. J. Pharmacol. Exp. Ther. 1992, 262, 692–698. [Google Scholar] [PubMed]
- Fantegrossi, W.E.; Kiessel, C.L.; Leach, P.T.; van Martin, C.; Karabenick, R.L.; Chen, X.; Ohizumi, Y.; Ullrich, T.; Rice, K.C.; Woods, J.H. Nantenine: An antagonist of the behavioral and physiological effects of MDMA in mice. Psychopharmacology 2004, 173, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Ortmann, R.; Biscoff, S.; Radeke, E.; Bueche, O.; Delini-Stula, A. Correlation between different measures of antiserotonin activity of drugs. Naunyn Schmiedeberg’s Arch. Pharmacology 1982, 321, 265–270. [Google Scholar]
- Lucki, I.; Nobler, M.S.; Frazer, A. Differential actions of serotonin antagonists on two behavioral models of serotonin receptor activation in the rat. J. Pharmacol. Exp. Ther. 1984, 228, 133–139. [Google Scholar] [PubMed]
- Corne, S.; Pickering, R.W. A possible correlation between druginduced hallucinations in man and a behavioural response in mice. Psychopharmacology 1967, 11, 65–68. [Google Scholar] [CrossRef]
- Handley, S.L.; Brown, J. Effects on the 5-hydroxytryptamine-induced head-twitch of drugs with selective actions on alpha1 and alpha2-adrenoceptors. Neuropharmacology 1982, 21, 507–510. [Google Scholar] [CrossRef]
- Corne, S.J.; Pickering, R.W.; Werner, B.T. A method for assessing the effects of drugs on the central actions of 5-hydroxytryptamine. Br. J. Pharmacol. 1963, 20, 106–120. [Google Scholar] [CrossRef]
- Chueh, F.S.; Chang, C.P.; Chio, C.C.; Lin, M.T. Puerarin acts through brain serotonergic mechanisms to induce thermal effects. J. Pharmacol. Sci. 2004, 96, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Kashaw, S.K.; Kashaw, V.; Mishra, P.; Jain, N.K.; Stables, J.P. Synthesis, anticonvulsant and CNS depressant activity of some new bioactive 1-(4-substituted-phenyl)-3-(4-oxo-2-phenyl/ethyl-4H-quinazolin-3-yl)-urea. Eur. J. Med. Chem. 2009, 44, 4335–4343. [Google Scholar] [CrossRef] [PubMed]
- Archer, T.; Fredriksson, A.; Jonsson, G.; Lewander, T.; Mohammed, A.K.; Ross, S.B.; Soderberg, U. Central noradrenaline depletion antagonizes aspects of d-amphetamine-induced hyperactivity in the rat. Psychopharmacology 1986, 88, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Archer, T.; Jonsson, G.; Ross, S.B. Active and passive avoidance following the administration of systemic DSP4, xylamine, or pchloroamphetamine. Behav. Neural. Biol. 1985, 43, 238–249. [Google Scholar] [CrossRef]
- Juhila, J.; Haapalinna, A.; Sirvio, J.; Sallinen, J.; Honkanen, A.; Korpi, E.R.; Scheinin, M. The α2-adrenoceptor antagonist atipamezole reduces the development and expression of d-amphetamine-induced behavioural sensitization. Naunyn-Schmiedeberg’s Arch. Pharmacology 2003, 367, 274–280. [Google Scholar]
- Juhila, J.; Honkanen, A.; Sallinen, J.; Haapalinna, A.; Korpi, E.R.; Scheinin, M. α2-Adrenoceptors regulate d-amphetamine-induced hyperactivity and behavioural sensitization in mice. Eur. J. Pharmacol. 2005, 517, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.; Danysz, W.; Ogren, S.O.; Archer, T. Central noradrenaline depletion attenuates amphetamine-induced locomotor behavior. Neurosci. Lett. 1986, 64, 139–144. [Google Scholar] [CrossRef]
- Ogren, S.O.; Archer, T.; Johansson, C. Evidence for a selective brain noradrenergic involvement in the locomotor stimulant effects of amphetamine in the rat. Neurosci. Lett. 1983, 43, 327–331. [Google Scholar] [CrossRef]
- Jackson, D.M.; Westlind-Danielsson, A. Dopamine receptors: Molecular biology, biochemistry and behavioural aspects. Pharmacol. Ther. 1994, 64, 291–370. [Google Scholar] [CrossRef]
- Abrahams, B.S.; Rutherford, J.D.; Mallet, P.E.; Beninger, R.J. Place conditioning with the dopamine D1-like receptor agonist SKF 82958 but not SKF 81297 or SKF 77434. Eur. J. Pharmacol. 1998, 343, 111–118. [Google Scholar] [CrossRef]
- Isacson, R.; Kull, B.; Wahlestedt, C.; Salmi, P. A 68930 and dihydrexidine inhibit locomotor activity and d-amphetamine- induced hyperactivity in rats: A role of inhibitory dopamine D1/5 receptors in the prefrontal cortex? Neuroscience 2004, 124, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Koster, R.; Anderson, M.; DeBeer, E.J. Acetic acid for analgesic screening. Fed. Proc. 1959, 18, 412–416. [Google Scholar]
- Le Bars, D.; Gozariu, M.; Cadden, S.W. Animal models of nociception. Pharmacol. Rev. 2001, 53, 597–652. [Google Scholar] [PubMed]
- Gutstein, H.B.; Akil, H. Opioid analgesics. W: Goodman I Gilman’s, The Pharmacological basis of therapeutics XIth edition; MacGrow Hill: City, NY, USA, 2006; pp. 547–590. [Google Scholar]
- LigPrep, version 2.4; Schrödinger; LLC: New York, NY, USA, 2010.
- Epik, version 2.1; Schrödinger; LLC: New York, NY, USA, 2010.
- Spartan 10. Available online: www.wavefun.com (accessed on 15 December 2015).
- Pedretti, A.; Villa, L.; Vistoli, G. VEGA-an open platform to develop chemo-bio-informatic applications, using plug-in architecture and script programming. J. Comput. Aided. Mol. Des. 2004, 18, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Dassault Systèmes BIOVIA. Discovery Studio Modeling Environment, Release 4.5; Dassault Systèmes: San Diego, CA, USA, 2015. [Google Scholar]
- Osiris Property Explorer. Available online: http://www.organic-chemistry.org/prog/peo/logS.html (accessed on 15 December 2015).
- Vogel, G.H.; Vogel, W.H. Drug Discovery and Evaluation: Pharmacological Assays; Springer-Verlag: Berlin, Germany, 1997. [Google Scholar]
- Talarek, S.; Fidecka, S. Role of nitric oxide in benzodiazepines-induced antinociception in mice. Pol. J. Pharmacol. 2002, 54, 27–34. [Google Scholar] [PubMed]
- Gross, F.; Tripod, J.; Meir, R. Zur pharmakologischen Charakterisierung des Schalafmittelsdoriden. Schweiz Med. Wochschr. 1955, 85, 305–309. [Google Scholar]
- Boissier, J.R.; Tardy, J.; Diverres, J.C. Une nouvelle méthode simple pour explorer l’actiontranquilisante: Le test de la cheminée. Med. Exp. 1960, 3, 81–84. [Google Scholar]
- Sample Available: Samples of the compounds are not available from the authors.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ED50 (mg/kg) | Dose |
|---|---|---|
| 3a | 330.2 | 0.1 ED50 |
| 3e | 1133.7 | 0.1; 0.05; 0.025 ED50 |
| 3m | 2000 | 0.1; 0.05; 0.025 ED50 |
| Compound | Clonic Seizures # | Tonic Convulsions # | Mortality ^ |
|---|---|---|---|
| 0.1 ED50 of 3a | 8/10 | 4/10 | 4/10 |
| 0.1 ED50 of 3e | 6/10 * | 5/10 | 3/10 |
| 0.05 ED50 of 3e | 9/10 | 5/10 | 5/10 |
| 0.1 ED50 of 3m | 9/10 | 8/10 | 7/10 |
| Control (PTZ 110 mg/kg) | 12/12 | 8/12 | 7/12 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szacoń, E.; Rządkowska, M.; Kaczor, A.A.; Kędzierska, E.; Orzelska-Górka, J.; Fidecka, S.; Matosiuk, D. Synthesis and Pharmacological Evaluation of Novel 1-(1,4-Alkylaryldisubstituted-4,5-dihydro-1H-imidazo)-3-substituted Urea Derivatives. Molecules 2016, 21, 582. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21050582
Szacoń E, Rządkowska M, Kaczor AA, Kędzierska E, Orzelska-Górka J, Fidecka S, Matosiuk D. Synthesis and Pharmacological Evaluation of Novel 1-(1,4-Alkylaryldisubstituted-4,5-dihydro-1H-imidazo)-3-substituted Urea Derivatives. Molecules. 2016; 21(5):582. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21050582
Chicago/Turabian StyleSzacoń, Elżbieta, Marzena Rządkowska, Agnieszka A. Kaczor, Ewa Kędzierska, Jolanta Orzelska-Górka, Sylwia Fidecka, and Dariusz Matosiuk. 2016. "Synthesis and Pharmacological Evaluation of Novel 1-(1,4-Alkylaryldisubstituted-4,5-dihydro-1H-imidazo)-3-substituted Urea Derivatives" Molecules 21, no. 5: 582. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21050582