
Flavin-Dependent Thymidylate Synthase as a New Antibiotic Target

Abstract
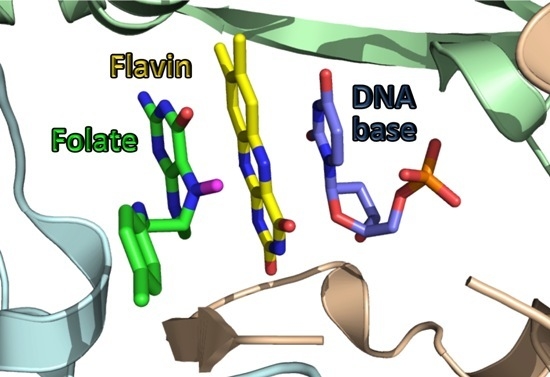
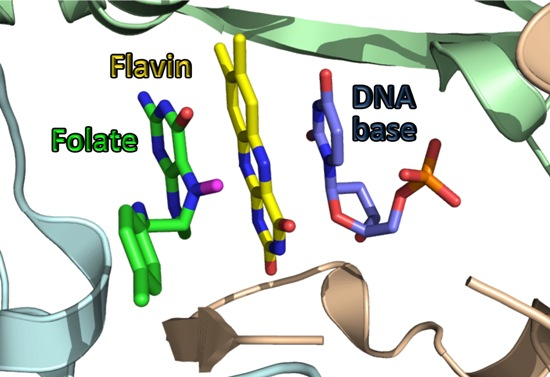
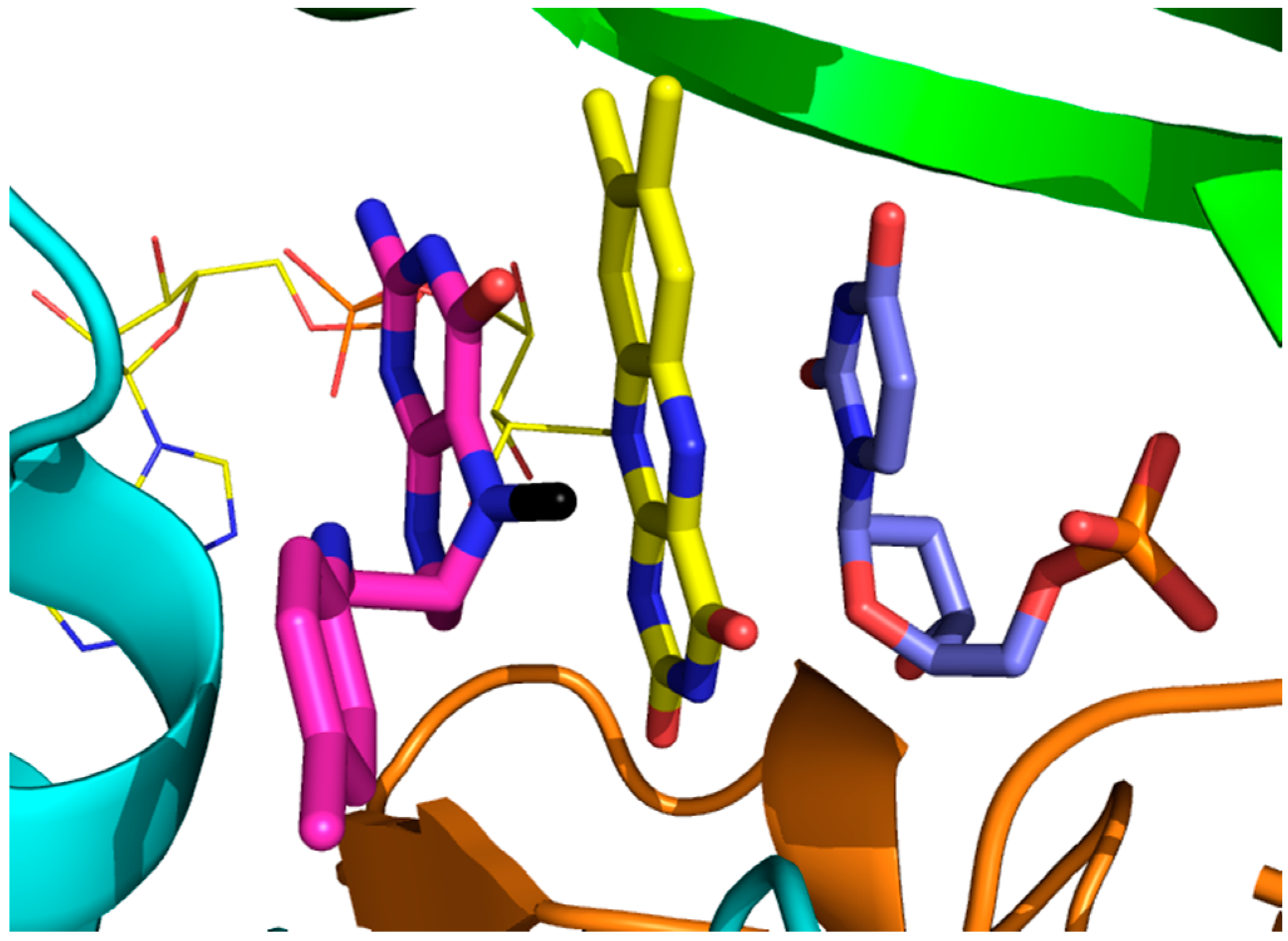
:
1. Introduction
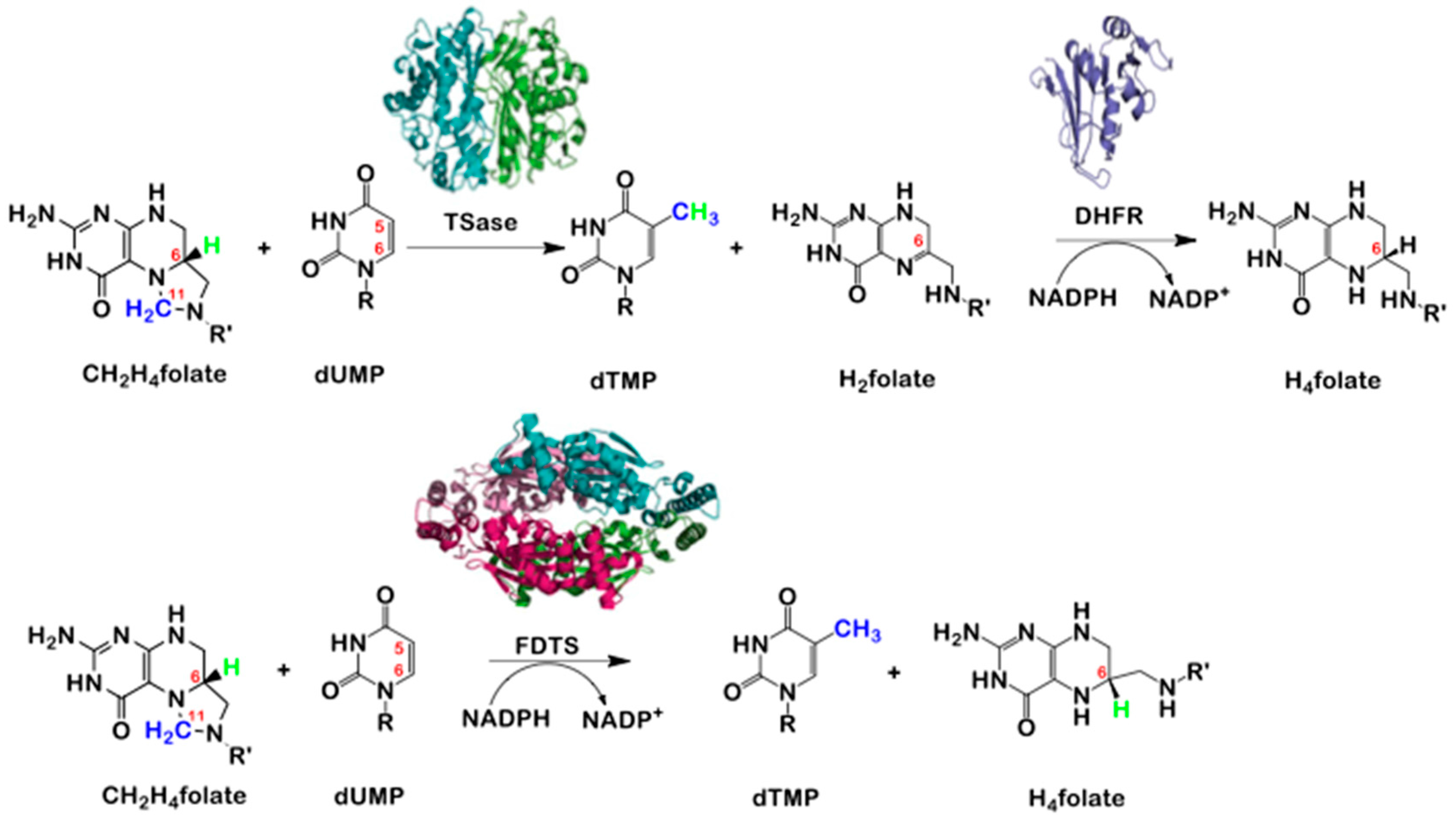
2. Thymidylate Synthase (TSase) vs. Flavin Dependent Thymidylate Synthase (FDTS)
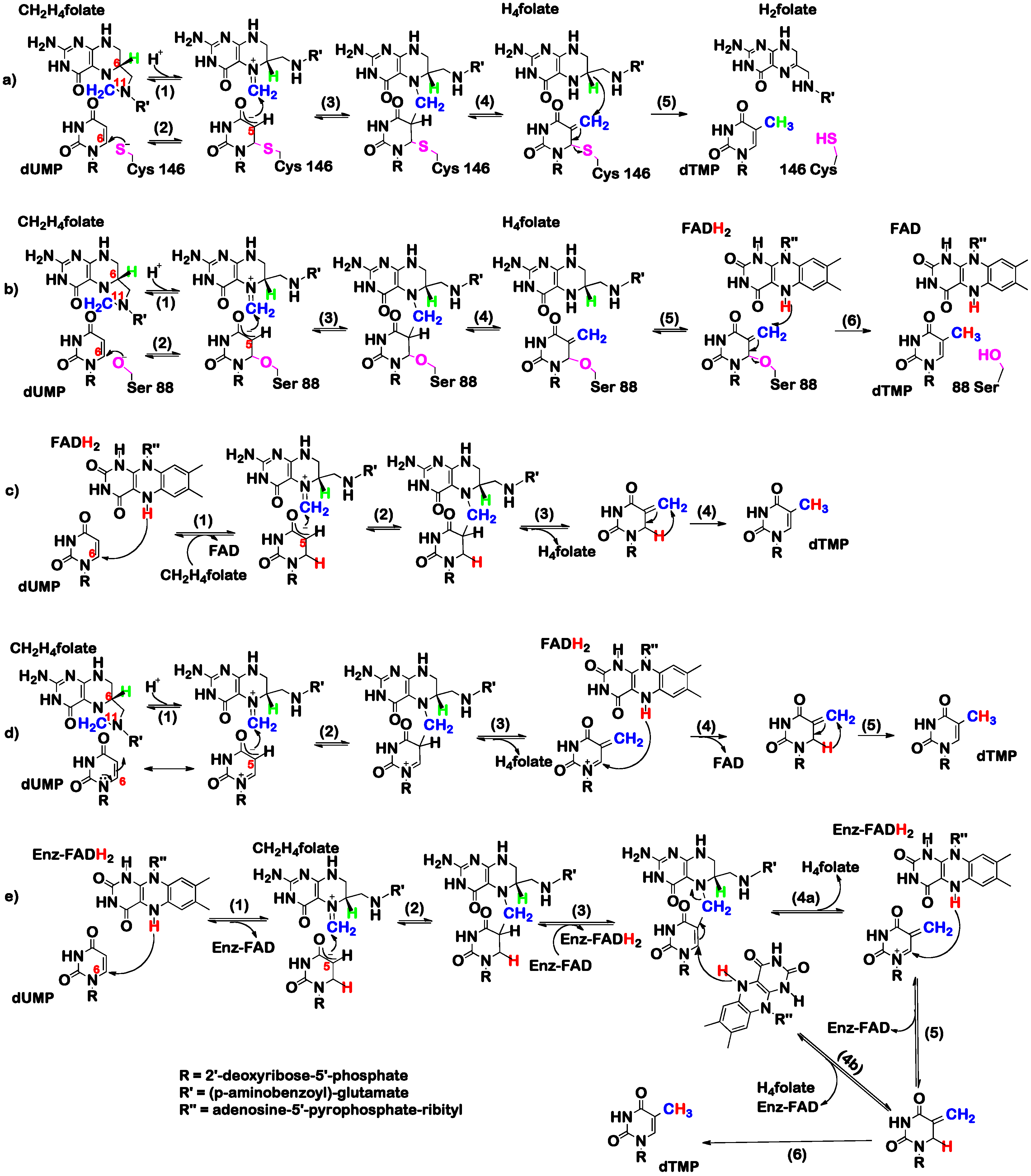
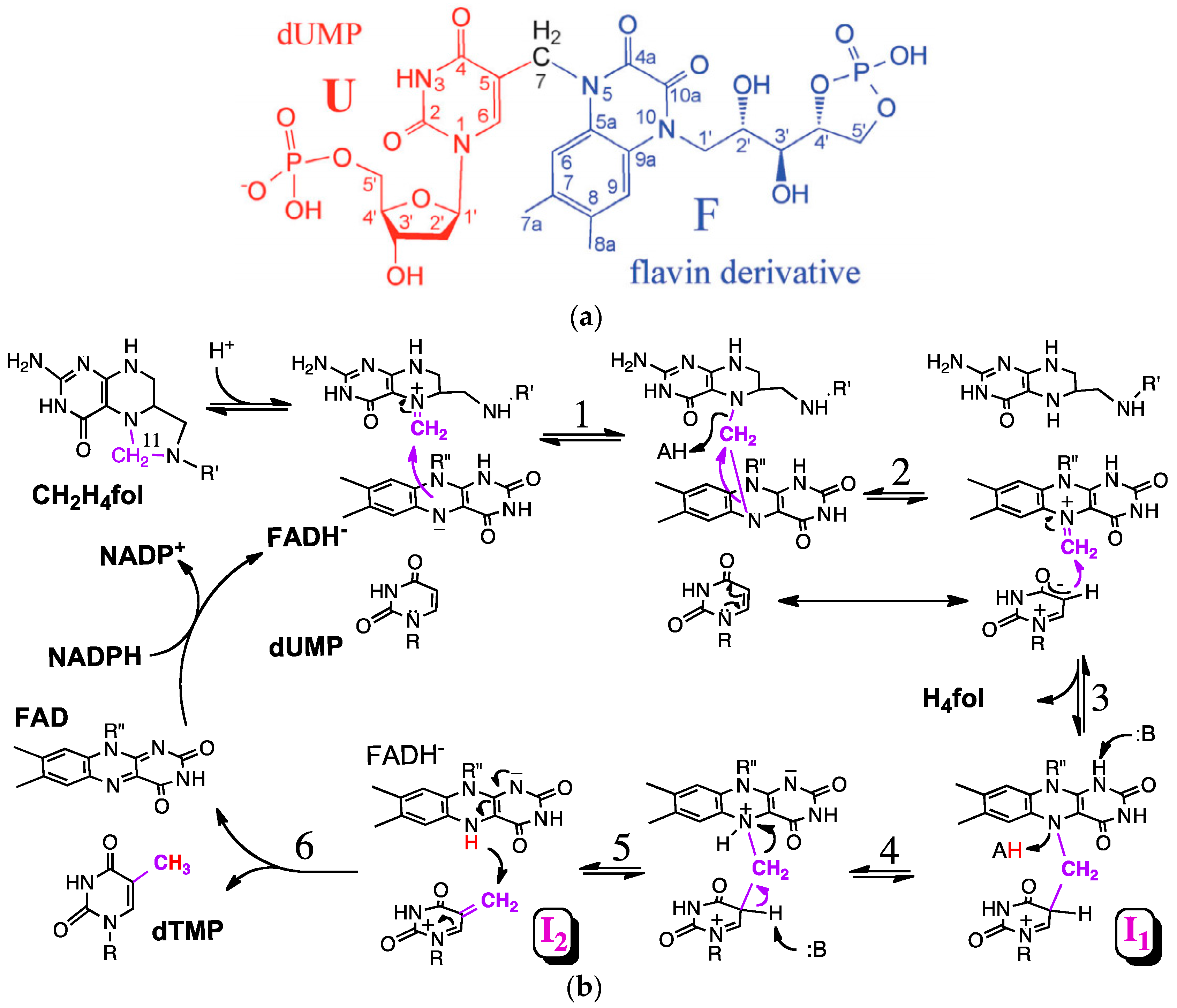
3. The Latest Proposed Mechanism for FDTS (April 2016)
4. FDTS Specific Inhibitors
5. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| dTMP | 2′-deoxythymidine-5′-monophosphate |
| dUMP | 2′-deoxyuridine-5′-monophosphate |
| TSase | Thymidylate synthase |
| DHFR | Dihydrofolate reductase |
| FDTS | Flavin dependent thymidylate synthase |
| MTHF, CH2H4folate | N5,N10-methylene-5,6,7,8,-tetrahydrofolate |
| DHF, H2folate | Dihydrofolate |
| THF, H4folate | Tetrahydrofolate |
| FAD | Flavin adenine dinucleotide |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
References
- Carreras, C.W.; Santi, D.V. The catalytic mechanism and structure of thymidylate synthase. Annu. Rev. Biochem. 1995, 64, 721–762. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.; Bhuva, N.; Harrison, M.; Buckley, A.; Saunders, M. Use of raltitrexed as an alternative to 5-fluorouracil and capecitabine in cancer patients with cardiac history. Eur. J. Cancer 2013, 49, 2303–2310. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Jolivet, J.; Cowan, K.H.; Curt, G.A.; Clendeninn, N.J.; Chabner, B.A. The pharmacology and clinical use of methotrexate. N. Engl. J. Med. 1983, 309, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Cronstein, B.N. Low-dose methotrexate: A mainstay in the treatment of rheumatoid arthritis. Pharmacol. Rev. 2005, 57, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Kielhofner, M.A. Trimethoprim- sulfamethoxazole: Pharmacokinetics, clinical uses, and adverse reactions. Tex. Heart Inst. J. 1990, 17, 86–93. [Google Scholar] [PubMed]
- Myllykallio, H.; Lipowski, G.; Leduc, D.; Filee, J.; Forterre, P.; Liebl, U. An alternative flavin-dependent mechanism for thymidylate synthesis. Science 2002, 297, 105–107. [Google Scholar] [CrossRef] [PubMed]
- Giladi, M.; Bitan-Banin, G.; Mevarech, M.; Ortenberg, R. Genetic evidence for a novel thymidylate synthase in the halophilic archaeon Halobacterium salinarum and in Campylobacter jejuni. FEMS Microbiol Lett 2002, 216, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Lesley, S.A.; Kuhn, P.; Kohen, A. Mechanistic studies of a flavin-dependent thymidylate synthase. Biochemistry 2004, 43, 10295–10301. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.; Lesley, S.A.; Mathews, I.I.; Canaves, J.M.; Brinen, L.S.; Dai, X.; Deacon, A.M.; Elsliger, M.A.; Eshaghi, S.; Floyd, R.; et al. Crystal structure of thy1, a thymidylate synthase complementing protein from Thermotoga maritima at 2.25 A resolution. Proteins 2002, 49, 142–145. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Q.; Yang, J. Bacterial thymidylate synthase with intein, group II Intron, and distinctive ThyX motifs. J. Bacteriol. 2004, 186, 6316–6319. [Google Scholar] [CrossRef] [PubMed]
- Fivian-Hughes, A.S.; Houghton, J.; Davis, E.O. Mycobacterium tuberculosis thymidylate synthase gene thyX is essential and potentially bifunctional, while thyA deletion confers resistance to p-aminosalicylic acid. Microbiology 2012, 158, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.R.; Moll, A.; Sturm, A.W.; Pawinski, R.; Govender, T.; Lalloo, U.; Zeller, K.; Andrews, J.; Friedland, G. Extensively drug-resistant tuberculosis as a cause of death in patients co-infected with tuberculosis and HIV in a rural area of South Africa. Lancet 2006, 368, 1575–1580. [Google Scholar] [CrossRef]
- Graham, D.Y.; Fischbach, L. Helicobacter pylori treatment in the era of increasing antibiotic resistance. Gut 2010, 59, 1143–1153. [Google Scholar] [CrossRef] [PubMed]
- Athamna, A.; Athamna, M.; Abu-Rashed, N.; Medlej, B.; Bast, D.J.; Rubinstein, E. Selection of Bacillus anthracis isolates resistant to antibiotics. J. Antimicrob. Chemother. 2004, 54, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Leduc, D.; Graziani, S.; Meslet-Cladiere, L.; Sodolescu, A.; Liebl, U.; Myllykallio, H. Two distinct pathways for thymidylate (dTMP) synthesis in (hyper)thermophilic Bacteria and Archaea. Biochem. Soc. Trans. 2004, 32, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Mishanina, T.V.; Yu, L.; Karunaratne, K.; Mondal, D.; Corcoran, J.M.; Choi, M.A.; Kohen, A. An unprecedented mechanism of nucleotide methylation in organisms containing thyX. Science 2016, 351, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Hardy, L.W.; Finer-Moore, J.S.; Montfort, W.R.; Jones, M.O.; Santi, D.V.; Stroud, R.M. Atomic structure of thymidylate synthase: target for rational drug design. Science 1987, 235, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Koehn, E.M.; Fleischmann, T.; Conrad, J.A.; Palfey, B.A.; Lesley, S.A.; Mathews, I.I.; Kohen, A. An unusual mechanism of thymidylate biosynthesis in organisms containing the thyX gene. Nature 2009, 458, 919–923. [Google Scholar] [CrossRef] [PubMed]
- Koehn, E.M.; Perissinotti, L.L.; Moghram, S.; Prabhakar, A.; Lesley, S.A.; Mathews, I.I.; Kohen, A. Folate binding site of flavin-dependent thymidylate synthase. Proc. Natl. Acad. Sci. USA 2012, 109, 15722–15727. [Google Scholar] [CrossRef] [PubMed]
- Mishanina, T.V.; Koehn, E.M.; Conrad, J.A.; Palfey, B.A.; Lesley, S.A.; Kohen, A. Trapping of an intermediate in the reaction catalyzed by flavin-dependent thymidylate synthase. J. Am. Chem. Soc. 2012, 134, 4442–4448. [Google Scholar] [CrossRef] [PubMed]
- Conrad, J.A.; Ortiz-Maldonado, M.; Hoppe, S.W.; Palfey, B.A. Detection of intermediates in the oxidative half-reaction of the FAD-dependent thymidylate synthase from Thermotoga maritima: Carbon transfer without covalent pyrimidine activation. Biochemistry 2014, 53, 5199–5207. [Google Scholar] [CrossRef] [PubMed]
- Stroud, R.M.; Finer-Moore, J.S. Conformational dynamics along an enzymatic reaction pathway: Thymidylate synthase, “the Movie”. Biochemistry 2003, 42, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Finer-Moore, J.S.; Santi, D.V.; Stroud, R.M. Lessons and conclusions from dissecting the mechanism of a bisubstrate enzyme: Thymidylate synthase mutagenesis, function and structure. Biochemistry 2003, 42, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Koehn, E.M.; Kohen, A. Flavin-dependent thymidylate synthase: A novel pathway towards thymine. Arch. Biochem. Biophys. 2010, 493, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Mishanina, T.V.; Corcoran, J.M.; Kohen, A. Substrate activation in flavin-dependent thymidylate synthase. J. Am. Chem. Soc. 2014, 136, 10597–10600. [Google Scholar] [CrossRef] [PubMed]
- Graziani, S.; Xia, Y.; Gurnon, J.R.; van Etten, J.L.; Leduc, D.; Skouloubris, S.; Myllykallio, H.; Liebl, U. Functional analysis of FAD-dependent thymidylate synthase ThyX from Paramecium bursaria Chlorella virus-1. J. Biol. Chem. 2004, 279, 54340–54347. [Google Scholar] [CrossRef] [PubMed]
- Parchina, A.; Froeyen, M.; Margamuljana, L.; Rozenski, J.; De Jonghe, S.; Briers, Y.; Lavigne, R.; Herdewijn, P.; Lescrinier, E. Discovery of an acyclic nucleoside phosphonate that inhibits Mycobacterium tuberculosis ThyX based on the binding mode of a 5-alkynyl substrate analogue. Chem. Med. Chem. 2013, 8, 1373–1383. [Google Scholar] [CrossRef] [PubMed]
- Kogler, M.; Vanderhoydonck, B.; de Jonghe, S.; Rozenski, J.; van Belle, K.; Herman, J.; Louat, T.; Parchina, A.; Sibley, C.; Lescrinier, E.; et al. Synthesis and evaluation of 5-substituted 2′-deoxyuridine monophosphate analogues as inhibitors of flavin-dependent thymidylate synthase in Mycobacterium tuberculosis. J. Med. Chem. 2011, 54, 4847–4862. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, C.; Derudas, M.; Gonczy, B.; Hinsinger, K.; Kandil, S.; Pertusati, F.; Serpi, M.; Snoeck, R.; Andrei, G.; Balzarini, J.; et al. ProTides of N-(3-(5-(2′-deoxyuridine))prop-2-ynyl)octanamide as potential anti-tubercular and anti-viral agents. Bioorg. Med. Chem. 2014, 22, 2816–2824. [Google Scholar] [CrossRef] [PubMed]
- Esra Onen, F.; Boum, Y.; Jacquement, C.; Spanedda, M.V.; Jaber, N.; Scherman, D.; Myllykallio, H.; Herscovici, J. Design, synthesis and evaluation of potent thymidylate synthase X inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 3628–3631. [Google Scholar] [CrossRef] [PubMed]





| Bacterial Species | Associated Disease (s) | Tdk | ThyA | FolA |
|---|---|---|---|---|
| Bacillus anthracis | anthrax | + | + | + |
| Borrelia burgdorferi | Lyme disease | + | − | − |
| Campylobacter jejuni | diarrhea | − | − | − |
| Chlamydia trachomatis | trachoma | − | − | + |
| Chlamydia pneumoniae | pneumonia | − | − | + |
| Clostridium botulinum | botulism | + | + | + |
| Helicobacter pylori | stomach ulcer, gastric cancer | − | − | − |
| Mycobacterium leprae | leprosy | − | + | + |
| Mycobacterium tuberculosis | tuberculosis | − | + | + |
| Rickettsia prowazeki | typhus | − | − | − |
| Rickettsia rickettsii | spotted fever | − | − | − |
| Treponema pallidum | syphilis | − | − | − |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, M.; Karunaratne, K.; Kohen, A. Flavin-Dependent Thymidylate Synthase as a New Antibiotic Target. Molecules 2016, 21, 654. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21050654
Choi M, Karunaratne K, Kohen A. Flavin-Dependent Thymidylate Synthase as a New Antibiotic Target. Molecules. 2016; 21(5):654. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21050654
Chicago/Turabian StyleChoi, Michael, Kalani Karunaratne, and Amnon Kohen. 2016. "Flavin-Dependent Thymidylate Synthase as a New Antibiotic Target" Molecules 21, no. 5: 654. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21050654