3.1. Chemistry
3.1.1. Reagents and Instruments
All non-aqueous reactions were performed under a dry atmosphere of nitrogen. The commercial reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA) or TCI (Tokyo, Japan). Solvents were purified and dried prior to use. Melting points were measured on Thomas-Hoover melting point apparatus (Thomas Scientific, Swedesboro, NJ, USA) and not corrected. 1H, 13C-NMR and HMBC spectra were taken on a Varian 400 MHz spectrometer (Thomas Scientific, Swedesboro, NJ, USA) in DMSO-d6. Chemical shifts (δ) are in parts per million (ppm) relative to tetramethylsilane, and coupling constants (J) are in Hertz. DIP-MS (EI) was obtained on an Agilent 7890A-5975C GC/MSD (Agilent Technologies, Santa Clara, CA, USA). GC/MS (EI) was obtained on a SHIMADZU QP 2010 model (Shimadzu, Kyoto, Japan) and FAB-MS was obtained on a JEOL JMS-700 Mstation (JEOL, Tokyo, Japan). Fraction collection was performed on an EYELA fraction collector DC-1500 (Tokyo Rikakikai, Tokyo, Japan). An analytical TLC was performed on pre-coated silica gel 60 F254 plates (Merck, Kenilworth, NJ, USA). Solvent systems for TLC were ethyl acetate/n-hexane mixtures and 10% methanol in dichloromethane. Column chromatography was carried out on Merck silica gel 9385 (Merck, Kenilworth, NJ, USA) (230–400 mesh).
3.1.2. General Experimental Procedures of 1i–3i
To a stirred solution of 3-nitrobenzoic acid (35.90 mmol) in anhydrous benzene (50 mL) was dropwise added: oxalyl chloride (46.67 mmol) and then triethylamine (39.49 mmol) at room temperature. The reaction mixture was refluxed for 1 h and the solvent and the unreacted oxalyl chloride were evaporated off under reduced pressure and the acid chloride was used without purification. To a solution of 3-aminobenzonitrile, 4-aminobenzonitrile, and 4-aminobenzyl cyanide (8.62 mmol) in anhydrous benzene (50 mL) was added: acyl chloride (10.78 mmol) and triethylamine (8.62 mmol) at room temperature and stirred for 3 h. To the reaction mixture, water was added and extracted with ethyl acetate (50 mL × 3), dried with anhydrous magnesium sulfate and filtrated. The filtrate was evaporated under reduced pressure to give crude compound, which was recrystallized with ethyl acetate and n-hexane to give a pure white or pale yellow compound, respectively.
3-Nitro-N-(3′-cyanophenyl) benzamide (1i). Yield: 81%; m.p.: 195–196 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 7.62 (2H, d, J = 8.0 Hz, H-6′,4′), 7.86 (1H, t, J = 8.0 Hz, H-5′), 8.06 (1H, t, J = 8.0 Hz, H-5), 8.26 (1H, s, H-2′), 8.41 (1H, ddd, J = 7.8, 1.5, 1.1 Hz, H-6), 8.47 (1H, ddd, J = 8.2, 2.3, 1.0 Hz, H-4), 8.81 (1H, t, J = 2.0 Hz, H-2), 10.88 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 111.5 (C-3′), 118.6 (C≡N), 122.5 (C-6′), 123.3 (C-2), 125.1(C-5′), 126.5 (C-4), 127.6 (C-2′), 130.3 (C-4′), 134.3 (C-6), 135.7 (C-1), 139.5 (C-1′), 147.8 (C-3), 163.8 (C=O); GC-MS (EI) m/z: 267 [M]+.
3-Nitro-N-(4′-cyanophenyl) benzamide (2i). Yield: 80%; m.p.: 225–226 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 7.85 (2H, d, J = 8.0 Hz, H-3′,5′), 7.86 (1H, t, J = 8.0 Hz, H-5), 7.99 (2H, d, J = 8.0 Hz, H-2′,6′), 8.41 (1H, d, J = 8.0 Hz, H-6), 8.46 (1H, ddd, J = 8.2, 2.3, 1.0 Hz, H-4), 8.67 (1H, t, J = 2.0 Hz, H-2), 10.96 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 106.6 (C-4′), 119.6 (C≡N), 121.1 (C-2′,6′), 123.3 (C-2), 127.2 (C-4), 131.0 (C-5), 133.9 (C-3′,5′), 135.0 (C-6), 136.4 (C-1), 143.7 (C-1′), 148.4 (C-3), 164.7 (C=O); GC-MS (EI) m/z: 267 [M]+.
3-Nitro-N-(4′-cyanomethylphenyl) benzamide (3i). Yield: 80%; m.p.: 197–198 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.97 (2H, s, CH2), 7.31 (2H, d, J = 8.8 Hz, H-3′,5′), 7.76 (2H, d, J = 8.8 Hz, H-2′,6′), 7.80 (1H, t, J = 8.0 Hz, H-5), 8.36 (1H, d, J = 8.0 Hz, H-6), 8.40 (1H, ddd, J = 8.2, 2.3, 1.0 Hz, H-4) 8.75 (1H, t, J = 2.0 Hz, H-2), 10.60 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 21.9 (CH2), 119.3 (C≡N), 121.0 (C-2′,6′), 122.4 (C-2), 126.2 (C-4′), 126.7 (C-4), 128.4 (C-3′,5′), 130.2 (C-5), 134.2 (C-6), 136.1 (C-1), 138.1 (C-1′), 147.7 (C-3), 163.3 (C=O); GC-MS (EI) m/z: 281 [M]+.
3.1.3. General Experimental Procedures of 1ii–3ii
To a solution of 1i–3i (1.79 mmol) in methanol (20 mL) was added: ammonium chloride (double amount of 1i–3i) and iron powder (3.58 mmol), and the reaction mixture was refluxed for 7 h. To the reaction mixture, ice water was added and rotary evaporated to remove methanol under reduced pressure. The aqueous residue was extracted with dichloromethane (30 mL × 3) and the organic phase was dried with anhydrous MgSO4, filtrated, and evaporated to give the crude solid, which was recrystallized with ethyl acetate and n-hexane to give a pure white or pale-yellow solid.
3-Amino-N-(3′-cyanophenyl) benzamide (1ii). Yield: 60%; m.p.: 166–168 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 5.35 (2H, s, NH2), 6.77 (1H, ddd, J = 8.2, 2.3, 1.0 Hz, H-4), 7.07 (1H, d, J = 7.6 Hz, H-6), 7.10 (1H, t, J = 1.9 Hz, H-2), 7.17 (1H, t, J = 7.6 Hz, H-5), 7.54-7.58 (2H, m, H-4′,5′), 8.04 (1H, dt, J = 7.2, 2.2 Hz, H-6′), 8.24 (1H, s, H-2′), 10.39 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 111.4 (C-3′), 112.9 (C-2), 114.7 (C-6), 117.1 (C-4), 118.8 (C≡N), 122.8 (C-6′), 124.7 (C-5′), 126.9 (C-2′), 128.9 (C-5), 130.1 (C-4′), 135.2 (C-1), 140.2 (C-1′), 148.9 (C-3), 166.8 (C=O); GC-MS (EI) m/z: 237 [M]+.
3-Amino-N-(4′-cyanophenyl) benzamide (2ii). Yield: 71%; m.p.: 182–183 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 5.36 (2H, s, NH2), 6.77 (1H, ddd, J = 8.0, 2.0, 1.2 Hz, H-4), 7.05 (1H, d, J = 7.6 Hz, H-6), 7.07 (1H, t, J = 1.9 Hz, H-2), 7.17 (1H, t, J = 8.0 Hz, H-5), 7.80 (2H, d, J = 8.0 Hz, H-3′,5′), 7.98 (2H, d, J = 8.0 Hz, H-2′,6′), 10.48 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 105.0 (C-4′), 112.9 (C-2), 114.8 (C-6), 117.2 (C-4), 119.1 (C≡N), 120.0 (C-2′,6′), 128.9 (C-5), 133.1 (C-3′,5′), 135.3 (C-1), 143.7 (C-1′), 148.9 (C-3), 167.0 (C=O); GC-MS (EI) m/z: 237 [M]+.
3-Amino-N-(4′-cyanomethylphenyl) benzamide (3ii). Yield: 68%; m.p.: 173–174 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.98 (2H, s, CH2), 5.31 (2H, s, NH2), 6.74 (1H, ddd, J = 7.8, 2.2, 1.2 Hz, H-4), 7.05 (1H, d, J = 7.6 Hz, H-6), 7.09 (1H, t, J = 1.9 Hz, H-2), 7.14 (1H, t, J = 8.0 Hz, H-5), 7.30 (2H, d, J = 8.0 Hz, H-3′,5′), 7.78 (2H, d, J = 8.0 Hz, H-2′,6′), 10.13 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 21.9 (CH2), 112.9 (C-2), 114.7 (C-6), 116.8 (C-4), 119.4 (C≡N), 120.6 (C-2′,6′), 125.9 (C-4′), 128.3 (C-3′,5′), 128.7 (C-5), 135.8 (C-1), 138.8 (C-1′), 148.8 (C-3), 166.4 (C=O); GC-MS (EI) m/z: 251 [M]+.
3.1.4. General Experimental Procedures of 1a–3a
To a solution of thiophene-2-carboxylic acid (7.8 mmol) in anhydrous dichloromethane (40 mL) was added: triethylamine (8.6 mmol) and dropwise added oxalyl chloride (10.1 mmol) at room temperature. The reaction mixture was refluxed for 1 h and rotary evaporated to remove dichloromethane and oxalyl chloride, which was used next reaction without purification. To a solution of 1ii–3ii (1.48 mmol) in anhydrous benzene (20 mL) was added: triethylamine (1.48 mmol) and thiophene-2-carbonyl chloride (1.85 mmol) at room temperature and the reaction mixture was refluxed for 1 h. To the reaction mixture, water was added and extracted with ethyl acetate (50 mL × 3), dried with anhydrous MgSO4, and filtrated. The filtrate was evaporated under reduced pressure to give a crude compound, which was recrystallized with ethyl acetate and n-hexane to give a pure white or pale yellow compound, respectively.
N-(3′-Cyanophenyl)-3-(thiophen-2′′-ylcarbonylamino) benzamide (1a). Yield: 64%; m.p.: 201–202 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 7.25 (1H, dd, J = 5.0, 4.0 Hz, H-4′′), 7.54 (1H, t, J = 8.0 Hz, H-5), 7.58-7.60 (2H, m, H-4′,5′), 7.71 (1H, d, J = 6.8 Hz, H-6), 7.89 (1H, dd, J = 5.0, 1.0 Hz, H-5′′), 8.03 (1H, dd, J = 8.0, 0.8 Hz, H-3′′), 8.07-8.09 (2H, m, H-4,6′), 8.26 (1H, t, J = 1.2 Hz, H-2′), 8.28 (1H, t, J = 1.8 Hz, H-2), 10.47 (1H, s, NHCO), 10.61 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 111.5 (C-3′), 118.7 (C≡N), 119.8 (C-2), 122.7 (C-6), 123.0 (C-4), 123.6 (C-6′), 124.9 (C-5′), 127.2 (C-2′), 128.2 (C-5), 128.9 (C-4′′), 129.4 (C-3′′), 130.2 (C-4′), 132.2 (C-5′′), 135.0 (C-1), 139.0 (C-3), 139.7 (C-2′′), 140.0 (C-1′), 160.1 (CONH), 165.9 (NHCO); DIP-MS (EI) m/z: 347 [M]+.
N-(4′-Cyanophenyl)-3-(thiophen-2′′-ylcarbonylamino) benzamide (2a). Yield: 71%; m.p.: 214–216 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 7.25 (1H, dd, J = 5.0, 4.0 Hz, H-4′′), 7.54 (1H, t, J = 8.0 Hz, H-5), 7.71 (1H, d, J = 6.8 Hz, H-6), 7.83 (2H, d, J = 8.0 Hz, H-3′,5′), 7.88 (1H, dd, J = 4.0, 0.8 Hz, H-5′′), 8.00 (2H, d, J = 8.0 Hz, H-2′,6′), 8.03 (1H, ddd, J = 8.0, 2.3, 1.0 Hz, H-4), 8.09 (1H, dd, J = 4.0, 0.8 Hz, H-3′′), 8.28 (1H, t, J = 1.9 Hz, H-2), 10.47 (1H, s, NHCO), 10.70 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 105.4 (C-4′), 119.2 (C≡N), 119.9 (C-2), 120.2 (C-2′,6′), 122.8 (C-6), 123.7 (C-4), 128.2 (C-5), 128.9 (C-4′′), 129.4 (C-3′′), 132.2 (C-5′′), 133.1 (C-3′,5′), 135.0 (C-1), 139.1 (C-3), 139.7 (C-2′′), 143.5 (C-1′), 160.1 (CONH), 166.1 (NHCO); DIP-MS (EI) m/z: 347 [M]+.
N-(4′-Cyanomethylphenyl)-2-(thiophen-2′′-ylcarbonylamino) benzamide (3a). Yield: 61%; m.p.: 202–203 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 4.01 (2H, s, CH2), 7.25 (1H, dd, J = 5.0, 4.0 Hz, H-4′′), 7.34 (2H, d, J = 8.0 Hz, H-3′,5′), 7.52 (1H, t, J = 8.0 Hz, H-5), 7.70 (1H, d, J = 8.0 Hz, H-6), 7.81 (2H, d, J = 8.0 Hz, H-2′,6′), 7.88 (1H, dd, J = 4.0, 0.8 Hz, H-5′′), 8.01 (1H, ddd, J = 8.0, 2.3, 1.0 Hz, H-4), 8.08 (1H, dd, J = 4.0, 0.8 Hz, H-3′′), 8.26 (1H, t, J = 2.0 Hz, H-2), 10.36 (1H, s, NHCO), 10.43 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 21.9 (CH2), 119.3 (C≡N), 119.8 (C-2), 120.7 (C-2′,6′), 122.7 (C-6), 123.3 (C-4), 126.2 (C-4′), 128.1 (C-5), 128.4 (C-3′,5′), 128.7 (C-4′′), 129.3 (C-3′′), 132.1 (C-5′′), 135.5 (C-1), 138.6 (C-3), 138.9 (C-1′), 139.7 (C-2′′), 160.0 (CONH), 165.5 (NHCO); DIP-MS (EI) m/z: 361 [M]+.
3.1.5. General Experimental Procedures of 1b–31ii–3ii
To a suspension of intermediate 1a–3a (0.86 mmol) in absolute ethanol (25 mL) and 1,4-dioxane (5 mL) was added: triethylamine (2.59 mmol) and refluxed to dissolve. The reaction mixture was added with hydroxylamine·HCl (3.45 mmol) and refluxed for 4–6 h. The mixture was evaporated to remove the ethanol and ice water was poured into the residue. The resulting precipitate was filtrated, washed with water, and dried under reduced pressure to give a pure white or pale yellow solid.
N-(3′-Amidoximephenyl)-3-(thiophen-2′′-ylcarbonylamino) benzamide (1b). Yield: 87%; m.p.: 234–235 °C; 1H-NMR (400MHz, DMSO-d6) δ: 5.75 (2H, s, NH2), 7.24 (1H, dd, J = 4.9, 3.8 Hz, H-4′′), 7.34–7.38 (2H, m, H-5, 4′), 7.51 (1H, t, J = 7.9 Hz, H-5′), 7.71 (1H, d, J = 7.8 Hz, H-4), 7.80 (1H, d, J = 7.6 Hz, H-6), 7.88 (1H, dd, J = 5.0, 0.8 Hz, H-5′′), 8.01 (1H, dd, J = 8.0, 1.2 Hz, H-6′), 8.05–8.10 (2H, m, H-2′,3′′), 8.27 (1H, t, J = 2.0 Hz, H-2), 9.62 (1H, s, NOH), 10.35 (1H, s, NHCO), 10.47 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 118.5 (C-2), 120.6 (C-2′), 121.6 (C-6′), 121.7 (C-6), 123.4 (C-4′), 124.0 (C-4), 128.8 (C-5), 128.9 (C-4′′), 129.4 (C-3′′), 130.0 (C-5′), 132.8 (C-5′′), 134.6 (C-1), 136.2 (C-1′), 139.6 (C-2′′), 140.5 (C-3), 151.6 (C=NOH), 160.7 (CONH), 166.1 (NHCO); FAB-MS m/z: 363 [M + 1 − H2O]+, 380 [M]+, 381 [M + 1]+, 403 [M + Na]+.
N-(4′-Amidoximephenyl)-3-(thiophen-2′′-ylcarbonylamino) benzamide (2b). Yield: 87%; m.p.: 241–242 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 5.76 (2H, s, NH2), 7.25 (1H, dd, J = 5.0, 4.0 Hz, H-4′′), 7.52 (1H, t, J = 8.0 Hz, H-5), 7.66 (2H, d, J = 8.0 Hz, H-3′,5′), 7.70 (1H, d, J = 7.6 Hz, H-6), 7.78 (2H, d, J = 8.0 Hz, H-2′,6′), 7.88 (1H, dd, J = 4.0, 0.8 Hz, H-5′′), 8.01 (1H, d, J = 7.6, Hz, H-4), 8.08 (1H, dd, J = 8.0, 0.8 Hz, H-3′′), 8.25 (1H, t, J = 2.0 Hz, H-2), 9.55 (1H, s, NOH), 10.37 (1H, s, NHCO), 10.44 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 119.6 (C-2′,6′), 119.8 (C-2), 122.7 (C-6), 123.3 (C-4), 125.7 (C-3′,5′), 128.2 (C-5), 128.7 (C-4′), 128.9 (C-4′′), 129.3 (C-3′′), 132.1 (C-5′′), 135.5 (C-1), 139.9 (C-3), 139.6 (C-1′), 139.7 (C-2′′), 150.5 (C=NOH), 160.0 (CONH), 165.5 (NHCO); FAB-MS m/z: 363 [M + 1 − H2O]+, 380 [M]+, 381 [M + 1]+, 403 [M + Na]+.
N-(4′-Amidoximemethylphenyl)-3-(thiophen-2′′-ylcarbonylamino) benzamide (3b). Yield: 90%; m.p.: 164–165 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.23 (2H, s, CH2), 5.37 (2H, s, NH2), 7.19–7.26 (3H, m, H-3′,5′,4′′), 7.50 (1H, t, J = 7.9 Hz, H-5), 7.67–7.69 (3H, m, H-2′,6′,4), 7.88 (1H, d, J = 4.9 Hz, H-5′′), 8.01 (1H, d, J = 8.0 Hz, H-6), 8.10 (1H, d, J = 3.5 Hz, H-3′′), 8.26 (1H, s, H-2), 8.88 (1H, s, NOH), 10.29 (1H, s, NHCO), 10.50 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 37.4 (CH2), 120.5 (C2′,6′), 122.9 (C-6), 123.3 (C-4), 128.9 (C-4′′), 129.4 (C-3′,5′), 129.5 (C-3′′), 130.0 (C-5), 132.8 (C-5′′), 133.9 (C-4′), 136.3 (C-1), 138.0 (C-3), 139.5 (C-2′′), 140.5 (C-1′), 152.6 (C=NOH), 160.7 (CONH), 165.9 (NHCO); FAB-MS m/z: 377 [M + 1 − H2O]+, 394 [M]+, 395 [M + 1]+.
3.1.6. General Experimental Procedures of 1–3
To a solution of amidoximes 1b–3b (0.53 mmol) in anhydrous dichloromethane (10 mL) was added: triethylamine (1.58 mmol) and acetyl chloride (0.6 mmol) at 0 °C. The reaction mixture was stirred for 1–2 h at room temperature and ice water was added. The aqueous layer was extracted with dichloromethane (30 mL × 3). The organic phase was washed with water, saturated NaHCO3, and water. The organic layer was dried with MgSO4, filtrated, and evaporated in reduced pressure to give the acetylated amidoximes (1c–3c), which were used next reaction without purification. To a solution of 10% Pd-C in ethanol (10 mL) was added: acetylated amidoximes (1.0 eq.) and c-HCl (1.0 eq.) and hydrogenated for 2 h at 60 psi, 45 °C. The reaction mixture was filtrated and concentrated under reduced pressure to give a crude oily compound, which was purified to yield a white or pale yellow solid by column chromatography.
N-(3′-Amidinophenyl)-3-(thiophen-2′′-ylcarbonylamino) benzamide·HCl (1). Yield: 46%; m.p.: 219–220 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 7.23 (1H, dd, J = 5.0, 3.8 Hz, H-4′′), 7.55–7.50 (2H, m, H-5,4′), 7.60 (1H, t, J = 7.9 Hz, H-5′), 7.76 (1H, d, J = 8.1 Hz, H-6), 7.87 (1H, dd, J = 5.0, 1.0 Hz, H-5′′), 8.03 (1H, ddd, J = 8.2, 2.3, 1.0 Hz, H-4), 8.08 (1H, dd, J = 8.0, 1.2 Hz, H-6′), 8.24 (1H, dd, J = 3.7, 0.7 Hz, H-3′′), 8.32 (1H, t, J = 1.8 Hz, H-2′), 8.38 (1H, t, J = 1.8 Hz, H-2), 9.45 (4H, br s, hydrogens of amidine·HCl), 10.68 (1H, s, NHCO), 10.72 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 119.5 (C-2′), 120.0 (C-2), 122.7 (C-6), 123.1 (C-6′), 123.7 (C-4), 125.1 (C-4′), 128.1 (C-5), 128.7 (C-4′′), 129.0 (C-3′′), 129.3, 129.5 (C-5′), 132.1 (C-5′′), 135.0 (C-1), 139.1 (C-3), 139.7 (C-2′′), 139.8 (C-1′), 160.1 (carbon of amidine), 165.9 (CONH), 166.3 (NHCO); FAB-MS m/z: 365 [M − Cl]+, 388 [M − Cl + Na]+; HRMS (EI): Calcd for C19H17N4O2S [M + H]+ 365.1072, found 365.1067.
N-(4′-Amidinophenyl)-3-(thiophen-2′′-ylcarbonylamino) benzamide·HCl (2). Yield: 37%; m.p.: 237–238 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 7.24 (1H, dd, J = 5.0, 3.8 Hz, H-4′′), 7.53 (1H, t, J = 7.9 Hz, H-5), 7.77 (1H, d, J = 7.9 Hz, H-6), 7.88 (1H, dd, J = 5.1, 1.1 Hz, H-5′′), 7.90 (2H, d, J = 9.0 Hz, H-3′,5′), 8.05 (3H, m, H-4, H-2′,6′), 8.21 (1H, d, J = 3.8 Hz, H-3′′), 8.36 (1H, t, J = 1.8 Hz, H-2), 9.17 (2H, br s, hydrogens of amidine·HCl), 9.36 (2H, br s, hydrogens of amidine·HCl), 10.63 (1H, s, NHCO), 10.78 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 119.7 (C-2′,6′), 120.0 (C-2), 122.1 (C-4′), 122.9 (C-6), 123.7 (C-4), 128.1 (C-5), 128.7 (C-4′′), 129.0 (C-3′′), 129.6 (C-3′,5′), 132.1 (C-5′′), 134.9 (C-1), 139.1(C-3), 139.7 (C-2′′), 144.2 (C-1′), 160.1 (carbon of amidine), 164.9 (CONH), 166.1 (NHCO); FAB-MS m/z: 365 [M − Cl]+, 388 [M − Cl + Na]+, 423 [M + Na]+; HRMS (EI): Calcd for C19H17N4O2S [M + H]+ 365.1072, found 365.1069.
N-(4′-Amidinomethylphenyl)-3-(thiophen-2′′-ylcarbonylamino) benzamide·HCl (3). Yield: 51%; m.p.: 219–220 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.71 (2H, s, CH2), 7.23 (1H, dd, J = 5.0, 3.8 Hz, H-4′′), 7.44 (2H, d, J = 8.6 Hz, H-3′,5′), 7.50 (1H, t, J = 7.9 Hz, H-5), 7.71 (1H, d, J = 7.9 Hz, H-6), 7.79 (2H, d, J = 8.6 Hz, H-2′,6′), 7.87 (1H, dd, J = 5.0, 0.9 Hz, H-5′′), 8.02 (1H, dd, J = 8.1, 1.3 Hz, H-4), 8.30 (1H, t, J = 1.8 Hz, H-2), 8.83 (2H, br s, hydrogens of amidine·HCl), 9.28 (2H, br s, hydrogens of amidine·HCl), 10.37 (1H, s, NHCO), 10.57 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 119.9 (C-2), 120.5 (C-2′,6′), 122.7 (C-6), 123.5 (C-2), 128.1 (C-4′), 128.6 (C-4′′), 129.2 (C-3′,5′), 129.5 (C-3′′), 132.1 (C-5′′), 135.5 (C-1), 138.6 (C-3), 139.0 (C-1′), 139.8 (C-2′′), 160.1 (carbon of amidine), 165.5 (CONH), 169.3 (NHCO); FAB-MS m/z: 379 [M − Cl]+, 402 [M − Cl + Na]+; HRMS (EI) Calcd. For C20H19N4O2S [M + H]+ 379.1229, found 379.1216.
3.1.7. General Experimental Procedures 4i–8i
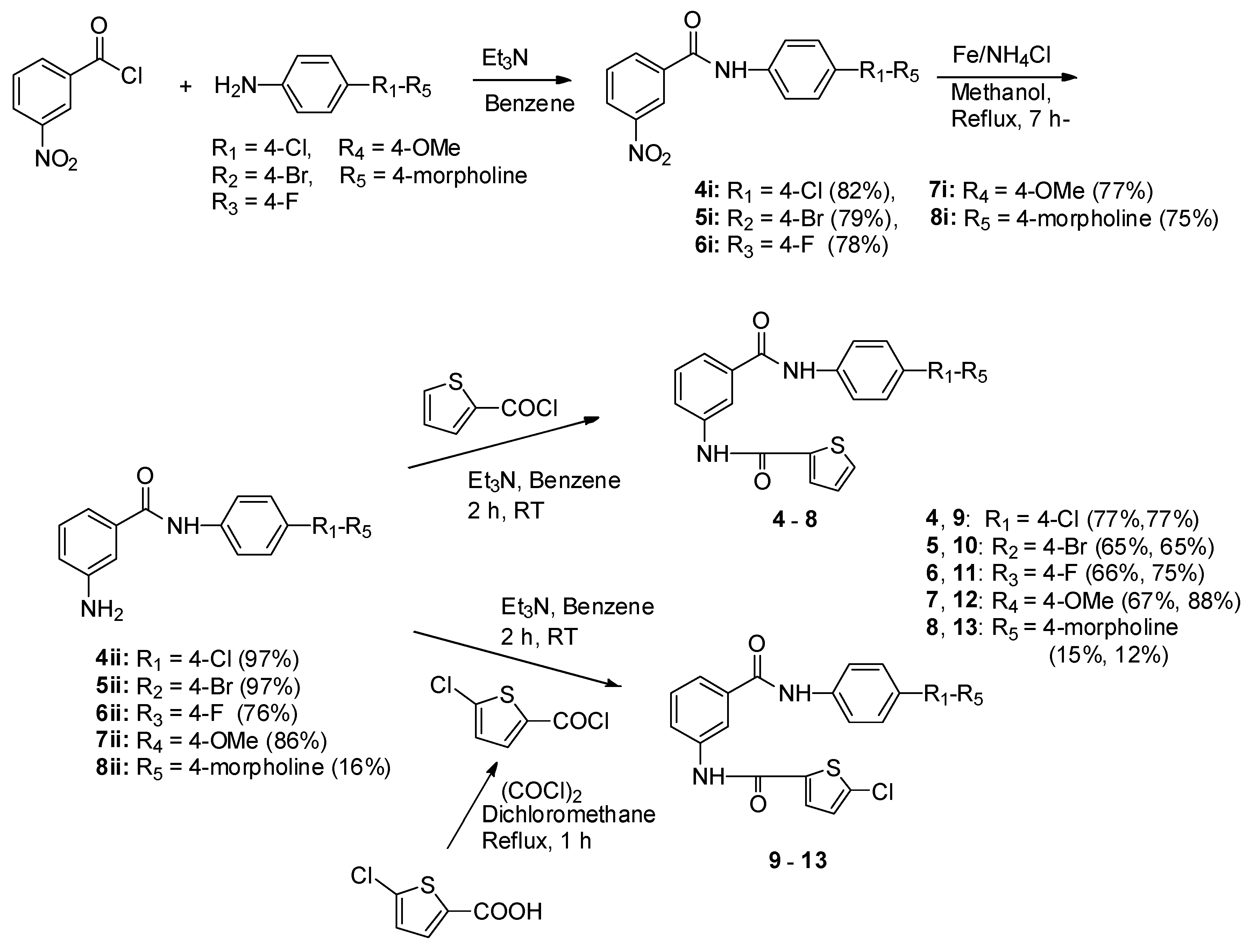
To a stirred solution of 3-nitrobenzoic acid (35.90 mmol) in anhydrous benzene (50 mL) was dropwise added: oxalyl chloride (46.67 mmol) and triethylamine (39.49 mmol) at room temperature. The reaction mixture was refluxed for 1 h, and benzene and unreacted oxalyl chloride were rotary evaporated off under reduced pressure and used without purification. To a solution of 4-chloroaniline, 4-bromoaniline, 4-fluoroaniline, 4-methoxyaniline, and 4-mor pholinoaniline (2.16 mmol) in anhydrous benzene (30 mL) was added: 3-nitrobenzoyl chloride (2.69 mmol) and triethylamine (2.16 mmol) at room temperature and stirred at the same temperature for 3 h. To the reaction mixture, water was added and extracted with ethylacetate (30 mL × 3), dried with anhydrous MgSO4, and filtrated. The filtrate was evaporated under reduced pressure to afford a crude product, which was recrystallized with ethyl acetate and n-hexane to give a pure white or pale yellow compound, respectively.
N-(4′-Chlorophenyl)-3-nitrobenzamide (4i). Yield: 82%; m.p.: 175–177 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 7.45 (2H, d, J = 8.9 Hz, H-3′,5′), 7.82 (2H, d, J = 8.9 Hz, H-2′,6′), 7.85 (1H, t, J = 8.0 Hz, H-5), 8.40 (1H, d, J = 8.0 Hz, H-6), 8.45 (1H, ddd, J = 8.2, 2.3, 1.0 Hz, H-4), 8.78 (1H, t, J = 1.9 Hz, H-2), 10.71 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 122.1 (C-2′,6′), 122.4 (C-2), 126.3 (C-4), 127.8 (C-4′), 128.7 (C-3′,5′), 130.3 (C-5), 134.2 (C-6), 136.0 (C-1), 137.7 (C-1′), 147.7 (C-3), 163.4 (CONH); GC-MS (EI) m/z: 276 [M]+.
N-(4′-Bromophenyl)-3-nitrobenzamide (5i). Yield: 79%; m.p.: 190–192 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 7.75 (2H, d, J = 8.8 Hz, H-3′,5′), 7.77 (2H, d, J = 8.8 Hz, H-2′,6′), 7.85 (1H, t, J = 8.0 Hz, H-5), 8.40 (1H, d, J = 7.8 Hz, H-6), 8.45 (1H, ddd, J = 8.2, 2.3, 1.0 Hz, H-4), 8.78 (1H, t, J = 1.9 Hz, H-2), 10.69 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 115.9 (C-4′), 122.4 (C-2,2′,6′), 126.3 (C-4), 130.2 (C-5), 131.5 (C-3′,5′), 134.2 (C-6), 136.0 (C-1), 138.1 (C-1′), 147.7 (C-3), 163.4 (CONH); GC-MS (EI) m/z: 320 [M − 1]+, 322 [M + 1]+.
N-(4′-Fluorophenyl)-3-nitrobenzamide (6i). Yield: 78%; m.p.: 190–191 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 7.23 (2H, t, J = 8.8 Hz, H-3′,6′), 7.80 (2H, dd, J = 9.0, 5.1 Hz, H-2′,6′), 7.85 (1H, t, J = 8.0 Hz, H-5), 8.40 (1H, d, J = 7.8 Hz, H-6), 8.44 (1H, dd, J = 8.2, 2.3 Hz, H-4), 8.79 (1H, t, J = 2.0 Hz, H-2), 10.63 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 115.3 (J = 22.0 Hz, C-3′,5′), 122.4(J = 7.2 Hz, H-2′,6′), 122.5 (C-2), 126.2 (C-4), 130.2 (C-5), 134.1 (C-6), 135.0 (J = 2.6 Hz, C-1′), 136.1 (C-1), 147.7 (C-3), 158.5 (J = 240.0 Hz, H-4′), 163.2 (CONH); GC-MS (EI) m/z: 260 [M]+.
N-(4′-Methoxyphenyl)-3-nitrobenzamide (7i). Yield: 77%; m.p.: 163–164 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.75 (3H, s, CH3), 6.96 (2H, d, J = 9.1 Hz, H-3′,5′), 7.68 (2H, d, J = 9.1 Hz, H-2′,6′), 7.83 (1H, t, J = 8.0 Hz, H-5), 8.39 (1H, d, J = 8.0 Hz, H-6), 8.43 (1H, ddd, J = 8.2, 2.3, 1.0 Hz, H-4), 8.78 (1H, t, J = 1.9 Hz, H-2), 10.47 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 55.9 (CH3), 114.5 (C-2′,6′), 122.9 (C-3′,5′), 123.0 (C-2), 126.7 (C-4), 130.9 (C-5), 132.4 (C-1′), 134.7 (C-6), 137.0 (C-1), 148.4 (C-3), 156.5 (C-4′), 163.5(CONH); GC-MS (EI) m/z: 272 [M]+.
N-(4′′-Morpholinophenyl)-3-nitrobenzamide (8i). Yield: 75%; m.p.: 204–205 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.20 (4H, t, J = 0.8 Hz, H-3′′,5′′), 3.82 (4H, t, J = 0.8 Hz, H-2′′,6′′), 7.16 (2H, d, J = 8.4 Hz, H-3′,5′), 7.71 (2H, J = 9.0 Hz, H-2′,6′), 7.83 (1H, t, J = 8.0 Hz, H-5), 8.39–8.44 (2H, m, H-4, H-6), 8.78 (1H, t, J = 1.2 Hz, H-2), 10.53 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 49.9 (C-3′′,5′′), 65.6 (C-2′′,6′′), 116.7 (C-3′,5′), 121.7 (C-2′,6′), 122.3 (C-2), 126.1 (C-4), 130.2 (C-5), 134.1 (C-6), 136.3 (C-1), 147.8 (C-3), 162.9 (CONH); GC-MS (EI) m/z: 327 [M]+.
3.1.8. General Experimental Procedures of 4ii–8ii
To a solution of 4i–8i (3.37 mmol) in methanol (20 mL) was added: ammonium chloride (double amounts of 4i–8i) and iron powder (6.03 mmol) and refluxed for 7 h. The reaction mixture was evaporated to give an oily residue, which was added with water and extracted with dichloromethane (30 mL × 3). The combined organic phase was dried with anhydrous MgSO4, filtrated, and the filtrate was evaporated to yield a crude solid, which recrystallized with ethyl acetate and n-hexane to give a pure white or pale yellow solid.
N-(4′-Chlorophenyl)-3-aminobenzamide (4ii). Yield: 97%; m.p.: 160–161 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 5.32 (2H, s, NH2), 6.75 (1H, ddd, J = 7.9, 2.3, 1.0 Hz, H-4), 7.04 (1H, d, J = 8.0Hz, H-6), 7.08 (1H, t, J = 2.0 Hz, H-2), 7.14 (1H, t, J = 7.7 Hz, H-5), 7.38 (2H, d, J = 8.9 Hz, 3′,5′), 7.80 (1H, d, J = 8.9 Hz, H-2′,6′), 10.19 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 112.9 (C-2), 114.7 (C-6), 116.9 (C-4), 121.5 (C-2′,6′), 126.9 (C-5), 128.4 (C-3′,5′), 129.8 (C-4′), 135.6 (C-1), 138.3 (C-1′), 148.8 (C-3), 166.9 (CONH); GC-MS (EI) m/z: 246 [M]+.
N-(4′-Bromophenyl)-3-aminobenzamide (5ii). Yield: 97%; m.p.: 176–177 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 5.32 (2H, s, NH2), 6.75 (1H, ddd, J = 7.9, 2.2, 0.9 Hz, H-4), 7.04 (1H, d, J = 8.0, Hz, H-6), 7.07 (1H, t, J = 2.0 Hz, H-2), 7.14 (1H, t, J = 7.7 Hz, H-5), 7.51 (2H, d, J = 8.9 Hz, H-3′,5′), 7.75 (2H, d, J = 8.9 Hz, H-2′,6′), 10.18 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 113.0 (C-2), 114.7 (C-6), 115.0 (C-4′), 116.9 (C-4), 122.0 (C-2′,6′), 128.8 (C-5), 131.3 (C-3′,5′), 135.6 (C-1), 138.8(C-1′), 148.8 (C-3), 166.5 (CONH); GC-MS (EI) m/z: 290 [M − 1]+, 292 [M + 1]+.
N-(4′-Fluorophenyl)-3-aminobenzamide (6ii). Yield: 76%; m.p.: 143–144 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 5.29 (2H, s, NH2), 6.74 (1H, ddd, J = 7.9, 2.3, 0.9 Hz, H-4), 7.05 (1H, d, J = 7.6 Hz, H-6), 7.08 (1H, t, J = 2.0 Hz, H-2), 7.12–7.23 (3H, m, H-5,3′,5′), 7.77 (2H, dd, J = 9.2, 5.1 Hz, H-2′, H-6′), 10.10 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 115.8 (C-2), 117.5 (C-6), 117.9 (J = 22.0 Hz, C-3′,5′), 119.6 (C-4), 124.8 (J = 7.8 Hz, C-2′,6′), 131.6 (C-5), 138.5 (J = 2.6 Hz, H-1′), 138.6 (C-1), 151.6 (C-3), 161.0 (J = 239.0 Hz, C-4′), 167.1 (CONH); GC-MS (EI) m/z: 230 [M]+.
N-(4′-Methoxyphenyl)-3-aminobenzamide (7ii). Yield: 86%; m.p.: 141–142 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.73 (3H, s, CH3), 5.28 (2H, s, NH2), 6.73 (1H, dd, J = 7.5, 1.8 Hz, H-4), 6.90 (2H, d, J = 9.1 Hz, H-3′, H-5′), 7.04 (1H, d, J = 7.9 Hz, H-6), 7.08 (1H, t, J = 2.0 Hz, H-2), 7.13 (1H, t, J = 7.7 Hz, H-5), 7.65 (2H, d, J = 9.0 Hz, H-2′,6′), 9.92 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 55.1 (CH3), 112.9 (C-2), 113.7 (C-3′,5′), 114.6 (C-6), 116.6 (C-4), 121.8 (C-2′,6′), 128.7 (C-5), 132.5 (C-1′), 136.0 (C-1), 148.7 (C-3), 155.3 (C-4′), 165.9 (CONH); GC-MS (EI) m/z: 242 [M]+.
N-(4′′-Morpholinophenyl)-3-aminobenzamide (8ii). Yield: 16%; m.p.: 173–174 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.06 (4H, t, J = 0.8 Hz, H-3′′, 5′′), 3.74 (4H, t, J = 0.8 Hz, H-2′′,6′′), 5.35 (2H, s, NH2), 6.73 (1H, d, J = 7.0 Hz, H-4), 6.91 (2H, d, J = 8.9 Hz, H-3′,5′), 7.12–7.21 (3H, m, H-2, H-5, H-6), 7.61 (2H, d, J = 8.9 Hz, H-2′,6′), 9.86 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 51.8 (C-3′′,5′′), 69.0 (C-2′′,6′′), 115.9 (C-2), 117.7 (C-6), 118.1 (C-2′,6′), 119.5 (C-4), 124.2 (C-3′, 5′), 131.5 (C-5), 134.5 (C-1′), 139.0 (C-1), 150.2 (C-4′), 151.3 (C-3), 168.6 (CONH); GC-MS (EI) m/z: 297 [M]+.
3.1.9. General Experimental Procedures of 4–13
To a solution of thiophene-2-carboxylic acid or 5-chlorothiophene-2-carboxylic acid (7.8 mmol) in anhydrous dichloromethane (40 mL) was added: triethylamine (8.6 mmol) and dropwise added oxalyl chloride (10.1 mmol) at room temperature. The reaction mixture was refluxed for 1 h and evaporated to remove the solvent and unreacted oxalyl chloride under reduced pressure. These acid chlorides were used for the next reaction without purification. To a solution of 4ii–8ii (0.41 mmol) in anhydrous benzene (20 mL) was added: triethylamine (0.41 mmol) and prepared thiophene-2-carbonyl chloride or 5-chlorothiophene-2-carbonyl chloride (0.51 mmol) at room temperature. The reaction mixture was stirred at room temperature for 2 h and evaporated to remove the benzene. To the residue, water was added and extracted with ethyl acetate (30 mL × 3). The organic phase was dried with anhydrous MgSO4, filtrated, and evaporated to prepare a crude compound, which was recrystallized with ethyl acetate and n-hexane to give a pure white or pale yellow solid.
N-(4′-Chlorophenyl)-3-(thiophen-2′′-ylcarbonylamino) benzamide (4). Yield: 77%; m.p.: 252–253 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 7.24 (1H, dd, J = 4.9, 3.8 Hz, H-4′′), 7.42 (2H, d, J = 8.8 Hz, H-3′, H-5′), 7.52 (1H, t, J = 7.9 Hz, H-5), 7.69 (1H, d, J = 7.8 Hz, H-6), 7.83 (2H, d, J = 8.9 Hz, H-2′,6′), 7.88 (1H, dd, J = 4.9, 0.9Hz, H-5′′), 8.01 (1H, dd, J = 8.1, 1.2 Hz, H-4), 8.08 (1H, dd, J = 3.7, 1.0 Hz, H-3′′), 8.25 (1H, s, H-2), 10.41 (1H, s, NHCO), 10.44 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 119.8 (C-2), 121.8 (C-2′,6′), 122.7 (C-6), 123.3 (C-4), 127.3 (C-4′), 128.2 (C-5), 128.5 (C-3′,5′), 128.8 (C-4′′), 129.3 (C-3′′), 132.2 (C-5′′), 135.4 (C-1), 138.1 (C-1′), 139.0 (C-3), 139.7 (C-2′′), 160.3 (CONH), 165.5 (NHCO); DIP-MS (EI) m/z: 356 [M]+; HRMS (EI) Calcd. For C18H14ClN2O2S 357.0465 [M + H]+, found 357.0457.
N-(4′-Bromophenyl)-3-(thiophen-2′′-ylcarbonylamino) benzamide (5). Yield: 65%; m.p.: 263–264 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 7.24 (1H, dd, J = 7.9, 3.8 Hz, H-4′′), 7.49–7.57 (3H, m, H-5,3′,5′), 7.69 (1H, d, J = 6.9 Hz, H-6), 7.77 (2H, d, J = 8.9 Hz, H-2′,6′), 7.88 (1H, dd, J = 5.0, 1.0 Hz, H-5′′), 8.01 (1H, dd, J = 8.1, 1.2 Hz, H-4), 8.07 (1H, dd, J = 3.8, 1.0 Hz, H-3′′), 8.25 (1H, t, J = 1.8 Hz, H-2), 10.41 (1H, s, NHCO), 10.44 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 115.3 (C-4′), 119.8 (C-2), 122.2 (C-2′,6′), 122.6 (C-6), 123.3 (C-4), 128.1 (C-5), 128.8 (C-4′′), 129.3 (C-3′′), 131.4 (C-3′,5′), 132.1 (C-5′′), 135.3 (C-1), 138.5 (C-1′), 139.0 (C-3), 139.7 (C-2′′), 160.0 (CONH), 165.5 (NHCO); DIP-MS (EI) m/z: 400 [M − 1]+, 402 [M + 1]+ ; HRMS (EI) Calcd. For C18H14BrN2O2S [M + H+] 400.9959, found 400.9947.
N-(4′-Fluorophenyl)-3-(thiophen-2′′-ylcarbonylamino) benzamide (6). Yield: 66%; m.p.: 236–237 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 7.20 (2H, t, J = 8.9 Hz, H-3′,5′), 7.24 (1H, dd, J = 5.0, 3.8 Hz, H-4′′), 7.52 (1H, t, J = 7.9 Hz, H-5), 7.69 (1H, d, J = 7.6 Hz, H-6), 7.80 (2H, dd, J = 9.2, 5.1 Hz, H-2′,6′), 7.88 (1H, dd, J = 5.0, 1.1Hz, H-5′′), 8.00 (1H, ddd, J = 8.1, 2.1, 0.8 Hz, H-4), 8.08 (1H, dd, J = 3.8, 1.1 Hz, H-3′′), 8.25 (1H, t, J = 1.9 Hz, H-2), 10.34 (1H, s, NHCO), 10.43 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 115.2 (J = 22.3 Hz, C-3′,5′), 119.8 (C-2), 122.1 (J = 7.4 Hz, C-2′,6′), 122.6 (C-6), 123.3 (C-4), 128.2 (C-5), 128.7 (C-4′′), 129.3 (C-3′′), 132.1 (C-5′′), 135.5 (J = 3.0 Hz, C-1′), 138.9 (C-3), 139.8 (C-2′′), 158.3 (J = 238.7 Hz, C-4′), 160.0 (CONH), 165.4 (NHCO); DIP-MS (EI) m/z: 340 [M]+ ; HRMS (EI) Calcd. For C18H14FN2O2S [M + H]+ 341.0760, found 341.0751.
N-(4′-Methoxyphenyl)-3-(thiophen-2′′-ylcarbonylamino) benzamide (7). Yield: 67%; m.p.: 236–237 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.75 (3H, s, CH3), 6.93 (2H, d, J = 9.0 Hz, H-3′,5′), 7.24 (1H, dd, J = 4.6, 4.0 Hz, H-4′′), 7.50 (1H, t, J = 7.9 Hz, H-5), 7.64–7.72 (3H, m, H-6,2′,6′), 7.88 (1H, d, J = 5.0 Hz, H-5′′), 8.00 (1H, dd, J = 8.0, 1.6 Hz, H-4), 8.11 (1H, d, J = 3.7 Hz, H-3′′), 8.25 (2H, t, J = 1.9 Hz, H-2), 10.17 (1H, s, NHCO), 10.46 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 55.2 (CH3), 113.7 (C-2′,6′), 119.8 (C-2), 122.0 (C-3′,5′), 122.8 (C-6), 123.1 (C-4), 128.4 (C-5), 128.7 (C-4′′), 129.4 (C-3′′), 132.2 (C-5′′), 134.1 (C-1′), 135.7 (C-1), 138.6 (C-3), 139.8 (C-2′′), 155.6 (C-4′), 159.0 (CONH), 164.9 (NHCO); DIP-MS (EI) m/z: 352 [M]+ ; HRMS (EI) Calcd. For C19H17N2O3S [M + H]+ 353.0960, found 353.0955.
N-(4′′-Morpholinophenyl)-3-(thiophen-2′′′-ylcarbonylamino) benzamide (8). Yield: 15%; m.p.: 254–255 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.07 (4H, t, J = 0.8 Hz, H-3′′ × 2, H-5′′ × 2), 3.74 (4H, t, J = 0.8 Hz, H-2′′,6′′), 6.94 (2H, d, J = 9.1 Hz, H-3′,5′), 7.24 (1H, dd, J = 4.9, 3.9 Hz, H-4′′′), 7.49 (1H, t, J = 7.9 Hz, H-5), 7.63 (2H, d, J = 9.0 Hz, H-2′,6′), 7.68 (1H, d, J = 7.8 Hz, H-6), 7.88 (1H, dd, J = 5.0, 0.9 Hz, H-5′′′), 7.98 (1H, dd, J = 8.1, 1.4 Hz, H-4), 8.07 (1H, dd, J = 3.8, 0.9 Hz, H-3′′′), 8.22 (1H, t, J = 1.7 Hz, H-2), 10.09 (1H, s, NHCO), 10.41 (CONH); 13C-NMR (100 MHz, D)MSO-d6) δ: 48.8 (C-3′′, C-5′′), 66.1 (C-2′′, C-6′′), 115.2 (C-2′,6′), 119.8 (C-2), 121.5 (C-3′,5′), 122.5 (C-6), 123.0 (C-4), 128.1 (C-5), 128.6 (C-4′′), 129.3 (C-3′′), 131.3 (C-1′), 132.1 (C-5′′), 135.8 (C-1), 138.8 (C-3), 139.8 (C-2′′), 147.5 (C-4′), 160.0 (CONH), 164.8 (NHCO); DIP-MS (EI) m/z: 407 [M]+; HRMS (EI) Calcd. For C22H22N2O3S [M + H]+ 408.1382, found 408.1372.
N-(4′-Chlorophenyl)-3-(5-chlorothiophen-2′′-ylcarbonylamino) benzamide (9). Yield: 77%; m.p.: 190–191 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 7.29 (1H, d, J = 4.1 Hz, H-4′′), 7.42 (2H, J = 8.9 Hz, H-3′,5′), 7.53 (1H, t, J = 7.9 Hz, H-5), 7.71 (1H, d, J = 7.8 Hz, H-6), 7.82 (2H, d, J = 8.9 Hz, H-2′,6′), 7.96–7.99 (2H, m, H-4,3′′), 8.22 (1H, t, J = 1.7 Hz, H-2), 10.41 (1H, s, NHCO), 10.51 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 119.8 (C-2), 121.8 (C-2′,6′), 122.9 (C-6), 123.4 (C-4), 127.3 (C-), 128.4 (C-5), 128.5 (C-3′,5′), 128.8 (C-4′′), 129.4 (C-3′′), 134.2 (C-1), 135.4 (C-3), 138.1 (C-1′), 138.6 (C-2′′), 139.8 (C-5′′), 159.0 (CONH), 165.4 (NHCO); DIP-MS (EI) m/z: 390 [M − 1]+ ; HRMS (EI) Calcd. For C18H13Cl2N2O2S [M + H]+ 391.0075, found 391.0066.
N-(4′-Bromophenyl)-3-(5-chlorothiophen-2′′-ylcarbonylamino) benzamide (10). Yield: 65%; m.p.: 271–272 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 7.29 (1H, d, J = 4.1 Hz, H-4′′), 7.49–7.57 (3H, m, H-5, H-3′,5′), 7.71 (1H, d, J = 8.9 Hz, H-6), 7.77 (2H, d, J = 7.9 Hz, H-2′,6′), 7.95–8.01 (2H, m, H-4,3′′), 8.21 (1H, t, J = 1.7 Hz, H-2), 10.41 (1H, s, NHCO), 10.51 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 116.1 (C-4′), 120.5 (C-2), 122.9 (C-2′,6′), 123.6(C-6), 124.1 (C-4), 129.1 (C-4′′), 129.5 (C-3′′), 130.0 (C-5), 132.1 (C-3′,5′), 134.9 (C-1), 136.1 (C-3), 139.2 (C-1′), 139.3 (C-2′′), 139.5 (C-5′′), 159.7 (CONH), 166.1 (NHCO); DIP-MS (EI) m/z: 434 [M − 1]+, 436 [M + 1]+; HRMS (EI) Calcd. For C18H14BrClN2O2S [M + H]+ 434.9570, found 434.9559.
N-(4′-Fluorophenyl)-3-(5-chlorothiophen-2′′-ylcarbonylamino) benzamide (11). Yield: 75%; m.p.: 257–258 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 7.20 (2H, t, J = 8.9 Hz, H-3′,5′), 7.29 (1H, d, J = 4.1 Hz, H-4′′), 7.52 (1H, t, J = 7.9 Hz, H-5), 7.71 (1H, d, J = 8.0 Hz, H-6), 7.79 (2H, dd, J = 9.2, 5.1 Hz, H-2′,6′), 7.96–7.99 (2H, m, H-4,3′′), 8.22 (1H, t, J = 1.8 Hz, H-2), 10.34 (1H, s, NHCO), 10.51 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 115.9 (J = 21.6 Hz, C-2′,6′), 119.9 (C-2), 122.2 (J = 7.5 Hz, C-3′,5′), 122.9 (C-6), 123.3 (C-4), 128.4 (C-5), 128.8 (C-4′′), 129.4 (C-3′′), 134.2 (C-1), 135.4 (C-3), 135.5 (J = 2.2 Hz, C-1′), 138.6 (C-2′′), 138.9 (C-5′′), 158.3 (J = 248.8 Hz, C-4′), 159.0 (CONH), 165.3 (NHCO); DIP-MS (EI) m/z: 374 [M]+; HRMS (EI) Calcd. For C18H14ClFN2O2S [M + H]+ 375.0370, found 375.0363.
N-(4′-Methoxyphenyl)-3-(5-chlorothiophen-2′′-ylcarbonylamino) benzamide (12). Yield: 88%; m.p.: 234–235 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.75 (3H, s, CH3), 6.93 (2H, d, J = 9.0 Hz, H-3′,5′), 7.29 (1H, d, J = 4.1 Hz, H-4′′), 7.50 (1H, t, J = 7.9 Hz, H-5), 7.66–7.71 (3H, m, H-6, H-2′,6′), 7.95–7.99 (2H, m, H-4,3′′), 8.21 (1H, t, J = 1.7 Hz, H-2), 10.16 (1H, s, NHCO), 10.52 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 55.2 (CH3), 113.7 (C-2′,6′), 119.8 (C-2), 121.9 (C-3′,5′), 122.8 (C-6), 123.1 (C-4), 128.4 (C-5), 128.7 (C-4′′), 129.3 (C-3′′), 132.2 (C-1′), 134.1 (C-1), 135.7 (C-3), 138.5 (C-2′′), 138.9(C-5′′), 155.5 (C-4′), 158.9 (CONH), 164.9 (NHCO); DIP-MS (EI) m/z: 386 [M]+ ; HRMS (EI) Calcd. For C19H16ClN2O3S [M + H]+ 387.0570, found 387.0565.
N-(4′′-Morpholinophenyl)-3-(5-chlorothiophen-2′′′-ylcarbonylamino) benzamide (13). Yield: 12%; m.p.: 269–270 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 3.07 (4H, t, J = 0.8 Hz, H-3′′,5′′×2), 3.74 (4H, t, J = 0.8 Hz, H-2′′,6′′), 6.94 (2H, d, J = 9.1 Hz, H-3′,5′), 7.29 (1H, d, J = 4.1 Hz, H-4′′′), 7.50 (1H, t, J = 7.9 Hz, H-5), 7.63 (2H, d, J = 9.1 Hz, H-2′,6′), 7.70 (1H, d, J = 7.8 Hz, H-6), 7.94–7.96 (2H, m, H-4,3′′′), 8.19 (1H, t, J = 1.8 Hz, H-2), 10.09 (1H, s, NHCO), 10.49 (1H, s, CONH); 13C-NMR (100 MHz, DMSO-d6) δ: 49.8 (C-3′′,5′′), 55.1 (C-2′′,6′′), 115.2 (C-2′,6′), 119.8 (C-2), 121.5 (C-3′,5′), 122.7 (C-6), 123.0 (C-4), 128.3 (C-5), 128.7 (C-5), 129.3 (C-3′′), 131.2 (C-1′), 134.1 (C-1), 135.8 (C-3), 138.5 (C-2′′), 138.9 (C-5′′), 147.5 (C-4′′), 159.9 (CONH), 164.7 (NHCO); DIP-MS (EI) m/z: 441 [M]+ ; HRMS (EI) Calcd. For C22H21ClN3O3S [M + H]+ 442.0992, found 442.0983.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}