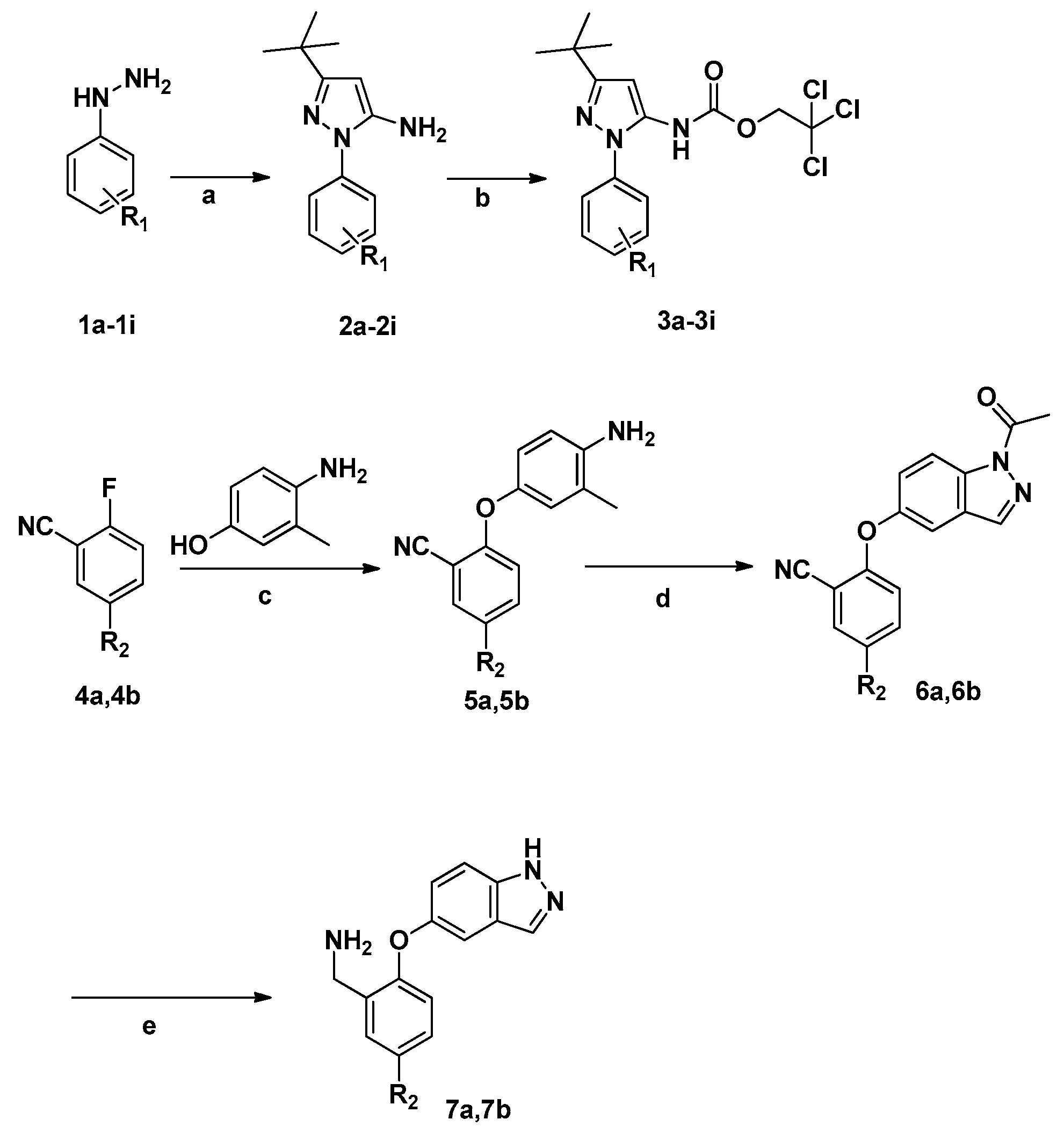
3.2. Synthesis
3.2.1. General Method for the Synthesis of 2a–2i
A solution of the corresponding substituted phenylhydrazine hydrochloride (1 equiv.) and pivaloylacetonitrile (1.2 equiv.) was stirred overnight at 80 °C in ethanol. The solution was extracted with EtOAc. The organic layer was washed with water and brine, dried (MgSO4), filtered, and evaporated to dryness.
3-(tert-Butyl)-1-(p-tolyl)-1H-pyrazol-5-amine (2a). From p-tolylhydrazine hydrochloride (1.59 g, 10 mmol) and pivaloylacetonitrile (1.5 g, 12 mmol), after work-up and without further purification, compound 2a was obtained as a white solid (1.8 g, 80%). IR (KBr): 3345, 1598, 1578 cm−1; 1H-NMR (CDCl3) δ: 7.40 (d, 2H, J = 8.8 Hz), 7.25 (d, 2H, J = 8.8 Hz), 5.50 (s, 1H), 3.72 (brs, 2H, NH2), 2.37 (s, 3H), 1.32 (s, 9H). MS (ESI): m/z 252.16 [M + Na]+. Mp 121.3–123.4 °C.
3-(tert-Butyl)-1-phenyl-1H-pyrazol-5-amine (2b). Yellow solid (1.74 g, 81%). IR (KBr): 3340, 1601, 1575 cm−1; 1H-NMR (CDCl3) δ: 7.60–7.69 (m, 2H), 7.50–7.57 (m, 2H), 7.36–7.41 (m, 1H), 5.56 (s, 1H), 3.79 (brs, 2H, NH2), 1.30 (s, 9H). MS (ESI): m/z 238.14 [M + Na]+. Mp 118.1–120.0 °C.
3-(tert-Butyl)-1-(m-tolyl)-1H-pyrazol-5-amine (2c). White solid (2.06 g, 90%). IR (KBr): 3343, 1600, 1579 cm−1; 1H-NMR (CDCl3) δ: 7.60–7.68 (m, 1H), 7.40–7.48 (m, 1H), 7.35–7.38 (m, 1H), 7.29–7.33 (m, 1H), 5.56 (s, 1H), 3.75 (brs, 2H, NH2), 2.37 (s, 3H), 1.30 (s, 9H). MS (ESI): m/z 252.16 [M + Na]+. Mp 120.3–122.4 °C.
3-(tert-Butyl)-1-(4-methoxyphenyl)-1H-pyrazol-5-amine (2d). White solid (1.97 g, 80%). IR (KBr): 3341, 1593, 1574 cm−1; 1H-NMR (CDCl3) δ: 7.41 (d, 2H), 6.97 (d, 2H), 5.43 (s, 1H), 3.83 (s, 3H), 3.72 (brs, 2H, NH2), 1.32 (s, 9H). MS (ESI): m/z 268.11 [M + Na]+. Mp 109.1–111.3 °C.
3-(tert-Butyl)-1-(4-chlorophenyl)-1H-pyrazol-5-amine (2e). White solid (1.87 g, 75%). IR (KBr): 3340, 1597, 1581 cm−1; 1H-NMR (CDCl3) δ: 7.59 (d, 2H, J = 8.8 Hz), 7.10 (d, 2H, J = 8.8 Hz), 5.47 (s, 1H), 3.74 (brs, 2H, NH2), 1.32 (s, 9H). MS (ESI): m/z 272.10 [M + Na]+. Mp 107.3–109.5 °C.
1-(4-Bromophenyl)-3-(tert-butyl)-1H-pyrazol-5-amine (2f). White solid (2.50 g, 85%). IR (KBr): 3337, 1596, 1575 cm−1; 1H-NMR (CDCl3) δ: 7.54 (d, 2H, J = 8.4 Hz), 7.14 (d, 2H, J = 8.4 Hz), 5.42 (s, 1H), 3.71 (brs, 2H, NH2), 1.31 (s, 9H). MS (ESI): m/z 316.05 [M + Na]+. Mp 109.1–112.7 °C.
3-(tert-Butyl)-1-(4-fluorophenyl)-1H-pyrazol-5-amine (2g). White solid (1.86 g, 79%). IR (KBr): 3333, 1594, 1582 cm−1; 1H-NMR (CDCl3) δ: 7.59 (d, 2H, J = 8.8 Hz), 7.10 (d, 2H, J = 8.8 Hz), 5.58 (s, 1H), 3.62 (brs, 2H, NH2), 1.31 (s, 9H). MS (ESI): m/z 256.34 [M + Na]+. Mp 102.3–104.6 °C.
3-(tert-Butyl)-1-(4-nitrophenyl)-1H-pyrazol-5-amine (2h). White solid (1.69 g, 65%). IR (KBr): 3333, 1594, 1582 cm−1; 1H-NMR (CDCl3) δ: 8.28 (d, 2H, J = 8.8 Hz), 7.93 (d, 2H, J = 8.8 Hz), 5.55 (s, 1H), 3.69 (brs, 2H, NH2), 1.30 (s, 9H). MS (ESI): m/z 283.13 [M + Na]+. Mp 105.1–107.3 °C.
4-(5-Amino-3-(tert-butyl)-1H-pyrazol-1-yl)benzenesulfonamide (2i). White solid (2.35 g, 80%). IR (KBr): 3367, 3342, 1592, 1588 cm−1; 1H-NMR (DMSO-d6) δ: 8.11 (d, 2H, J = 8.8 Hz), 7.83 (d, 2H, J = 8.8 Hz), 7.47 (s, 2H), 5.51 (s, 1H), 3.73 (brs, 2H, NH2), 1.32 (s, 9H).MS (ESI): m/z 317.12 [M + Na]+. Mp 103.1–105.2 °C.
3.2.2. General Method for the Synthesis of 3a–3i
A solution of the corresponding 2a–2i (1 equiv.) and 2,2,2-trichloroethyl carbonochloridate (1.1 equiv.) in THF was stirred 6 h at 0 °C, then the mixture was filtered and extracted three times with ethyl acetate. The solution was dried with MgSO4 and concentrated to dryness.
2,2,2-Trichloroethyl(3-(tert-butyl)-1-(p-tolyl)-1H-pyrazol-5-yl)carbamate (3a). From 2a (2.30 g, 10 mmol) and 2,2,2-trichloroethyl carbonochloridate (2.3 g, 11 mmol), after work-up and without further purification, 3a was obtained as a white solid (3.02 g, 75%). IR (KBr): 3169, 1597, 1580 cm−1; 1H-NMR (DMSO-d6) δ: 9.47 (s, 1H), 7.41 (d, 2H, J = 8.8 Hz), 7.23 (d, 2H, J = 8.8 Hz), 5.51 (s, 1H), 4.85(s, 2H), 1.32 (s, 9H). MS (ESI): m/z 426.06 [M + Na]+. Mp 153.3–155.7 °C.
2,2,2-Trichloroethyl(3-(tert-butyl)-1-phenyl-1H-pyrazol-5-yl)carbamate (3b). White solid (3.50 g, 90%). IR (KBr): 3170, 1593, 1577 cm−1; 1H-NMR (DMSO-d6) δ: 9.47(s, 1H), 7.65 (m, 2H), 7.57 (m, 2H), 7.40 (m, 1H), 5.52 (s, 1H), 4.85 (s, 2H), 1.32 (s, 9H). MS (ESI): m/z 412.11 [M + Na]+. Mp 151.1–153.2 °C.
2,2,2-Trichloroethyl(3-(tert-butyl)-1-(m-tolyl)-1H-pyrazol-5-yl)carbamate (3c). White solid (3.62 g, 90%). IR (KBr): 3168, 1601, 1582 cm−1; 1H-NMR (DMSO-d6) δ: 9.43 (s, 1H), 7.60–7.69 (m, 1H), 7.39–7.44 (m, 1H), 7.35–7.38 (m, 1H), 7.29–7.33 (m, 1H), 5.49 (s, 1H), 4.88 (s, 2H), 2.37 (s, 3H), 1.30 (s, 9H). MS (ESI): m/z 426.06 [M + Na]+. Mp 148.3–150.2 °C.
2,2,2-Trichloroethyl(3-(tert-butyl)-1-(4-methoxyphenyl)-1H-pyrazol-5-yl)carbamate (3d). White solid (3.56 g, 85%). IR (KBr): 3170, 1600, 1578 cm−1; 1H-NMR (DMSO-d6) δ: 9.40 (s, 1H), 7.41 (d, 2H, J = 8.8 Hz), 6.97 (d, 2H, J = 8.8 Hz), 5.44 (s, 1H), 4.81(s, 2H), 1.30 (s, 9H). MS (ESI): m/z 442.09 [M + Na]+. Mp 150.1–152.2 °C.
2,2,2-Trichloroethyl(3-(tert-butyl)-1-(4-chlorophenyl)-1H-pyrazol-5-yl)carbamate (3e). White solid (3.60 g, 85%). IR (KBr): 3166, 1600, 1577 cm−1; 1H-NMR (DMSO-d6) δ: 9.42 (s, 1H), 7.57 (d, 2H, J = 8.8 Hz), 7.12 (d, 2H, J = 8.8 Hz), 5.41 (s, 1H), 4.80 (s, 2H), 1.32 (s, 9H). MS (ESI): m/z 446.01 [M + Na]+. Mp 147.2–149.3 °C.
2,2,2-Trichloroethyl(1-(4-bromophenyl)-3-(tert-butyl)-1H-pyrazol-5-yl)carbamate (3f). White solid (3.54 g, 76%). IR (KBr): 3169, 1602, 1579 cm−1; 1H-NMR (DMSO-d6) δ: 9.42 (s, 1H), 7.54 (d, 2H, J = 8.8 Hz), 7.16 (d, 2H, J = 8.8 Hz), 5.37 (s, 1H), 4.78 (s, 2H), 1.30 (s, 9H). MS (ESI): m/z 490.00 [M + Na]+. Mp 145.1–147.6 °C.
2,2,2-Trichloroethyl(3-(tert-butyl)-1-(4-fluorophenyl)-1H-pyrazol-5-yl)carbamate (3g). White solid (3.25 g, 80%). IR (KBr): 3166, 1593, 1570 cm−1; 1H-NMR (DMSO-d6) δ: 9.40 (s, 1H), 7.48 (d, 2H, J = 8.8 Hz), 7.18 (d, 2H, J = 8.8 Hz), 5.33 (s, 1H), 4.64 (s, 2H), 1.29 (s, 9H). MS (ESI): m/z 430.04 [M + Na]+. Mp 148.7–150.1 °C.
2,2,2-Trichloroethyl(3-(tert-butyl)-1-(4-nitrophenyl)-1H-pyrazol-5-yl)carbamate (3h). Yellow solid (3.47 g, 80%). IR (KBr): 3165, 1595, 1573 cm−1; 1H-NMR (DMSO-d6) δ: 9.39 (s, 1H), 8.28 (d, 2H, J = 8.8 Hz), 7.93 (d, 2H, J = 8.8 Hz), 5.37 (s, 1H), 4.73 (s, 2H), 1.30 (s, 9H). MS (ESI): m/z 457.13 [M + Na]+. Mp 149.3–151.2 °C.
2,2,2-Trichloroethyl(3-(tert-butyl)-1-(4-sulfamoylphenyl)-1H-pyrazol-5-yl)carbamate (3i). White solid (3.27 g, 70%). IR (KBr): 3367, 3160, 1599, 1580 cm−1; 1H-NMR (DMSO-d6) δ: 9.39 (s, 1H), 8.15 (d, 2H, J = 8.8 Hz), 7.80 (d, 2H, J = 8.8 Hz), 7.45 (s, 2H), 5.35 (s, 1H), 4.70 (s, 2H), 1.29 (s, 9H); MS (ESI): m/z 491.02 [M + Na]+. Mp 143.4–145.6 °C.
3.2.3. General Method for the Synthesis of 5a–5b
A solution of the corresponding 4a–4b (1 equiv.) and 4-amino-3-methylphenol (10 mmol) was stirred 2 h at 90 °C in the presence of K2CO3 (4.14g 30 mmol) in DMSO (50 mL). Then the solution was extracted with EtOAc. The organic layer was washed with water and brine, dried (MgSO4), filtered and evaporated to dryness. The product was separated by column chromatography using PE/EA (3:1) as eluent.
2-(4-Amino-3-methylphenoxy)benzonitrile (5a). From 4a (1.21 g, 10 mmol), and 4-amino-3-methylphenol (1.23 g, 10 mmol), after work-up and purification, 5a was obtained as a white solid. (1.56 g, 70%). IR (KBr): 3275, 2250, 1595, 1575 cm−1; 1H-NMR (DMSO-d6) δ: 8.10–8.16 (m, 1H), 8.00–8.06 (m, 1H), 7.77–7.81 (m, 1H), 7.39–7.42 (m, 1H), 7.29–7.32 (m, 1H), 7.22–7.27 (m, 1H), 7.09–7.11 (m, 1H), 4.39 (s, 2H, NH2), 2.55 (s, 3H); MS (ESI): m/z 447.09 [M + Na]+. Mp 191.3–193.7 °C.
2-(4-Amino-3-methylphenoxy)-5-fluorobenzonitrile (5b). White solid (1.81 g, 75%). IR (KBr): 3274, 2250, 1593, 1574 cm−1; 1H-NMR (DMSO-d6) δ: 8.10–8.16 (m, 1H), 8.00–8.06 (m, 1H), 7.74–7.78 (m, 1H), 7.39–7.42 (m, 1H), 7.19–7.24 (m, 1H), 7.10–7.14 (m, 1H), 4.38 (s, 2H, NH2), 2.54 (s, 3H); MS (ESI): m/z 465.25 [M + Na]+. Mp 191.3–193.7 °C.
3.2.4. General Method for the Synthesis of 6a–6b
A solution of the corresponding 5a–5b (1 equiv.) and isoamyl nitrite (1.5 equiv.) was stirred 8 h at 90 °C in the presence of potassium acetate (0.98 g, 10 mmol) and acetic anhydride (1.75 g, 15 mmol) in toluene (50 mL). The solution was extracted with EtOAc. The organic layer was washed with water and brine, dried (MgSO4), filtered, and evaporated to dryness. The product was separated by column chromatography using PE/EA (8:1) as eluent.
2-((1-Acetyl-1H-indazol-5-yl)oxy)benzonitrile (6a). From 5a (2.24 g, 10 mmol), acetic anhydride (3 g, 30 mmol), potassium acetate (0.98 g, 10 mmol) and isoamyl nitrite (1.75 g, 15 mmol), after work-up and purification, 6a was obtained as a white solid. (1.66 g, 60%). IR (KBr): 2250, 1698, 1593, 1574 cm−1; 1H-NMR (DMSO-d6) δ: 8.46 (s, 1H), 8.34–8.39 (m, 1H), 7.90–7.95 (m, 1H), 7.66–7.69 (m, 1H), 7.61–7.65 (m, 1H), 7.44–7.51 (m, 1H), 7.27–7.32 (m, 1H), 6.92–6.99 (m, 1H), 2.74 (s, 3H); MS (ESI): m/z 310.09 [M + Na]+. Mp 201.7–203.1 °C.
2-((1-Acetyl-1H-indazol-5-yl)oxy)-5-fluorobenzonitrile (6b). White solid (1.62 g, 55%). IR (KBr): 2250, 1696, 1599, 1579 cm−1; 1H-NMR (DMSO-d6) δ: 8.49 (s, 1H), 8.29–8.36 (m, 1H), 7.87–7.92 (m, 1H), 7.60–7.65 (m, 1H), 7.50–7.57 (m, 1H), 7.40–7.46 (m, 1H), 6.91–6.99 (m, 1H), 2.76 (s, 3H); MS (ESI): m/z 318.22 [M + Na]+. Mp 205.1–207.3 °C.
3.2.5. General Method for the Synthesis of 7a–7b
A solution of the corresponding 6a or 6b (1 equiv.) and LiAlH4 (3 equiv.) in THF was stirred 1 h at 65 °C. Then the solution was extracted with EtOAc. The organic layer was washed with water and brine, dried (MgSO4), filtered and evaporated to dryness. Products were purified by column chromatography using DCM/MeOH (50:1) as eluent.
(2-((1H-Indazol-5-yl)oxy)phenyl)methanamine (7a). From 6a (2.77 g, 10 mmol) and LiAlH4 (1.14 g, 15 mmol), after work-up and purification, 7a was obtained as a solid a white solid. (1.20 g, 50%). IR (KBr): 3277, 1603, 1579 cm−1; 1H-NMR (DMSO-d6) δ: 13.12 (s, 1H), 8.08 (s, 1H), 7.84–7.91 (m, 1H), 7.60–7.65 (m, 3H), 7.23–7.28 (m, 2H), 6.81–6.87 (m, 1H), 4.23–4.25 (t, 2H), 3.79–3.83 (m, 2H); MS (ESI): m/z 262.11 [M + Na]+. Mp 178.3–180.5 °C.
(2-((1H-Indazol-5-yl)oxy)-5-fluorophenyl)methanamine (7b) White solid solid (1.41 g, 55%). IR (KBr): 3279, 1599, 1576 cm−1; 1H-NMR (DMSO-d6) δ: 13.11 (s, 1H), 8.08 (s, 1H), 7.84–7.89 (m, 1H), 7.59–7.65 (m, 2H), 7.21–7.25 (m, 2H), 6.81–6.88 (m, 1H), 4.25–4.27 (t, 2H), 3.80–3.86 (m, 2H); MS (ESI): m/z 280.26 [M + Na]+. Mp 180.1–182.7 °C.
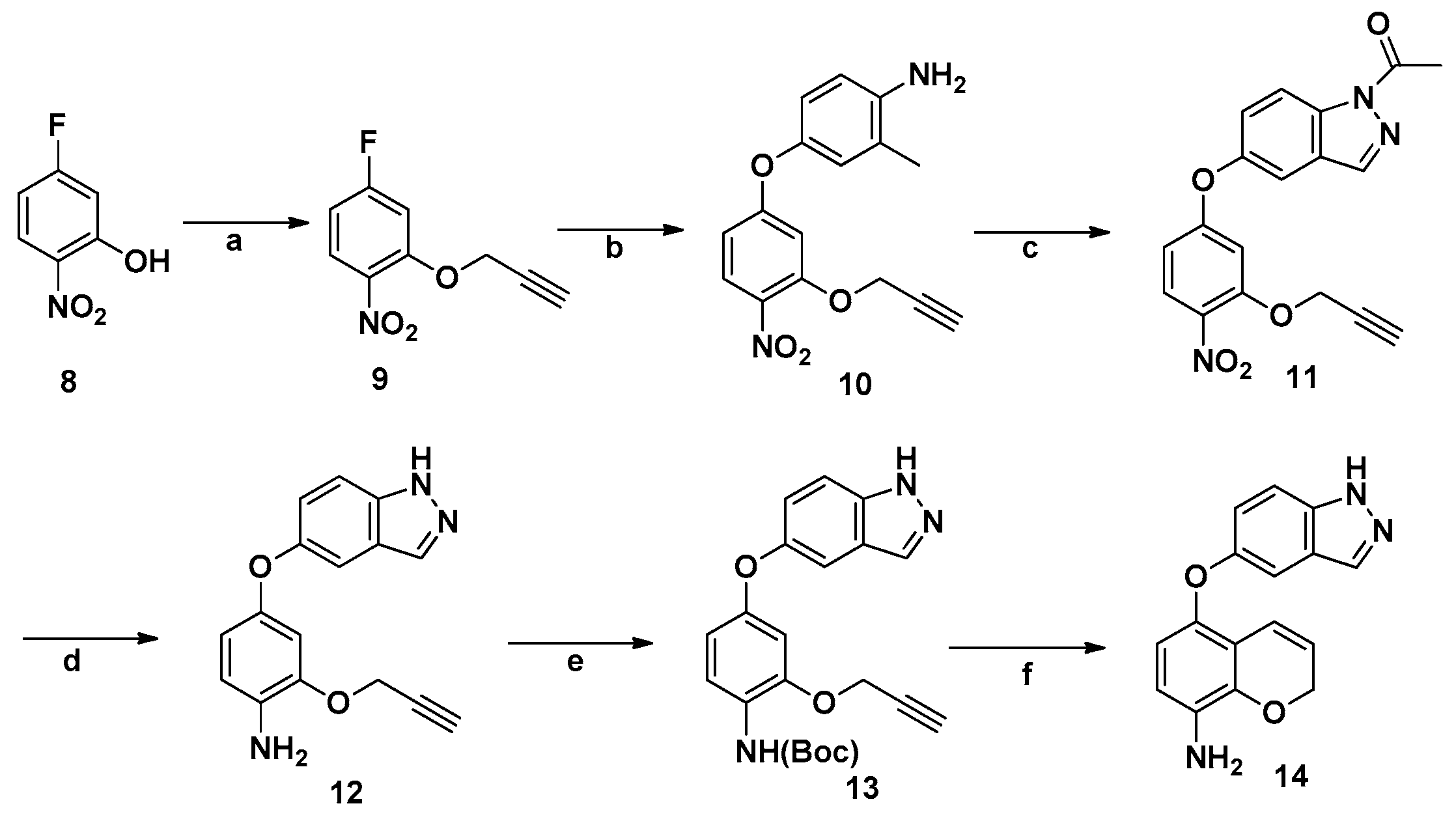
3.2.6. Synthesis of 14
4-Fluoro-1-nitro-2-(prop-2-yn-1-yloxy)benzene (9). Compound 8 (6.7 g, 40 mmol) was dissolved in DMF (60 mL), and K2CO3 (8.3 g, 60 mmol) was added while stirring vigorously. 3-Bromoprop-1-yne (50 mmol, 5.9 g) was then added dropwise while the mixture was on ice, and the reaction was allowed to proceed at room temperature for 12 h. The mixture was poured into ice water and kept in fridge for a day. The yellow solid 9 that appeared was filtered off and dried (yield 97%). IR (KBr): 3309, 2160, 1593, 1573 cm−1; 1H-NMR (DMSO-d6) δ: 8.00–8.06 (m, 1H), 7.31–7.37 (m, 1H), 7.01–7.07 (m, 1H), 5.06 (s, 2H), 3.75 (s, 1H); MS (ESI): m/z 218.15 [M + Na]+. Mp 110.4–112.7 °C.
2-Methyl-1-nitro-4-(4-nitro-3-(prop-2-yn-1-yloxy)phenoxy)benzene (10). To DMF (40 mL) compound 9 (1.95 g, 10 mmol) and K2CO3 (4.0 g, 30 mmol) were added and the mixture was stirred at room temperature for 4 h, followed by the addition of 4-amino-3-methylphenol (1.23 g, 10 mmol). The suspension formed was stirred at 100 °C for 3 h. The reaction mixture was cooled, and poured into ice water (100 mL), then the product was briefly placed in a fridge whereby a white solid appeared. The product was recovered by suction filtration and the filter cake was dried to give 2.55 g (90%) of compound 10. IR (KBr): 3309, 3267, 2160, 1593, 1573 cm−1; 1H-NMR (DMSO-d6) δ: 8.00–8.08 (m, 1H), 7.30–7.37 (m, 1H), 7.20–7.26 (m, 1H), 7.10–7.15 (m, 1H), 7.00–7.07 (m, 1H), 6.08–6.12 (m, 1H), 5.06 (s, 2H), 3.75 (s, 1H), 2.95 (s, 3H); MS (ESI): m/z 321.10 [M + Na]+. Mp 150.1–152.4 °C.
1-(5-(3-(Ethynyloxy)-4-nitrophenoxy)-1H-indazol-1-yl)ethanone (11). Compound 10 (0.8 g, 2.7 mmol) and acetic anhydride (1.14 g) were added to dry toluene (50 mL), followed by potassium acetate (0.3 g). The suspension was stirred at 80 °C and then isopentyl nitrite (0.6 g, 5 mmol) was added. The reaction was continued for 12 h, then the suspension was cooled down and filtered. Product 11 can be purified by column chromatography using DCM/MeOH (100: 1) as eluent, yield (75%). IR (KBr): 3309, 2160, 1670, 1593, 1573 cm−1; 1H-NMR (DMSO-d6) δ: 8.04 (s, 1H), 7.90–7.94 (m, 1H), 7.40–7.45 (m, 1H), 7.21–7.26 (m, 1H), 7.11–7.14 (m, 1H), 7.06–7.11 (m, 1H), 6.10–6.15 (m, 1H), 5.06 (s, 2H), 3.75 (s, 1H), 2.25 (s, 3H); MS (ESI): m/z 374.09 [M + Na]+. Mp 169.7–171.3 °C.
4-((1H-Indazol-5-yl)oxy)-2-(ethynyloxy)aniline (12). Compound 11 (2.0 g, 6 mmol) was placed in a 100 mL flask and then dissolved in ethyl acetate (40 mL). Ethanol (40 mL) was then added, followed by SnCl2 (4.5 g, 21 mmol). The mixture was heated to reflux for 6 h. Next saturated NaHCO3 solution was added until bubbles ceased to come out, and the mixture was filtered. The filtrate was extracted with ethyl acetate and then dried and then separated by column chromatography using DCM/MeOH (100:1) as eluent, yield (54%). IR (KBr): 3309, 3277, 2160, 1593, 1573 cm−1; 1H-NMR (DMSO-d6) δ: 13.01 (s, 1H), 8.05 (s, 1H), 7.90–7.99 (m, 1H), 7.40–7.45 (m, 1H), 7.21–7.24 (m, 1H), 7.11–7.15 (m, 1H), 7.02–7.12 (m, 1H), 6.11–6.14 (m, 1H), 5.04 (s, 2H), 3.77 (s, 1H), 3.28 (s, 2H); MS (ESI): m/z 302.09 [M + Na]+. Mp 192.1–194.7 °C.
tert-Butyl(4-((1H-indazol-5-yl)oxy)-2-(ethynyloxy)phenyl)carbamate (13). Compound 12 (2 g, 7 mmol) was dissolved in CH2Cl2 (25 mL) in a 100 mL flask. The flask was placed in a low temperature reaction tank and Et3N (3 mL) was added while the temperature was kept under −10 °C. Then (Boc)2O (3.9 g, 0.017 mol) was added to the flask in portions. The reaction was carried out for 48 h, then the solution was washed with water, dried, then separated by column chromatography using DCM/MeOH (50:1) as eluent, yield (63%). IR (KBr): 3309, 3250, 2160, 1690, 1593, 1573 cm−1; 1H-NMR (DMSO-d6) δ: 13.01 (s, 1H), 8.01 (s, 1H), 7.92–7.99 (m, 1H), 7.52–7.57 (m, 2H), 7.30–7.35 (m, 1H), 7.11–7.15 (m, 2H), 6.83–6.88 (m, 1H), 6.48–6.53 (m, 1H), 4.80 (s, 2H), 3.61 (s, 1H), 2.30 (s, 1H), 1.45 (s, 9H); MS (ESI): m/z 412.15 [M + Na]+. Mp 177.4–179.1 °C.
5-((1H-Indazol-5-yl)oxy)-2H-chromen-8-amine (14). Compound 13 (1.6 g, 0.004 mol) was dissolved in diphenyl ether (20 mL) in a 50 mL flask. The method using microwave was carried out as follows: temperature: 220 °C, pressure: 8.0 bar, time: 30 min, power: 250 w. Then the oxydibenzene was removed through vacuum distillation. Product 14 was then separated by column chromatography using DCM/MeOH (80:1) as eluent, yield (25%). IR (KBr): 3310, 3245, 1680, 1593, 1573 cm−1; 1H-NMR (DMSO-d6) δ: 12.99 (s, 1H), 7.92 (s, 1H), 7.48–7.52 (m, 1H), 7.05–7.09 (m, 1H), 6.98–7.02 (m, 1H), 6.45–6.49 (m, 1H), 6.40–6.43 (m, 1H), 6.30–6.34 (m, 1H), 5.80–5.83 (m, 1H), 4.73–4.76 (m, 2H), 4.56 (s, 2H); MS (ESI): m/z 312.15 [M + Na]+. Mp 181.3–183.7 °C.
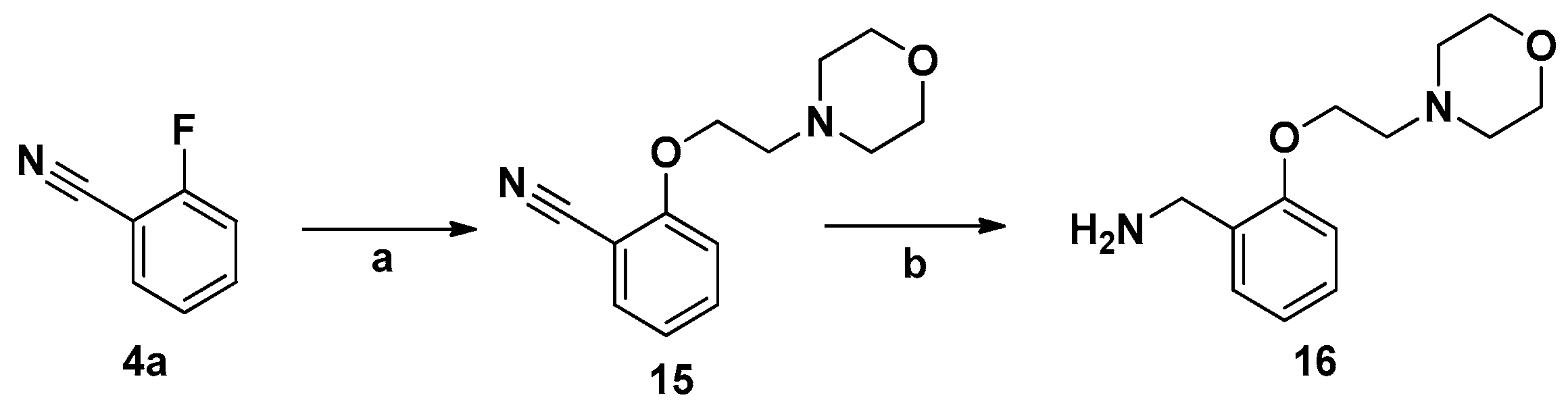
3.2.7. Synthesis of 16
2-(2-Morpholinoethoxy)benzonitrile (15). 2-Morpholinoethanol (4.2 g) was added to DMF (50 mL) in a 250 mL flask, followed by addition of sodium hydride (3 g) at −35 °C. An amount of 4a (3.6 g) was dissolved in DMF (15 mL). This solution was added dropwise to the flask and the mixture was stirred at −35 °C for 12 h. The mixture was poured into ice water (200 mL), and the solution was extracted with ethyl acetate. A gray oil was collected after drying and concentrating; yield 64%. IR (KBr): 2250, 1598, 1578 cm−1; 1H-NMR (DMSO-d6) δ: 7.22–7.25 (m, 1H), 7.13–7.16 (m, 1H), 6.85–6.88 (m, 1H), 6.70–6.74 (m, 1H), 4.07–4.09 (t, 2H, J = 5.6), 3.57–3.59 (t, 4H, J = 4.8), 2.72–2.74 (t, 2H, J = 5.6), 2.44–2.46 (t, 4H, J = 4.8); MS (ESI): m/z 255.12 [M + Na]+.
(2-(2-Morpholinoethoxy)phenyl)methanamine (16). LiAlH4 (1.5 g) was added to anhydrous THF (40 mL) in a 150 mL flask, which was kept in an ice bath. Compound 15 (2.32 g, 10 mmol) was dissolved in anhydrous THF (15 mL) and this solution was added dropwise to LiAlH4 suspension and the mixture then stirred at 60 °C for 2 h. Ethanol was added to the mixture until gas evolution stopped, then the suspension was filtered and concentrated. The product 16 was obtained as a solid by column chromatography (DCM/MeOH (100:1)), yield (64%). IR (KBr): 3270, 1597, 1577 cm−1; 1H-NMR (DMSO-d6) δ: 7.23–7.27 (m, 1H), 7.16–7.18 (m, 1H), 6.93–6.99 (m, 1H), 6.80–6.87 (m, 1H), 4.28–4.30 (t, 2H, J = 7.6), 4.08–4.10 (t, 2H, J = 5.6), 3.56–3.58 (t, 4H, J = 4.8), 2.70–2.72 (t, 2H, J = 5.6), 2.46–2.48 (t, 4H, J = 4.8); MS (ESI): m/z 259.12 [M + Na]+. Mp 181.3–183.7 °C.
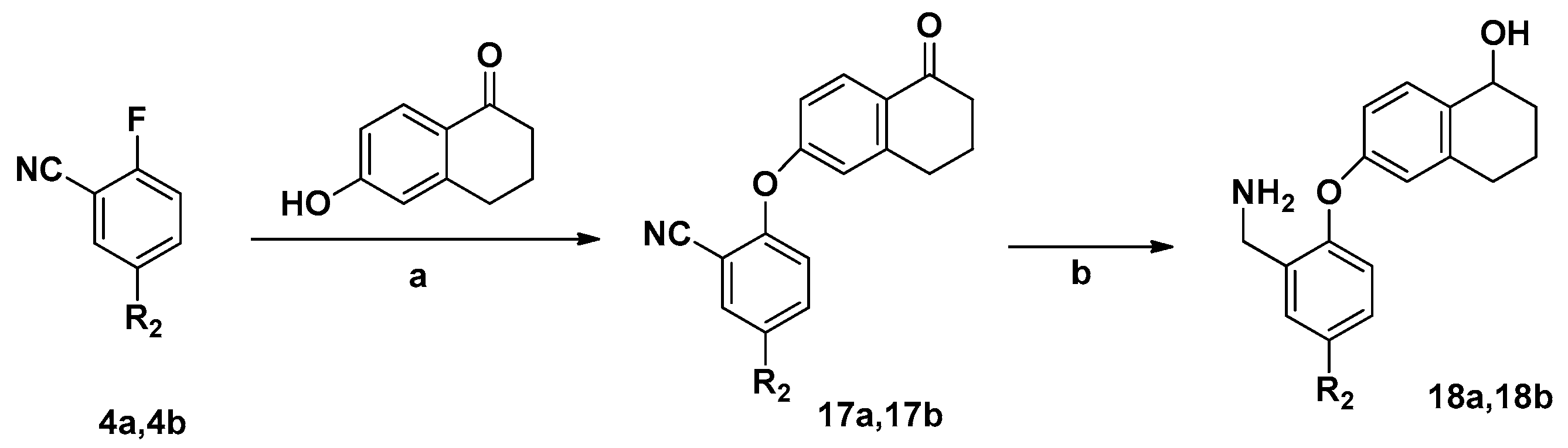
3.2.8. General Method for the Synthesis of 18a and 18b
2-((5-Oxo-5,6,7,8-tetrahydronaphthalen-2-yl)oxy)benzonitrile (17a). 6-Hydroxy-1-tetralone (5 g, 30.86 mmol) was placed in a 250 mL three-necked flask and dissolved in DMSO (100 mL). The flask was heated to 90 °C, and K2CO3 (12.78 g) was added under stirring. Compound 4a (4.48 g, 37.03 mmol) was added dropwise into the flask and the reaction mixture was heated to 90 °C for 4 h. The mixture was poured into water (200 mL), and the water phase was extracted twice with ethyl acetate. The organic phases were combined, washed with water and brine, dried with Na2SO4, and the solvent removed under vacuum to yield the crude product, which was purified by column chromatography using petroleum ether/ethyl acetate (5:1) as eluent to give compound 17a as a yellow solid (75.3%). IR (KBr): 2253, 1688, 1597, 1577 cm−1; 1H-NMR (CDCl3), δ: 8.32–8.35 (m, 1H), 7.66–7.69 (m, 1H), 7.59–7.61 (m, 1H), 7.35–7.38 (m, 3H), 7.00–7.04 (m, 1H), 2.90–2.94 (m, 2H), 2.71–2.74 (m, 2H), 2.00–2.03 (m, 2H). MS (ESI): m/z 286.34 [M + Na]+. Mp 178.3–180.5 °C.
5-Fluoro-2-((5-oxo-5,6,7,8-tetrahydronaphthalen-2-yl)oxy)benzonitrile (17b). White solid, yield 71.1%. IR (KBr): 2250, 1687, 1597, 1577 cm−1; 1H-NMR (CDCl3), δ: 8.40–8.43 (m, 1H), 7.59–7.62 (m, 1H), 7.35–7.39 (m, 2H), 7.19–7.24 (m, 1H), 7.07–7.11 (m, 1H), 2.93–2.96 (m, 2H), 2.73–2.77 (m, 2H), 2.01–2.05 (m, 2H). MS (ESI): m/z 304.22 [M + Na]+. Mp 171.7–173.2 °C.
6-(2-(Aminomethyl)phenoxy)-1,2,3,4-tetrahydronaphthalen-1-ol (18a). LiAlH4 (0.35 g, 9 mmol) was added to anhydrous THF (15 mL) in a 50 mL flask, which was placed in an ice bath. Compound 17a (0.74 g, 3 mmol) was dissolved in anhydrous THF (15 mL) and this solution was added dropwise into the LiAlH4 suspension and the mixture then stirred at 65 °C for 30–40 min. Ethanol was added until bubble evolution ceased, then the suspension was filtered and concentrated. Compound 18a was separated by column chromatography (dichloromethane/methanol 25:1), yield 77.7%. IR (KBr): 3350, 1597, 1577 cm−1; 1H-NMR (CDCl3), δ: 7.20–7.25 (m, 1H), 7.14–7.17 (m, 1H), 7.05–7.09 (m, 2H), 7.00–7.02 (m, 1H), 6.87–6.93 (m, 1H), 6.86–6.90 (m, 1H), 4.55–4.60 (m, 1H), 4.20–4.22 (t, 2H, J = 7.6 Hz), 2.90–2.94 (m, 2H), 2.73–2.76 (m, 2H), 2.01–2.04 (m, 2H). MS (ESI): m/z 292.01 [M + Na]+. Mp 165.2–167.1 °C.
6-(2-(Aminomethyl)-4-fluorophenoxy)-1,2,3,4-tetrahydronaphthalen-1-ol (18b). White solid, yield 77.7%. IR (KBr): 3347, 1595, 1575 cm−1; 1H-NMR (CDCl3), δ: 7.21–7.24 (m, 1H), 7.16–7.20 (m, 1H), 7.08–7.12 (m, 2H), 6.89–6.92 (m, 1H), 6.85–6.89 (m, 1H), 4.54–4.59 (m, 1H), 4.19–4.21 (t, 2H, J = 7.6 Hz), 2.95–2.99 (m, 2H), 2.75–2.80 (m, 2H), 2.04–2.09 (m, 2H). MS (ESI): m/z 310.13 [M + Na]+. Mp 166.6–168.3 °C.
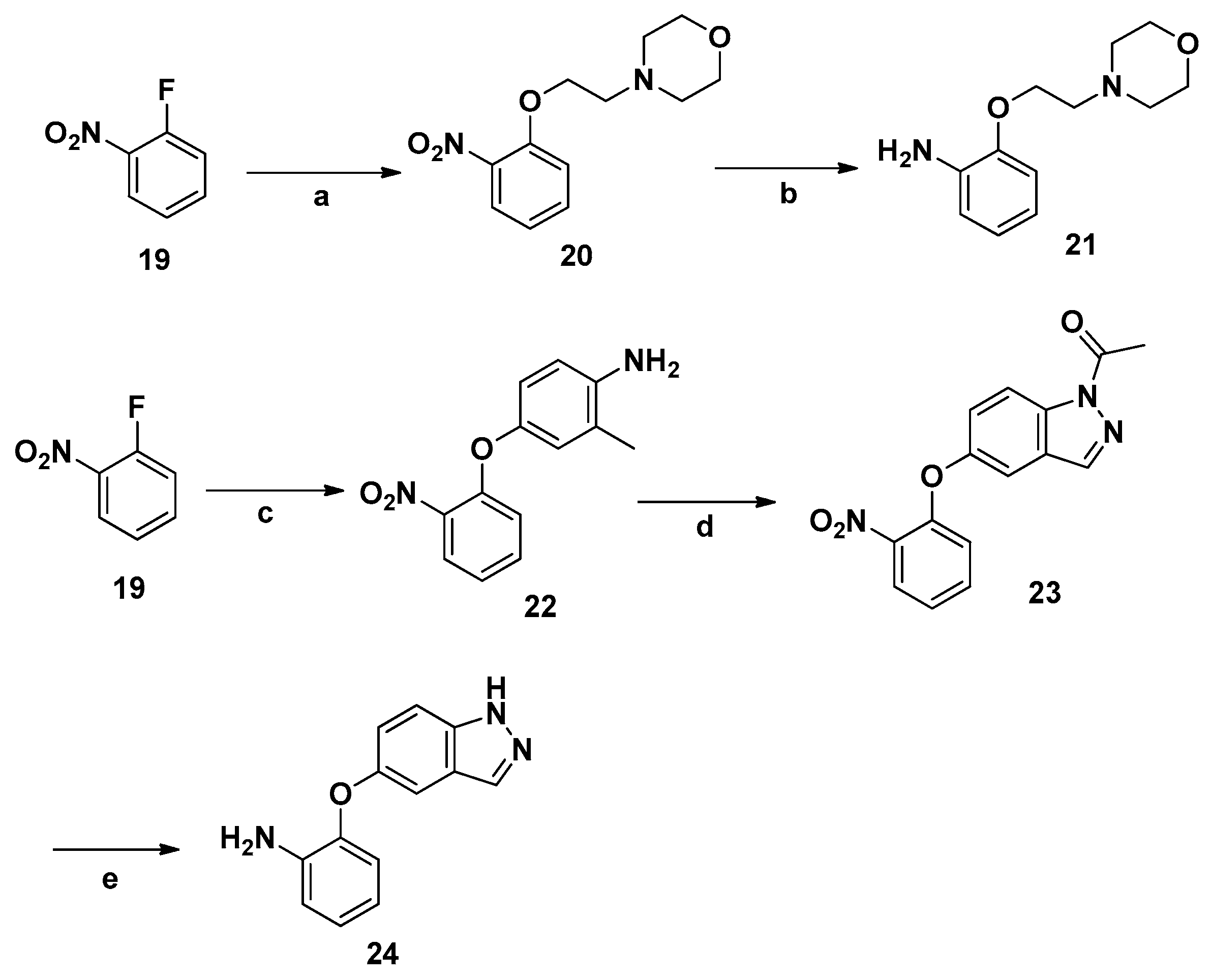
3.2.9. Synthesis of 21
4-(2-(2-Nitrophenoxy)ethyl)morpholine (20). 2-Morpholinoethanol (1.3 g,10 mmol) was added to DMF (30 mL) in a 50-mL flask, followed by addition of sodium hydride (0.48 g, 20 mmol) at −35 °C. An amount of 1-fluoro-2-nitrobenzene (19, 3.26 g, 12 mmol) was dissolved in DMF (15 mL) and the solution was added dropwise to the flask and the reaction mixture stirred at −35 °C for 12 h. The mixture was poured into ice water (200 mL), and extracted with ethyl acetate. A gray oil was collected after drying and concentrating; yield 64%. IR (KBr): 1598, 1578 cm−1; 1H-NMR (DMSO-d6) δ: 8.29–8.32 (m, 1H), 7.87–7.89 (m, 1H), 7.57–7.61 (m, 1H), 7.43–7.48 (m, 1H), 4.07–4.09 (t, 2H, J = 5.6 Hz), 3.59–3.61 (t, 4H, J = 4.8 Hz), 2.76–2.78 (t, 2H, J = 5.6 Hz), 2.46–2.48 (t, 4H, J = 4.8 Hz); MS (ESI): m/z 275.12 [M + Na]+.
2-(2-Morpholinoethoxy)aniline (21). Compound 20 (2.52 g, 10 mmol) was added to EtOH (50 mL) in a 100-mL flask, followed by SnCl2·2H2O (11.28 g (50 mmol) and the mixture was heated at 80 °C for 2 h. The mixture was then poured into 10% aqueous NaOH solution (200 mL), and the solid was filtered. The water phase was extracted twice with ethyl acetate, and the organic phases were combined and washed with water and brine and dried with Na2SO4. The crude product was obtained after concentration; yield 60%. IR (KBr): 3267, 1597, 1577 cm−1; 1H-NMR (DMSO-d6) δ: 8.29–8.32 (m, 1H), 7.87–7.89 (m, 1H), 7.57–7.61 (m, 1H), 7.43–7.48 (m, 1H), 6.12 (s, 1H) 4.05–4.07 (t, 2H, J = 5.6 Hz), 3.58–3.60 (t, 4H, J = 4.8 Hz), 2.73–2.75 (t, 2H, J = 5.6 Hz), 2.49–2.51 (t, 4H, J = 4.8 Hz); MS (ESI): m/z 245.12 [M + Na]+. Mp 165.3–167.7 °C.
3.2.10. Synthesis of 24
2-Methyl-4-(2-nitrophenoxy)aniline (22). Compound 19 (1.41 g, 10 mmol) and 4-amino-3-methylphenol (1.23 g, 10 mmol) were stirred 2 h at 90 °C in the presence of K2CO3 (4.14g 30 mmol) in DMSO (50 mL). The mixture was poured into ice water (200 mL), and the solution was extracted with ethyl acetate. The organic layer was washed with water and brine, dried (MgSO4), filtered, and evaporated to dryness. After work-up and purification by column chromatography using PE/EA (3:1) as eluent, intermediate 20 was obtained as a white solid; yield 74%. IR (KBr): 3275, 1595, 1575 cm−1; 1H-NMR (DMSO-d6) δ: 8.17–8.21 (m, 1H), 8.01–8.04 (m, 1H), 7.72–7.76 (m, 1H), 7.44–7.47 (m, 1H), 7.33–7.36 (m, 1H), 7.27–7.31 (m, 1H), 7.12–7.15 (m, 1H), 4.41(s, 2H, NH2), 2.53(s, 3H); MS (ESI): m/z 267.08 [M + Na]+. Mp 153.1–155.5 °C.
1-(5-(2-Nitrophenoxy)-1H-indazol-1-yl) ethanone (23). Compound 22 (2.44 g, 10 mmol, and acetic anhydride (3.06 g, 30 mmol) were added to dry toluene (50 mL) followed by potassium acetate (0.98 g, 10 mmol). The suspension was stirred at 80 °C, and isopentyl nitrite (1.76 g, 15 mmol) was added. The reaction was continued for 18 h. The suspension was cooled and filtered. The product was separated by column chromatography using PE/EA (10:1) as an eluent; yield 60%. IR (KBr): 1696, 1594, 1573 cm−1; 1H-NMR (DMSO-d6) δ: 8.43–8.45 (s, 1H), 8.31–8.34 (m, 1H), 7.90–7.92 (m, 1H), 7.64–7.67 (m, 1H), 7.63–7.66 (m, 1H), 7.55–7.58 (m, 1H), 7.33–7.36 (m, 1H), 6.99 (m, 1H), 2.76 (s, 3H); MS (ESI): m/z 320.07 [M + Na]+. Mp 167.4–169.6 °C.
2-((1H-Indazol-5-yl)oxy)aniline (24). Compound 23 (2.97 g, 10 mmol) was added to EtOH (50 mL) in a 100-mL flask, followed by SnCl2·2H2O (11.30 g, 50 mmol) and the mixture was reacted at 80 °C for 2 h. The mixture was poured into 10% aqueous NaOH solution (200 mL), and the solid formed was filtered. The water phase was extracted twice with ethyl acetate, and the organic phases were combined, washed with water and brine, and dried with Na2SO4. A yellow solid was collected after concentration; yield 57%. IR (KBr): 3272, 1600, 1578 cm−1; 1H-NMR (DMSO-d6) δ: 13.11 (s, 1H), 8.04 (s, 1H), 7.84–7.89 (m, 1H), 7.63–7.66 (m, 3H), 7.20–7.24 (m, 2H), 6.86–6.89 (m, 1H), 3.80–3.82 (m, 2H); MS (ESI): m/z 248.09 [M + Na]+. Mp 180.1–182.4 °C.
3.2.11. General Method for the Synthesis of 25a–25k
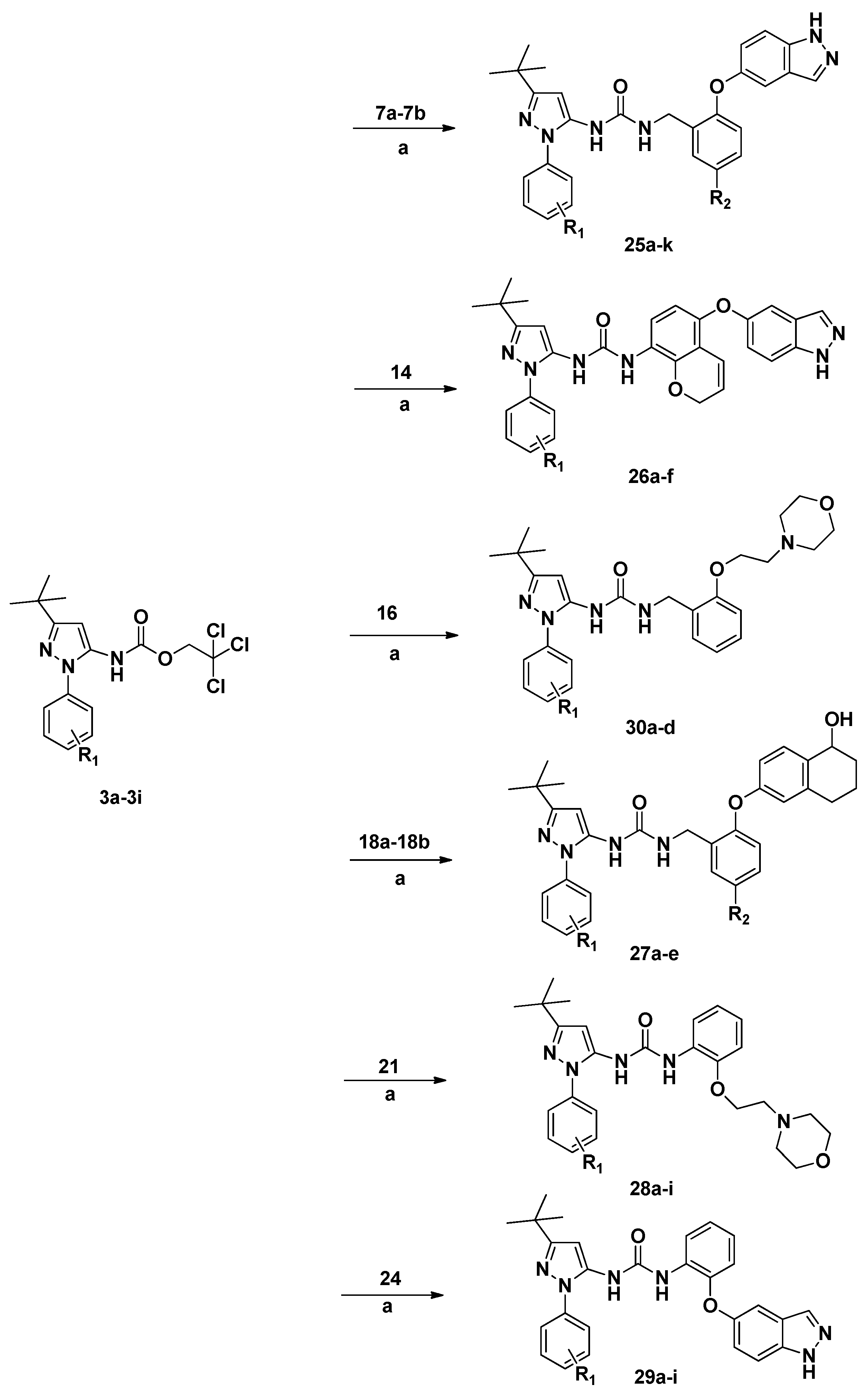
A solution of the corresponding 3a–3i (1.1 equiv.) and 7a–7b (1 equiv.) was stirred for 2 h at 90 °C in the presence of Et3N (1 mL) in DMSO (50 mL). The mixture was poured into ice water, and the solution was extracted with EtOAc. The organic layer was washed with water and brine, dried (MgSO4), filtered and evaporated to dryness. The product was separated by column chromatography using DCM/MeOH (100:1) as an eluent.
1-(2-((1H-Indazol-5-yl)oxy)benzyl)-3-(3-(tert-butyl)-1-(p-tolyl)-1H-pyrazol-5-yl)urea (25a). From 3a (4.03 g, 10 mmol), and 7a (2.39 g, 10 mmol), after work-up and purification 25a (3.46 g, 70%) was obtained as a white solid. HPLC purity 98.1% (tR =11.97 min). IR (KBr): 3257, 1670, 1594, 1576 cm−1; 1H-NMR (DMSO-d6) δ: 13.13 (s, 1H), 8.22 (s, 1H), 8.01 (s, 1H), 7.58 (d, 1H, J = 8.8 Hz), 7.37–7.40 (m, 2H), 7.25–7.27 (m, 3H), 7.06–7.10 (m, 4H), 6.95–6.97 (t, 1H, J = 5.6 Hz), 6.75(d, 1H, J = 8.0 Hz), 6.23(s, 1H), 4.31(d, 2H, J = 5.6 Hz), 3.80 (s, 3H), 1.25(s, 9H). 13C-NMR (DMSO-d6) δ: 160.4 (1C, pyrazole), 159.2 (1C, pyrazole), 156.8 (1C, CO), 154.6 (1C, pyrazole), 150.8, 137.6, 136.9, 136.5, 136.3, 133.4, 133.0, 132.9, 129.6, 124.1, 123.2, 119.6, 119.3, 115.0, 111.7, 107.6 (18C, Ar-C), 95.8 (1C, pyrazole), 38.1 (1C, CH2), 32.0 (1C, CH3-C), 30.2 (3C, C-CH3), 20.6 (1C, Ar-CH3); MS (ESI): m/z 517.24 [M + Na]+. Mp 201.3–203.5 °C.
1-(2-((1H-Indazol-5-yl)oxy)benzyl)-3-(3-(tert-butyl)-1-phenyl-1H-pyrazol-5-yl)urea (25b). White solid (3.12 g, 65%). HPLC purity 97.2% (tR = 11.13 min). IR (KBr): 3254, 1670, 1596, 1578 cm−1; 1H-NMR (DMSO-d6) δ: 13.09 (s, 1H), 8.30 (s, 1H), 7.99 (s, 1H), 7.57 (d, 1H, J = 8.8 Hz), 7.48–7.51 (m, 4H), 7.39–7.43 (m, 1H), 7.26–7.29 (m, 3H), 7.10–7.12 (m, 2H), 6.94–6.96 (t, 1H, J = 5.6 Hz), 6.75 (d, 2H, J = 8.0 Hz), 4.32 (d, 2H, J = 5.6 Hz), 1.27 (s, 9H). 13C-NMR (DMSO-d6) δ: 160.6 (1C, pyrazole), 159.4 (1C, pyrazole), 157.1 (1C, CO), 154.8 (1C, pyrazole), 151.0, 153.2, 150.9, 138.0, 136.9, 136.8, 133.8, 133.4, 130.0, 124.5, 121.0, 120.7, 117.4, 115.3, 115.0, 111.1 (18C, Ar-C), 96.2 (1C, pyrazole), 38.5 (1C, CH2), 32.6 (1C, CH3-C), 30.6 (3C, C-CH3); MS (ESI): m/z 503.14 [M + Na]+. Mp 197.1–199.2 °C.
1-(2-((1H-Indazol-5-yl)oxy)benzyl)-3-(3-(tert-butyl)-1-(m-tolyl)-1H-pyrazol-5-yl)urea (25c). White solid (3.70 g, 75%). HPLC purity 95.5% (tR = 11.82 min). IR (KBr): 3255, 1670, 1599, 1574 cm−1; NMR (DMSO-d6) δ: 13.11 (s, 1H), 8.30 (s, 1H), 7.99 (s, 1H), 7.57 (d, 1H, J = 8.8 Hz), 7.36–7.39 (m, 1H), 7.25–7.28 (m, 6H), 7.11–7.14 (m, 2H), 6.97–6.99 (t, 1H, J = 5.6 Hz), 6.74 (d, 1H, J = 8.0 Hz), 6.25 (s, 1H), 4.31 (d, 2H, J = 5.6 Hz), 2.46 (s, 3H), 1.24 (s, 9H); 13C-NMR (DMSO-d6) δ: 160.4 (1C, pyrazole), 154.4 (1C, pyrazole), 153.7 (1C, CO), 152.7 (1C, pyrazole), 152.1, 149.5, 146.5, 137.7, 136.5, 136.3, 130.9, 129.6, 129.1, 128.6, 124.2, 124.0, 122.5, 120.0, 119.1, 105.3 (18C, Ar-C), 95.1 (1C, pyrazole), 38.6 (1C, CH2), 31.0 (1C, CH3-C), 30.2 (3C, C-CH3), 22.1 (1C, Ar-CH3); MS (ESI): m/z 517.24 [M + Na]+. Mp 200.4–201.9 °C.
1-(2-((1H-Indazol-5-yl)oxy)benzyl)-3-(3-(tert-butyl)-1-(4-methoxyphenyl)-1H-pyrazol-5-yl)urea (25d). White solid (3.16 g, 62%). HPLC purity 96.3% (tR = 12.13 min). IR (KBr): 3259, 1670, 1600, 1579 cm−1; 1H-NMR (DMSO-d6) δ: 13.09 (s, 1H), 8.23 (s, 1H), 7.99 (s, 1H), 7.57 (d, 1H, J = 8.8 Hz), 7.34–7.37 (m, 3H), 7.25–7.27 (m, 4H), 7.10–7.14 (m, 2H), 6.93–6.95 (t, 1H, J = 5.6 Hz), 6.74 (d, 1H, J = 8.0 Hz), 6.23 (s, 1H), 4.31 (d, 2H, J = 5.6 Hz), 2.35 (s, 3H), 1.25 (s, 9H); 13C-NMR (DMSO-d6) δ: 161.6 (1C, pyrazole), 155.3 (1C, pyrazole), 154.5 (1C, CO), 150.3 (1C, pyrazole), 142.0, 141.3, 138.2, 137.1, 133.4, 130.0, 128.7, 128.4, 126.8, 123.6, 123.2, 123.0, 119.8, 117.4, 111.7, 108.4 (18C, Ar–C), 97.0 (1C, pyrazole), 59.8 (1C, O-CH3), 37.5 (1C, CH2), 31.0 (1C, CH3-C), 30.1 (3C, C-CH3); MS (ESI): m/z 533.23 [M + Na]+. Mp 198.0–200.7 °C.
1-(2-((1H-Indazol-5-yl)oxy)benzyl)-3-(3-(tert-butyl)-1-(4-chlorophenyl)-1H-pyrazol-5-yl)urea (25e). White solid (2.83 g, 55%). HPLC purity 97.7% (tR = 12.18 min). IR (KBr): 3257, 1670, 1596, 1577 cm−1; 1H-NMR (DMSO-d6) δ: 13.10 (s, 1H), 8.34 (s, 1H), 7.99(s, 1H), 7.57 (d, 1H, J = 8.8 Hz), 7.51–7.54 (m, 4H), 7.25–7.28 (m, 3H), 7.10–7.13 (m, 2H), 6.91–6.93 (t, 1H, J = 5.6 Hz), 6.75 (d, 1H, J = 8.0 Hz), 6.25 (s, 1H), 4.31 (d, 2H, J = 5.6 Hz), 1.25 (s, 9H); 13C-NMR (DMSO-d6) δ: 160.6 (1C, pyrazole), 159.4 (1C, pyrazole), 157.0 (1C, CO), 154.8 (1C, pyrazole), 151.1, 137.7, 137.1, 136.6, 136.4, 133.6, 133.1, 133.0, 129.8, 124.3, 123.4, 119.8, 119.4, 115.2, 111.9, 107.7 (18C, Ar–C), 96.1 (1C, pyrazole), 38.3 (1C, CH2), 32.2 (1C, CH3-C), 30.4 (3C, C-CH3); MS (ESI): m/z 537.19 [M + Na]+. Mp 199.6–201.4 °C.
1-(2-((1H-Indazol-5-yl)oxy)benzyl)-3-(1-(4-bromophenyl)-3-(tert-butyl)-1H-pyrazol-5-yl)urea (25f). White solid (4.01 g, 72%). HPLC purity 97.5% (tR = 12.01 min). IR (KBr): 3256, 1670, 1593, 1575 cm−1; 1H-NMR (DMSO-d6) δ: 13.13 (s, 1H), 8.38 (s, 1H), 8.00 (s, 1H), 7.66 (d, 2H, J = 8.4 Hz), 7.58 (d, 1H, J = 8.8), 7.25–7.28 (m, 3H), 7.11–7.13 (m, 2H), 6.94–6.96 (t, 1H, J = 5.6 Hz), 6.74 (d, 1H, J = 8.0 Hz), 6.27 (s, 1H), 4.31 (d, 2H, J = 5.6 Hz), 1.25 (s, 9H); 13C-NMR (DMSO-d6) δ: 161.6 (1C, pyrazole), 155.8 (1C, pyrazole), 155.0 (1C, CO), 150.8 (1C, pyrazole), 138.5, 138.2, 137.5, 133.8, 131.8, 130.5, 129.6, 129.3, 128.8, 126.1, 123.7, 123.4, 120.3, 117.9, 112.1, 108.9 (18C, Ar-C), 96.8 (1C, pyrazole), 38.8 (1C, CH2), 32.6 (1C, CH3-C), 30.6 (3C, C-CH3); MS (ESI): m/z 581.14 [M + Na]+. Mp 203.6–205.7 °C.
1-(2-((1H-Indazol-5-yl)oxy)benzyl)-3-(3-(tert-butyl)-1-(4-fluorophenyl)-1H-pyrazol-5-yl)urea (25g). White solid (3.23 g, 65%). HPLC purity 96.6% (tR = 12.13 min). IR (KBr): 3258, 1670, 1597, 1577 cm−1; 1H-NMR (DMSO-d6) δ: 13.12 (s, 1H), 8.31 (s, 1H), 8.00 (s, 1H), 7.58 (d, 1H, J = 8.8 Hz), 7.51–7.54 (m, 2H), 7.25–7.28 (m, 5H), 7.10–7.13 (m, 2H), 6.93–6.95 (t, 1H, J = 5.6 Hz), 6.74 (d, 1H, J = 8.0 Hz), 6.25 (s, 1H), 4.31 (d, 2H, J = 5.6 Hz), 1.25 (s, 9H); 13C-NMR (DMSO-d6), δ: 160.7 (1C, pyrazole), 155.2 (1C, pyrazole), 154.4 (1C, CO), 150.2 (1C, pyrazole), 138.0, 135.1, 133.3, 129.9, 128.7, 128.3, 126.5, 126.4, 123.1, 122.9, 119.7, 117.3, 116.0, 115.8, 111.5, 108.0 (18C, Ar-C), 95.5 (1C, pyrazole), 38.2 (1C, CH2), 32.0 (1C, CH3-C), 30.1 (3C, C-CH3); MS (ESI): m/z 521.11 [M + Na]+. Mp 191.1–193.3 °C.
1-(2-((1H-Indazol-5-yl)oxy)benzyl)-3-(3-(tert-butyl)-1-(4-nitrophenyl)-1H-pyrazol-5-yl)urea (25h). White solid (4.20 g, 80%). HPLC purity 97.3% (tR = 11.86 min). IR (KBr): 3256, 1670, 1595, 1578 cm−1; 1H-NMR (DMSO-d6) δ: 13.12 (s, 1H), 8.62 (s, 1H), 8.31 (d, 2H, J = 8.8 Hz), 8.00 (s, 1H), 7.83 (d, 2H, J = 8.8 Hz), 7.57 (d, 1H, J = 8.8 Hz), 7.26–7.30 (m, 3H), 7.08–7.11 (m, 3H), 6.74 (d, 1H, J = 8.0 Hz), 6.32 (s, 1H), 4.31 (d, 2H, J = 5.6 Hz), 1.26 (s, 9H); 13C-NMR (DMSO-d6), δ: 161.6 (1C, pyrazole), 155.8 (1C, pyrazole), 155.0 (1C, CO), 150.8 (1C, pyrazole), 138.6, 138.4, 137.5, 133.9, 132.5, 130.4, 129.3, 128.8, 126.4, 123.7, 123.4, 120.3, 120.1, 117.8, 112.1, 109.9 (18C, Ar-C), 96.9 (1C, pyrazole), 38.8 (1C, CH2), 32.6 (1C, CH3-C), 30.6 (3C, C-CH3); MS (ESI): m/z 548.21 [M + Na]+. Mp 177.3–179.9 °C.
1-(2-((1H-Indazol-5-yl)oxy)-5-fluorobenzyl)-3-(3-(tert-butyl)-1-(4-nitrophenyl)-1H-pyrazol-5-yl)urea (25i). White solid (3.80 g, 70%). HPLC purity 98.7% (tR = 11.91 min). IR (KBr): 3260, 1670, 1599, 1578 cm−1; 1H-NMR (DMSO-d6) δ: 13.11 (s, 1H), 8.61 (s, 1H), 8.30 (d, 2H, J = 8.8 Hz), 8.01(s, 1H), 7.81 (d, 2H, J = 8.8 Hz), 7.51 (d, 1H, J = 8.8 Hz), 7.22–7.25 (m, 3H), 7.05–7.08 (m, 2H), 6.72 (d, 1H, J = 8.0 Hz), 6.31 (s, 1H), 4.30 (d, 2H, J = 5.6 Hz), 1.25 (s, 9H); 13C-NMR (DMSO-d6), δ: 162.7 (1C, pyrazole), 156.9 (1C, pyrazole), 156.1 (1C, CO), 152.0 (1C, pyrazole), 139.6, 139.3, 138.6, 135.0, 132.9, 131.6, 130.7, 130.4, 130.0, 127.2, 124.8, 124.5, 121.4, 119.0, 113.2, 110.0 (18C, Ar-C), 98.0 (1C, pyrazole), 39.9 (1C, CH2), 33.7 (1C, CH3-C), 31.5 (3C, C-CH3); MS (ESI): m/z 566.20 [M + Na]+. Mp 189.1–191.6 °C.
1-(2-((1H-Indazol-5-yl)oxy)-5-fluorobenzyl)-3-(3-(tert-butyl)-1-(p-tolyl)-1H-pyrazol-5-yl)urea (25j). White solid (3.84 g, 75%). HPLC purity 96.5% (tR = 11.84 min). IR (KBr): 3257, 1670, 1596, 1576 cm−1; 1H-NMR (DMSO-d6) δ: 13.11 (s, 1H) ,8.20 (s, 1H), 8.00 (s, 1H), 7.53 (d, 1H, J = 8.8 Hz), 7.33–7.37 (m, 1H), 7.26–7.30 (m, 3H), 7.04–7.07 (m, 4H), 6.93–6.95 (t, 1H, J = 5.6 Hz), 6.71 (d, 1H, J = 8.0 Hz), 6.20 (s, 1H), 4.30 (d, 2H, J = 5.6 Hz), 3.77(s, 3H), 1.24 (s, 9H). 13C-NMR (DMSO-d6), δ: 160.4 (1C, pyrazole), 159.2 (1C, pyrazole), 156.8 (1C, CO), 154.6 (1C, pyrazole), 150.8, 137.6, 136.9, 136.5, 136.3, 133.4, 133.0, 129.9, 129.5, 124.1, 123.1, 119.6, 119.3, 115.0, 111.7, 107.6, 111.1 (18C, Ar-C), 95.8 (1C, pyrazole), 38.0 (1C, CH2), 32.0 (1C, CH3-C), 30.2 (3C, C-CH3), 20.6 (1c, Ar-CH3); MS (ESI): m/z 535.23 [M + Na]+. Mp 188.7–190.3 °C.
4-(5-(3-(2-((1H-Indazol-5-yl)oxy)benzyl)ureido)-3-(tert-butyl)-1H-pyrazol-1-yl)benzenesulfonamide (25k). White solid (2.96 g, 53%). HPLC purity 97.3% (tR = 9.57 min). IR (KBr): 3257, 1670, 1595, 1575 cm−1; 1H-NMR (DMSO-d6) δ: 13.10 (s, 1H), 8.49 (s, 1H), 8.00 (s, 1H), 7.92 (d, 1H, J = 8.8 Hz), 7.71 (d, 2H, J = 8.8 Hz), 7.59 (d, 1H, J = 8.8 Hz), 7.47 (s, 2H), 7.29–7.31 (m, 3H), 7.13–7.16 (m, 2H), 6.99–7.02 (m, 1H), 6.77 (d, 1H, J = 6.4 Hz), 6.36 (s, 1H), 4.70 (s, 1H), 4.34 (d, 2H, J = 5.6 Hz), 1.27 (s, 9H); 13C-NMR (DMSO-d6) δ: 163.5 (1C, pyrazole), 157.7 (1C, pyrazole), 156.9 (1C, CO), 152.7 (1C, pyrazole), 140.5, 140.3, 139.4, 135.8, 134.4, 132.3, 131.2, 130.7, 128.3, 125.6, 125.3, 122.2, 122.0, 119.7, 114.0, 110.8 (18C, Ar-C), 96.3 (1C, pyrazole), 38.6 (1C, CH2), 34.8 (1C, CH3-C), 32.4 (1C, C-CH3); MS (ESI): m/z 582.20 [M + Na]+. Mp 194.7–196.8 °C.
3.2.12. General Method for the Synthesis of 26a–26f
A solution of the corresponding 3a–3i (1.1 equiv.) and 14 (1 equiv.) was stirred 2 h at 90 °C in the presence of Et3N (1 mL) in DMSO (50 mL). The mixture was poured into ice water, and the solution was extracted with EtOAc. The organic layer was washed with water and brine, dried (MgSO4), filtered, and evaporated to dryness. The product was separated by column chromatography using DCM/MeOH (150:1) as eluent.
1-(5-((1H-Indazol-5-yl)oxy)-2H-chromen-8-yl)-3-(3-(tert-butyl)-1-(p-tolyl)-1H-pyrazol-5-yl)urea (26a). From 3a (4.03 g, 10 mmol), and 14 (2.79 g, 10 mmol), after work-up and purification the title compound (3.73 g, 70%) was obtained as a white solid. HPLC purity 95.4% (tR = 10.32 min). IR (KBr): 3256, 1670, 1640, 1595, 1578 cm−1; 1H-NMR (DMSO-d6) δ: 13.07 (s, 1H), 8.92 (s, 1H), 8.51 (s, 1H), 7.97 (s, 1H), 7.87 (d, 1H, J = 8.8 Hz), 7.54 (d, 2H, J = 8.8 Hz), 7.37–7.40 (m, 4H), 7.17 (d, 1H, J = 2.4 Hz), 7.11 (dd, 1H, J = 2.4, 8.8 Hz), 6.61–6.64 (m, 1H), 6.38(s, 1H), 6.34(d, 1H, J = 8.8 Hz), 5.93–5.95 (m, 1H), 4.85 (d, 2H, J = 1.6 Hz), 2.36 (s, 3H), 1.24 (s, 9H); 13C-NMR (DMSO-d6) δ: 160.4 (1C, pyrazole), 159.8 (1C, pyrazole), 157.5 (1C, CO), 154.6 (1C, pyrazole), 153.1, 149.5, 149.2, 146.5, 137.5, 136.5, 136.3, 134.7, 129.5, 124.0, 123.9, 122.5, 121.3, 120.1, 115.7, 115.0, 114.7, 104.5, 95.8 (1C, pyrazole), 73.5 (1C, O-CH2), 32.0 (1C, CH3-C), 30.2 (3C, C-CH3), 20.6 (1C, Ar-CH3); MS (ESI): m/z 557.20 [M + Na]+. Mp 210.3–212.7 °C.
1-(5-((1H-Indazol-5-yl)oxy)-2H-chromen-8-yl)-3-(3-(tert-butyl)-1-phenyl-1H-pyrazol-5-yl)urea (26b). White solid (2.86 g, 55%). HPLC purity 95.9% (tR = 10.43 min). IR (KBr): 3259, 1670, 1644, 1599, 1579 cm−1; 1H-NMR (DMSO-d6) δ: 13.07 (s, 1H), 8.99 (s, 1H), 8.51 (s, 1H), 7.97 (s, 1H), 7.86 (d, 1H, J = 8.8 Hz), 7.53 (m, 5H), 7.41 (m, 1H), 7.16 (d, 1H, J = 2.4 Hz), 7.12 (dd, 1H, J = 2.4, 8.8 Hz), 6.61–6.64 (m, 1H), 6.39 (s, 1H), 6.36 (d, 1H, J = 8.8 Hz), 5.93–5.96 (m, 1H), 4.85 (d, 2H, J = 1.6 Hz), 1.24 (s, 9H); 13C-NMR (DMSO-d6) δ: 162.5 (1C, pyrazole), 161.2 (1C, pyrazole), 160.3 (1C, CO), 160.1 (1C, pyrazole), 157.9, 155.1, 153.5, 149.7, 149.3, 147.0, 138.2, 135.7, 134.4, 134.3, 126.8, 126.7, 123.1, 121.8, 116.5, 116.2, 115.4, 105.0, 96.6 (1C, pyrazole), 74.3 (1C, O-CH2), 32.4 (1C, CH3-C), 30.6 (3C, C-CH3); MS (ESI): m/z 543.22 [M + Na]+. Mp 209.9–212.1 °C.
4-(5-(3-(5-((1H-Indazol-5-yl)oxy)-2H-chromen-8-yl)ureido)-3-(tert-butyl)-1H-pyrazol-1-yl)benzenesulfonamide (26c). White solid (2.99 g, 50%). HPLC purity 96.6% (tR = 8.33 min). IR (KBr): 3253, 1670, 1641, 1595, 1575 cm−1; 1H-NMR (DMSO-d6) δ: 13.08 (s, 1H), 9.14 (s, 1H), 8.52 (s, 1H), 7.99–8.01 (m, 3H), 7.86–7.89 (d, 1H, J = 8.8 Hz), 7.73–7.75 (d, 2H, J = 8.8 Hz), 7.54–7.57 (m, 3H), 7.17 (s, 1H), 7.10 (d, 1H, J = 8.8 Hz), 6.62 (d, 1H, J = 10.0 Hz), 6.41–6.44 (m, 2H), 5.93–5.95 (m, 1H), 4.87 (d, 2H, J = 1.6 Hz), 1.24 (s, 9H); 13C-NMR (DMSO-d6) δ: 162.5 (1C, pyrazole), 161.2 (1C, pyrazole), 160.3 (1C, CO), 160.1 (1C, pyrazole), 158.0, 155.1, 153.6, 150.4, 149.6, 147.1, 138.2, 135.7, 134.5, 134.4, 126.8, 126.7, 124.6, 123.2, 121.9, 116.5, 116.2, 104.9, 96.7 (1C, pyrazole), 74.9 (1C, O-CH2), 32.5 (1C, CH3-C), 30.6 (3C, C-CH3); MS (ESI): m/z 622.20 [M + Na]+. Mp 213.4–215.2 °C.
1-(5-((1H-Indazol-5-yl)oxy)-2H-chromen-8-yl)-3-(3-(tert-butyl)-1-(4-methoxyphenyl)-1H-pyrazol-5-yl)urea (26d). White solid (2.47 g, 45%). HPLC purity 96.2% (tR = 10.04 min). IR (KBr): 3256, 1670, 1640, 1593, 1574 cm−1; 1H-NMR (DMSO-d6) δ: 13.06 (s, 1H), 8.22 (s, 1H), 7.96 (s, 1H), 7.86 (d, 2H, J = 8.8 Hz), 7.71 (s, 1H), 7.53 (d, 1H, J = 8.8 Hz), 7.33 (d, 2H, J = 8.0 Hz), 7.25 (d, 3H, J = 8.0 Hz), 6.58–6.61 (m, 1H), 6.38 (d, 1H, J = 8.8 Hz), 5.88–5.93 (m, 1H), 4.84 (d, 2H, J = 1.6 Hz), 2.41 (s, 3H), 1.24 (s, 9H). 13C-NMR (DMSO-d6) δ: 163.2 (1C, pyrazole), 161.9 (1C, pyrazole), 161.1 (1C, CO), 160.8 (1C, pyrazole), 158.7, 155.8, 154.2, 150.4, 150.0, 147.6, 138.9, 136.4, 135.1, 135.0, 127.5, 127.4, 123.8, 122.5, 117.2, 117.0, 116.1, 105.7, 97.4 (1C, pyrazole), 71.2 (1C, O-CH2), 51.2 (1C, O-CH3), 33.1 (1C, CH3-C), 31.3 (3C, C-CH3); MS (ESI): m/z 573.23 [M + Na]+. Mp 215.1–217.1 °C.
1-(5-((1H-Indazol-5-yl)oxy)-2H-chromen-8-yl)-3-(3-(tert-butyl)-1-(4-fluorophenyl)-1H-pyrazol-5-yl)urea (26e). White solid (2.69 g, 45%). HPLC purity 98.3% (tR = 10.65 min). IR (KBr): 3253, 1670, 1637, 1599, 1579 cm−1; 1H-NMR (DMSO-d6) δ: 13.07 (s, 1H), 8.95 (s, 1H), 8.46 (s, 1H), 7.97 (s, 1H), 7.86 (d, 1H, J = 8.8 Hz), 7.53–7.56 (m, 3H), 7.38–7.41 (m, 2H), 7.17 (d, 1H, J = 2.4 Hz), 7.11 (dd, 1H, J = 2.4, 8.8 Hz), 6.61–6.65 (m, 1H), 6.39 (s, 1H), 6.35 (d, 1H, J = 8.8 Hz), 5.92–5.95 (m, 1H), 4.85 (d, 2H, J = 1.6 Hz), 1.24 (s, 9H); 13C-NMR (DMSO-d6) δ: 162.8 (1C, pyrazole), 161.5 (1C, pyrazole), 160.6 (1C, CO), 160.4 (1C, pyrazole), 158.2, 155.4, 153.8, 150.0, 149.6, 147.2, 138.5, 136.0, 134.7, 134.6, 127.1, 127.0, 123.4, 122.1, 116.7, 116.5, 115.7, 105.3, 96.9 (1C, pyrazole), 74.6 (1C, O-CH2), 32.8 (3C, CH3-C), 32.3 (1C, C-CH3); MS (ESI): m/z 561.21 [M + Na]+. Mp 197.3–199.7 °C.
1-(5-((1H-Indazol-5-yl)oxy)-2H-chromen-8-yl)-3-(3-(tert-butyl)-1-(4-nitrophenyl)-1H-pyrazol-5-yl)urea (26f). Yellow solid (3.50 g, 62%). HPLC purity 97.6% (tR = 10.54 min). IR (KBr): 3254, 1670, 1633, 1601, 1581 cm−1; 1H-NMR (DMSO-d6) δ: 13.03 (s, 1H), 9.15 (s, 1H), 8.43 (s, 1H), 8.35 (dd, 1H, J = 2.0, 8.8 Hz), 7.93 (s, 1H), 7.81–7.84 (m, 3H), 7.50–7.53 (d, 1H, J = 8.8 Hz), 7.12 (d, 1H, J = 2.0 Hz), 7.06 (d, 1H, J = 2.0, 8.8 Hz), 6.58 (m, 1H), 6.40 (s, 1H), 6.34 (d, 1H, J = 8.8 Hz), 5.90–5.95 (m, 1H), 4.84 (d, 2H, J = 1.6 Hz), 1.24 (s, 9H); 13C-NMR (DMSO-d6) δ: 162.0 (1C, pyrazole), 154.5 (1C, pyrazole), 153.0 (1C, CO), 149.2 (1C, pyrazole), 148.0, 146.2, 137.7, 136.9, 135.1, 133.8, 126.2, 126.1, 123.8, 122.1, 121.7, 121.1, 120.0, 115.9, 115.7, 115.4, 115.1, 104.5, 96.1 (1C, pyrazole), 74.4 (1C, O-CH2), 31.3 (1C, CH3-C), 30.1 (3C, C-CH3); 588.21 [M + Na]+. Mp 207.1–209.6 °C.
3.2.13. General Method for the Synthesis of 27a–27e
A solution of the corresponding 3a–3i (1.1 equiv.) and 18a–18b (1 equiv.) was stirred 2 h at 90 °C in the presence of Et3N (1 mL) in DMSO (50 mL). Then the mixture was poured into ice water and the solution was extracted with EtOAc. The organic layer was washed with water and brine, dried (MgSO4), filtered and evaporated to dryness. The product was separated by column chromatography using DCM/MeOH (80:1) as eluent.
1-(3-(tert-Butyl)-1-(p-tolyl)-1H-pyrazol-5-yl)-3-(2-((5-hydroxy-5,6,7,8-tetrahydronaphthalen-2-yl)oxy)-benzyl)urea (27a). From 3a (4.03 g, 10 mmol), and 18a (2.69 g, 10 mmol), after work-up and purification, 27a (3.66 g, 70%) was obtained as a white solid. HPLC purity 96.6% (tR = 8.46 min). IR (KBr): 3252, 1670, 1595, 1575 cm−1; 1H-NMR (DMSO-d6) δ: 8.25 (s, 1H), 7.38–7.41 (m, 2H), 7.35–7.38 (m, 2H), 7.11–7.15 (m, 3H), 7.15–7.18 (m, 1H), 6.91–6.93 (t, 1H, J = 8.0 Hz), 6.86–6.89 (m, 1H), 6.74–6.77 (m, 1H), 6.61 (d, 1H, J = 8.8 Hz), 6.24 (s, 1H), 5.09–5.12 (m, 1H), 4.54 (s, 1H), 4.28 (d, 2H, J = 5.6 Hz), 2.65–2.68 (m, 2H), 2.37 (s, 3H), 1.87–1.91 (m, 2H), 1.63–1.67 (m, 2H), 1.29 (s, 9H). 13C-NMR (DMSO-d6) δ: 160.9 (1C, pyrazole), 156.0 (1C, CO), 154.9 (1C, pyrazole), 154.5, 139.0, 138.3, 137.0, 136.7, 135.9, 131.3, 130.7, 130.1, 129.3, 128.9, 124.7, 124.1, 119.2, 117.6, 116.0 (18C, Ar-C), 95.8 (1C, pyrazole), 66.4 (1C, HO-C), 38.6 (1C, N-CH2), 32.9 (1C, CH2), 32.5 (1C, C), 31.5, (1C, CH2), 30.7 (3C, CH3), 21.3 (1C, Ar–CH3), 19.4 (1C, CH2). MS (ESI): m/z 547.20 [M + Na]+. Mp 201.6–203.7 °C.
1-(3-(tert-Butyl)-1-(4-nitrophenyl)-1H-pyrazol-5-yl)-3-(2-((5-hydroxy-5,6,7,8-tetrahydronaphthalen-2-yl)-oxy)benzyl)urea (27b). White solid (2.78 g, 50%). HPLC purity 96.7% (tR = 8.39 min). IR (KBr): 3251, 1667, 1593, 1573 cm−1; 1H-NMR (DMSO-d6) δ: 8.59 (s, 1H), 8.33 (s, 2H), 7.83 (d, 2H, J = 8.8 Hz), 7.38 (d, 1H, J = 8.8 Hz), 7.25–7.28 (m, 2H), 7.09–7.11 (m, 1H), 6.96–6.98 (t, 1H, J = 8.0 Hz), 6.81–6.85 (m, 1H), 6.74–6.77 (m, 1H), 6.61 (d, 1H, J = 8.8 Hz), 6.33 (s, 1H), 5.08–5.12 (m, 1H), 4.53 (s, 1H), 4.24 (d, 2H, J = 5.6 Hz), 2.64–2.67 (m, 2H), 1.87–1.90 (m, 2H), 1.63–1.66 (m, 2H), 1.27 (s, 9H); 13C-NMR (DMSO-d6) δ: 162.9 (1C, pyrazole), 155.9 (1C, CO), 155.1 (1C, pyrazole), 154.4, 145.3, 144.8, 139.1, 139.0, 135.9, 131.2, 130.7, 129.2, 128.9, 125.3, 124.0, 123.6, 119.1, 117.6, 116.0 (18C, Ar-C), 99.1 (1C, pyrazole), 66.4 (1C, HO-C), 38.7 (1C, N-CH2), 32.8 (1C, CH2), 32.7 (1C, C), 30.5 (3C, CH3), 29.5 (1C, CH2), 19.4 (1C, CH2); MS (ESI): m/z 578.05 [M + Na]+. Mp 202.1–204.3 °C.
1-(3-(tert-Butyl)-1-phenyl-1H-pyrazol-5-yl)-3-(2-((5-hydroxy-5,6,7,8-tetrahydronaphthalen-2-yl)oxy)-benzyl)urea (27c). White solid (2.14 g, 42%). HPLC purity 98.8% (tR = 8.41 min). IR (KBr): 3250, 1667, 1593, 1573 cm−1; 1H-NMR (DMSO-d6) δ: 8.31 (s, 1H), 7.49–7.52 (m, 4H), 7.39–7.42 (m, 2H), 7.26–7.28 (m, 2H), 7.11–7.14 (m, 1H), 6.92–6.94 (t, 1H, J = 8.0 Hz), 6.83–6.86 (m, 1H), 6.72–6.75 (m, 1H), 6.61 (d, 1H), 6.26 (s, 1H), 5.08–6.12 (m, 1H), 4.53 (s, 1H), 4.24 (d, 2H, J = 5.6 Hz), 2.64–2.68 (m, 2H), 1.87–1.91 (m, 2H), 1.67–1.71 (m, 2H), 1.25–1.29 (s, 9H); 13C-NMR (DMSO-d6) δ: 160.7 (1C, pyrazole), 155.5 (1C, CO), 154.5 (1C, pyrazole), 153.9, 138.8, 138.5, 137.8, 135.4, 130.8, 130.2, 129.2, 128.8, 128.4, 127.1, 124.2, 123.7, 118.7, 117.1, 115.5 (18C, Ar-C), 95.7 (1C, pyrazole), 65.9 (1C, HO-C), 38.1 (1C, N-CH2), 32.4 (1C, CH2), 32.0 (1C, C), 30.2 (3C, CH3), 29.0 (1C, CH2), 18.9 (1C, CH2); MS (ESI): m/z 533.07 [M + Na]+. Mp 202.5–204.8 °C.
1-(3-(tert-Butyl)-1-(4-nitrophenyl)-1H-pyrazol-5-yl)-3-(5-fluoro-2-((5-hydroxy-5,6,7,8-tetrahydro-naphthalen-2-yl)oxy)benzyl)urea (27d). White solid (3.72 g, 65%). HPLC purity 97.4% (tR = 8.51 min). IR (KBr): 3251, 1668, 1593, 1573 cm−1; 1H-NMR (DMSO-d6) δ: 8.59 (s, 1H), 8.33–8.36 (m, 2H), 7.83–7.88 (m, 2H), 7.38–7.41 (m, 1H), 7.25–7.29 (m, 1H), 7.09–7.13 (m, 1H), 6.96–6.98 (t, 1H, J = 8.0 Hz), 6.81–6.85 (m, 1H), 6.72–6.76 (m, 1H), 6.61 (d, 1H, J = 8.8 Hz), 6.31 (s, 1H), 5.05–5.07 (m, 1H), 4.53 (s, 1H), 4.23 (d, 2H, J = 5.6 Hz), 2.64–2.68 (m, 2H), 1.86–1.89 (m, 2H), 1.63–1.66 (m, 2H), 1.25 (s, 9H). 13C-NMR (DMSO-d6) δ: 162.8 (1C, pyrazole), 160.0 (1C, CO), 157.7 (1C, pyrazole), 156.2, 155.3, 150.0, 145.3, 144.8, 139.0, 138.8, 135.8, 134.2, 130.7, 125.2, 123.5, 121.4, 116.9, 115.3, 115.0 (18C, Ar-C), 99.8 (1C, pyrazole), 66.4 (1C, HO-C), 38.7 (1C, N-CH2), 32.8 (1C, CH2), 32.7 (1C, C), 30.4 (3C, CH3), 29.5 (1C, CH2), 19.4 (1C, CH2); MS (ESI): m/z 596.04 [M + Na]+. Mp 191.4–193.9 °C.
1-(3-(tert-Butyl)-1-(p-tolyl)-1H-pyrazol-5-yl)-3-(5-fluoro-2-((5-hydroxy-5,6,7,8-tetrahydronaphthalen-2-yl)-oxy)benzyl)urea (27e). White solid (2.81 g, 52%). HPLC purity 97.3% (tR = 8.43 min). IR (KBr): 3251, 1667, 1597, 1576 cm−1; 1H-NMR (DMSO-d6) δ: 8.29 (s, 1H), 7.37–7.40 (m, 2H), 7.35–7.39 (m, 2H), 7.11–7.14 (m, 2H), 6.95–6.97 (t, 1H, J = 8.0 Hz), 6.86–6.91 (m, 1H), 6.98–7.01 (m, 1H), 6.70–6.77 (m, 1H), 6.58 (d, 1H, J = 8.8 Hz), 6.23 (s, 1H), 5.07–5.09 (m, 2H), 4.52 (s, 1H), 4.18 (d, 2H, J = 5.6 Hz), 2.61–2.65 (m, 2H), 2.35 (s, 3H), 1.84–1.87 (m, 2H), 1.63–1.66 (m, 2H), 1.21 (s, 9H). 13C-NMR (DMSO-d6) δ: 160.9 (1C, pyrazole), 160.1 (1C, CO), 157.7 (1C, pyrazole), 156.3, 155.0, 150.0, 139.0, 138.0, 136.8, 135.8, 130.7, 130.1, 124.6, 121.5, 121.4, 117.0, 115.5, 115.4, 115.3 (18C, Ar-C), 96.2 (1C, pyrazole), 66.4 (1C, HO-C), 38.4 (1C, N-CH2), 32.9 (1C, CH2), 32.5 (1C, C), 30.7 (3C, CH3), 29.5 (1C, CH2), 21.1 (1C, Ar-CH3), 19.4 (1C, CH2).MS (ESI): m/z 565.09 [M + Na]+. Mp 200.1–203.8 °C.
3.2.14. General Method for the Synthesis of 28a–28i
A solution of the corresponding 3a–3i (1.1 equiv.) and 21 (1 equiv.) was stirred 2 h at 90 °C in the presence of Et3N (1 mL) in DMSO (50 mL). Then the mixture was poured into ice water and the solution was extracted with EtOAc. The organic layer was washed with water and brine, dried (MgSO4), filtered and evaporated to dryness. The product was separated by column chromatography using DCM/MeOH (100:1) as eluent.
1-(3-(tert-Butyl)-1-(p-tolyl)-1H-pyrazol-5-yl)-3-(2-(2-morpholinoethoxy)phenyl)urea (28a). From 3a (4.03 g, 10 mmol), and 21 (2.22 g, 10 mmol), 28a (3.43 g, 72%) was obtained as a white solid. HPLC purity 95.9% (tR = 8.02 min). IR (KBr): 3247, 1666, 1593, 1573cm−1; 1H-NMR (DMSO-d6) δ: 9.04 (s, 1H), 8.29 (d, 1H, J = 8.0 Hz), 7.38–7.41 (m, 4H), 7.02 (t, 1H), 6.93–6.96 (m, 2H), 6.34 (s, 1H), 4.13–4.15 (t, 2H, J = 5.6 Hz), 3.54–3.57 (m, 4H), 2.70–2.76 (m, 2H), 2.44–2.48 (m, 4H), 2.36 (s, 3H), 1.25 (s, 9H); MS (ESI): m/z 500.27 [M + Na]+. Mp 166.3–168.2 °C.
1-(3-(tert-Butyl)-1-phenyl-1H-pyrazol-5-yl)-3-(2-(2-morpholinoethoxy)phenyl)urea (28b). White solid (2.31 g, 50%). HPLC purity 98.9% (tR = 8.11 min). IR (KBr) 3244, 1663, 1598, 1577 cm−1; 1H-NMR (DMSO-d6) δ: 9.06 (s, 1H), 8.23 (d, 1H, J = 8.0 Hz), 7.35–7.39 (m, 4H), 7.00–7.04 (m, 2H), 6.91–6.95 (m, 2H), 6.30 (s, 1H), 4.13–4.15 (t, 2H, J = 5.6 Hz), 3.55–3.58 (m, 4H), 2.73–2.76 (m, 2H), 2.45–2.48 (m, 4H), 1.26 (s, 9H); MS (ESI): m/z 486.26 [M + Na]+. Mp 179.2–182.3 °C.
1-(3-(tert-Butyl)-1-(m-tolyl)-1H-pyrazol-5-yl)-3-(2-(2-morpholinoethoxy)phenyl)urea (28c). White solid (3.00 g, 63%). HPLC purity 97.7% (tR = 8.05 min). IR (KBr): 3245, 1664, 1591, 1571 cm−1; 1H-NMR (DMSO-d6) δ: 9.05 (s, 1H), 8.23 (d, 1H, J = 8.0 Hz), 7.36–7.40 (m, 4H), 7.03–7.07 (m, 1H), 6.91–6.94 (m, 2H), 6.36 (s, 1H), 4.13–4.15 (t, 2H, J = 5.6 Hz), 3.55–3.59 (m, 4H), 2.72–2.76 (m, 2H), 2.42–2.46 (m, 4H), 2.33 (s, 3H), 1.26 (s, 9H); MS (ESI): m/z 500.27 [M + Na]+. Mp 191.6–193.7 °C.
1-(3-(tert-Butyl)-1-(4-methoxyphenyl)-1H-pyrazol-5-yl)-3-(2-(2-morpholinoethoxy)phenyl)urea (28d). White solid (2.71 g, 55%). HPLC purity 96.6% (tR = 8.13 min). IR (KBr): 3246, 1659, 1594, 1575 cm−1; 1H-NMR (DMSO-d6) δ: 9.06 (s, 1H), 8.25 (d, 1H, J = 8.0 Hz), 7.34–7.37 (m, 4H), 7.05–7.08 (m, 1H), 6.93–6.97 (m, 2H), 6.35 (s, 1H), 4.13–4.15 (t, 2H, J = 5.6 Hz), 3.84 (3H, O–CH3), 3.57–3.61 (m, 4H), 2.72–2.76 (m, 2H), 2.42–2.48 (m, 4H), 1.25 (s, 9H); MS (ESI): m/z 516.24 [M + Na]+. Mp 154.2–156.4 °C.
1-(3-(tert-Butyl)-1-(4-chlorophenyl)-1H-pyrazol-5-yl)-3-(2-(2-morpholinoethoxy)phenyl)urea (28e). White solid (2.23 g, 45%). HPLC purity 99.1% (tR = 8.15 min). IR (KBr): 3243, 1657, 1597, 1577 cm−1; 1H-NMR (DMSO-d6) δ: 9.12 (s, 1H), 8.23 (d, 1H, J = 8.0), 7.33–7.36 (m, 4H), 7.07–7.11 (m, 1H), 6.94–6.99 (m, 2H), 6.37 (s, 1H), 4.13–4.15 (t, 2H, J = 5.6), 3.59–3.62 (m, 4H), 2.74–2.78 (m, 2H), 2.44–2.48 (m, 4H), 1.26 (s, 9H); MS (ESI): m/z 520.22 [M + Na]+. Mp 171.3–173.2 °C.
1-(1-(4-Bromophenyl)-3-(tert-butyl)-1H-pyrazol-5-yl)-3-(2-(2-morpholinoethoxy)phenyl)urea (28f). White solid (3.78 g, 70%). HPLC purity 96.4% (tR = 8.12 min). IR (KBr): 3243, 1657, 1597, 1577 cm−1; 1H-NMR (DMSO-d6) δ: 9.13 (s, 1H), 8.20 (d, 1H, J = 8.0 Hz), 7.30–7.33 (m, 4H), 7.05–7.09 (m, 1H), 6.95–7.00 (m, 2H), 6.35 (s, 1H), 4.13–4.15 (t, 2H, J = 5.6 Hz), 3.57–4.02 (m, 4H), 2.76–2.81 (m, 2H), 2.42–2.45 (m, 4H), 1.24 (s, 9H); MS (ESI): m/z 564.17 [M + Na]+. Mp 189.6–191.3 °C.
1-(3-(tert-Butyl)-1-(4-fluorophenyl)-1H-pyrazol-5-yl)-3-(2-(2-morpholinoethoxy)phenyl)urea (28g). White solid (2.84 g, yield 69%). HPLC purity 97.2% (tR = 8.13 min). IR (KBr): 3246, 1655, 1594, 1575 cm−1; 1H-NMR (DMSO-d6), δ: 9.13 (s, 1H), 8.23 (d, 1H, J = 8.0 Hz), 7.35–7.39 (m, 4H), 7.08–7.12 (m, 1H), 6.97–7.02 (m, 2H), 6.38 (s, 1H),4.16–4.18 (t, 2H, J = 5.6 Hz), 3.60–3.65 (m, 4H), 2.77–2.81 (m, 2H), 2.44–2.49 (m, 4H), 1.25 (s, 9H); MS (ESI): m/z 504.17 [M + Na]+. Mp 170.3–172.0 °C.
1-(3-(tert-Butyl)-1-(4-nitrophenyl)-1H-pyrazol-5-yl)-3-(2-(2-morpholinoethoxy)phenyl)urea (28h). White solid (2.54 g, 50%). HPLC purity 98.8% (tR =8.10 min). IR (KBr): 3244, 1656, 1600, 1578 cm−1; 1H-NMR (DMSO-d6), δ: 9.10 (s, 1H), 8.21 (d, 1H, J = 8.0 Hz), 7.36–7.42 (m, 4H), 7.09–7.12 (m, 1H), 6.98–7.02 (m, 2H), 6.39 (s, 1H), 4.17–4.19 (t, 2H, J = 5.6 Hz), 3.61–3.66 (m, 4H), 2.78–2.82 (m, 2H), 2.46–2.51 (m, 4H), 1.26 (s, 9H); MS (ESI): m/z 531.24 [M + Na]+. Mp 163.3–165.6 °C.
4-(3-(tert-Butyl)-5-(3-(2-(2-morpholinoethoxy)phenyl)ureido)-1H-pyrazol-1-yl)benzenesulfonamide (28i). White solid (2.81 g, 52%). HPLC purity 98.4% (tR = 7.53 min). IR (KBr): 3246, 1657, 1601, 1579 cm−1; 1H-NMR (DMSO-d6) δ: 9.09 (s, 1H), 8.23 (d, 1H, J = 8.0 Hz), 7.35–7.41 (m, 4H), 7.08–7.12 (m, 1H), 6.97–7.02 (m, 2H), 6.37 (s, 1H), 4.19–4.21 (t, 2H, J = 5.6 Hz), 3.63–3.67 (m, 4H), 2.75–2.77 (m, 2H), 2.43–2.48 (m, 4H), 1.25 (s, 9H); MS (ESI): m/z 565.23 [M + Na]+. Mp 172.2–174.1 °C.
3.2.15. General Method for the Synthesis of 29a–29i
A solution of the corresponding 3a–3i (1.1 equiv.) and 24 (1 equiv.) was stirred 2 h at 90 °C in the presence of Et3N (1 mL) in DMSO (50 mL). Then the mixture was poured into ice water and the solution was extracted with EtOAc. The organic layer was washed with water and brine, dried (MgSO4), filtered and evaporated to dryness. The product was separated by column chromatography using DCM/MeOH (100:1) as eluent.
1-(2-((1H-Indazol-5-yl)oxy)phenyl)-3-(3-(tert-butyl)-1-(p-tolyl)-1H-pyrazol-5-yl)urea (29a). From 3a (4.03 g, 10 mmol), and 24 (2.25 g, 10 mmol), after work-up and purification 29a (2.88 g, 60%) was obtained as a white solid. HPLC purity 96.6% (tR = 11.32 min). IR (KBr): 3247, 1656, 1600, 1578 cm−1; 1H-NMR (DMSO-d6) δ: 13.16 (s, 1H), 9.07 (s, 1H), 8.87 (s, 1H), 8.20 (d, 1H, J = 8.8 Hz), 8.04 (s, 1H), 7.53–7.59 (m, 3H), 7.40–7.44 (m, 3H), 7.37–7.39 (m, 1H), 7.35–7.37 (m, 1H), 7.30–7.34 (m, 1H), 7.13–7.16 (m, 1H), 6.39 (s, 1H), 2.36 (s, 3H), 1.25 (s, 9H); MS (ESI): m/z 503.13 [M + Na]+. Mp 201.1–203.4 °C.
1-(2-((1H-Indazol-5-yl)oxy)phenyl)-3-(3-(tert-butyl)-1-phenyl-1H-pyrazol-5-yl)urea (29b). White solid (1.95 g, 42%). HPLC purity 98.1% (tR = 11.41 min). IR (KBr): 3245, 1657, 1601, 1579 cm−1; 1H-NMR (DMSO-d6) δ: 13.07 (s, 1H), 8.32 (s, 1H), 7.97 (s, 1H), 7.59 (d, 1H, J = 8.8 Hz), 7.49–7.55 (m, 4H), 7.37–7.42 (m, 1H), 7.27–7.32 (m, 3H), 7.12–7.15 (m, 2H), 6.95–6.97 (t, 1H, J = 5.6 Hz), 6.76 (d, 2H, J = 8.0 Hz), 1.25 (s, 9H). MS (ESI): m/z 489.13 [M + Na]+. Mp 188.2–190.3 °C.
1-(2-((1H-Indazol-5-yl)oxy)phenyl)-3-(3-(tert-butyl)-1-(m-tolyl)-1H-pyrazol-5-yl)urea (29c). White solid (2.64 g, 55%). HPLC purity 98.8% (tR =11.37 min). IR (KBr): 3246, 1652, 1594, 1574 cm−1; 1H-NMR (DMSO-d6) δ: 13.08 (s, 1H), 8.31 (s, 1H), 7.97 (s, 1H), 7.54 (d, 1H, J = 8.8 Hz), 7.37–7.42 (m, 1H), 7.24–7.29 (m, 6H), 7.13–7.16 (m, 2H), 6.95–6.97 (t, 1H, J = 5.6 Hz), 6.73 (d, 1H, J = 8.0 Hz), 6.23 (s, 1H), 2.45 (s, 3H), 1.26 (s, 9H); MS (ESI): m/z 503.56 [M + Na]+. Mp 194.3–196.5 °C.
1-(2-((1H-Indazol-5-yl)oxy)phenyl)-3-(3-(tert-butyl)-1-(4-methoxyphenyl)-1H-pyrazol-5-yl)urea (29d). White solid (2.13 g, 43%). HPLC purity 97.3% (tR = 11.11 min). IR (KBr): 3243, 1650, 1591, 1574 cm−1; 1H-NMR (DMSO-d6) δ:13.07 (s, 1H), 8.235 (s, 1H), 7.97 (s, 1H), 7.59 (d, 1H, J = 8.8 Hz), 7.36–7.42 (m, 3H), 7.27–7.31 (m, 4H), 7.13–7.16 (m, 2H), 6.95–6.97 (t, 1H, J = 5.6 Hz), 6.76 (d, 1H, J = 8.0 Hz), 6.25 (s, 1H), 2.37(s, 3H), 1.27 (s, 9H); MS (ESI): m/z 519.23 [M + Na]+. Mp 197.1–199.6 °C.
1-(2-((1H-Indazol-5-yl)oxy)phenyl)-3-(3-(tert-butyl)-1-(4-chlorophenyl)-1H-pyrazol-5-yl)urea (29e). White solid (3.20 g, 63%). HPLC purity 95.9% (tR = 11.24 min). IR (KBr): 3252, 1671, 1600, 1580 cm−1; 1H-NMR (DMSO-d6) δ: 13.08 (s, 1H), 8.33 (s, 1H), 7.97 (s, 1H), 7.53 (d, 1H, J = 8.8 Hz), 7.47–7.50 (m, 4H), 7.26–7.31 (m, 3H), 7.12–7.16 (m, 2H), 6.92–6.94 (t, 1H, J = 5.6 Hz), 6.76 (d, 1H, J = 8.0 Hz), 6.23 (s, 1H), 1.26 (s, 9H); MS (ESI): m/z 523.17 [M + Na]+.
1-(2-((1H-Indazol-5-yl)oxy)phenyl)-3-(1-(4-bromophenyl)-3-(tert-butyl)-1H-pyrazol-5-yl)urea (29f). White solid (2.61 g, 48%). HPLC purity 97.7% (tR = 11.08 min). IR (KBr): 3250, 1667, 1602, 1579 cm−1; 1H-NMR (DMSO-d6) δ: 13.06 (s, 1H), 8.34 (s, 1H), 7.94 (s, 1H), 7.54 (d, 1H, J = 8.8 Hz), 7.46–7.52 (m, 4H), 7.24–7.27 (m, 3H), 7.13–7.16 (m, 2H), 6.93–6.95 (t, 1H, J = 5.6 Hz), 6.74 (d, 1H, J = 8.0 Hz), 6.24 (s, 1H), 1.25 (s, 9H); MS (ESI): m/z 567.17 [M + Na]+. Mp 200.8.6–202.3 °C.
1-(2-((1H-Indazol-5-yl)oxy)phenyl)-3-(3-(tert-butyl)-1-(4-fluorophenyl)-1H-pyrazol-5-yl)urea (29g). White solid (2.66 g, yield 55%). HPLC purity 96.6% (tR = 11.14 min). IR (KBr): 3250, 1675, 1595, 1581 cm−1; 1H-NMR (DMSO-d6) δ: 13.05 (s, 1H), 8.35 (s, 1H), 7.99 (s, 1H), 7.54 (d, 1H, J = 8.8 Hz), 7.48–7.52 (m, 4H), 7.27–7.32 (m, 3H), 7.13–7.16 (m, 2H), 6.95–6.97 (t, 1H, J = 5.6 Hz), 6.74 (d, 1H, J = 8.0 Hz), 6.25 (s, 1H), 1.25(s, 9H); MS (ESI): m/z 507.20 [M + Na]+. Mp 189.6–191.7 °C.
1-(2-((1H-Indazol-5-yl)oxy)phenyl)-3-(3-(tert-butyl)-1-(4-nitrophenyl)-1H-pyrazol-5-yl)urea (29h). White solid (3.42 g, 67%). HPLC purity 98.9% (tR = 11.40 min). IR (KBr): 3247, 1673, 1591, 1579 cm−1; 1H-NMR (DMSO-d6) δ: 13.09 (s, 1H), 8.60 (s, 1H), 8.33 (d, 2H, J = 8.8 Hz), 8.03 (s, 1H), 7.85 (d, 2H, J = 8.8 Hz), 7.56 (d, 1H, J = 8.8 Hz), 7.24–7.28 (m, 3H), 7.09–7.12 (m, 3H), 6.75 (d, 1H, J = 8.0 Hz), 6.33 (s, 1H), 1.24 (s, 9H); MS (ESI): m/z 534.09 [M + Na]+. Mp 193.2–195.5 °C.
4-(5-(3-(2-((1H-Indazol-5-yl)oxy)phenyl)ureido)-3-(tert-butyl)-1H-pyrazol-1-yl)benzenesulfonamide (29i). White solid (3.59 g, yield 66%). HPLC purity 97.3% (tR = 9.96 min). IR (KBr): 3255, 1668, 1593, 1577 cm−1; 1H-NMR (DMSO-d6) δ: 13.07 (s, 1H), 8.44 (s, 1H), 8.01 (s, 1H), 7.93 (d, 1H, J = 8.8 Hz), 7.73 (d, 2H, J = 8.8 Hz), 7.54 (d, 1H, J = 8.8 Hz), 7.48 (s, 2H), 7.28–7.31 (m, 3H), 7.15–7.18 (m, 2H), 6.96–7.01 (m, 1H), 6.75(d, 1H, J = 6.4 Hz), 6.34(s, 1H), 4.78 (s, 1H), 1.25(s, 9H); MS (ESI): m/z 568.18 [M + Na]+. Mp 199.2–201.4 °C.
3.2.16. General Method for the Synthesis of 30a–30d
A solution of the corresponding 3a–3i (1.1 equiv.) and 16 (1 equiv.) was stirred 2 h at 90 °C in the presence of Et3N (1 mL) and DMSO (50 mL). Then the mixture was poured into ice water and the solution was extracted with EtOAc. The organic layer was washed with water and brine, dried (MgSO4), filtered and evaporated to dryness. The product was separated by column chromatography using DCM/MeOH (80:1) as eluent.
1-(3-(tert-Butyl)-1-(4-chlorophenyl)-1H-pyrazol-5-yl)-3-(2-(2-morpholinoethoxy)benzyl)urea (30a) From 3e (4.23 g, 10 mmol), and 16 (2.36 g, 10 mmol), after work-up and purification 30a (3.83 g, 75%) was obtained as a white solid. HPLC purity 98.4% (tR = 8.27 min). IR (KBr) 3254, 1669, 1596, 1578 cm−1; 1H-NMR (DMSO-d6) δ: 8.32 (s, 1H), 7.53–7.56 (m, 4H), 7.21–7.24 (m, 1H), 7.10 (dd, 1H, J = 1.6, 8.0 Hz), 6.98 (d, 1H, J = 8.0 Hz), 6.89–6.93 (m, 1H), 6.73–6.75 (t, 1H, J = 5.6 Hz), 6.27 (s, 1H), 4.19 (d, 2H, J = 5.6), 4.09–4.11 (t, 2H, J = 5.6 Hz), 3.55–3.57 (t, 4H, J = 4.8 Hz), 2.70–2.72 (t, 2H, J = 5.6 Hz), 2.50–2.52 (t, 4H, J = 4.0 Hz), 1.25 (s, 9H); MS (ESI): m/z 534.24 [M + Na]+. Mp 163.6–165.7 °C.
1-(3-(tert-Butyl)-1-(p-tolyl)-1H-pyrazol-5-yl)-3-(2-(2-morpholinoethoxy)benzyl)urea (30b). White solid (2.94 g, 60%). HPLC purity 95.6% (tR = 8.16 min). IR (KBr): 3257, 1668, 1595, 1575 cm−1; 1H-NMR (DMSO-d6) δ: 8.17 (s, 1H), 7.35 (d, 2H, J = 8.8 Hz), 7.21–7.26 (m, 1H), 7.11 (d, 1H, J = 8.0 Hz), 7.03 (d, 2H, J = 8.8 Hz), 6.98 (d, 1H, J = 8.0 Hz), 6.89–6.91 (t, 1H, J = 7.6 Hz), 6.74–6.76 (t, 1H, J = 5.6 Hz), 6.24 (s, 1H), 4.20 (d, 2H, J = 5.6 Hz), 4.09–4.11 (t, 2H, J = 5.6 Hz), 3.80 (s, 3H), 3.55–3.57 (t, 4H, J = 4.8 Hz), 2.71–2.73 (t, 2H, J = 5.6 Hz), 2.50–2.52 (t, 4H, J = 5.6 Hz), 1.24 (s, 9H). MS (ESI): m/z 514.24 [M + Na]+. Mp 171.1–173.7 °C.
4-(3-(tert-Butyl)-5-(3-(2-(2-morpholinoethoxy)benzyl)ureido)-1H-pyrazol-1-yl)benzenesulfonamide (30c). White solid (3.05 g, 55%). HPLC purity 97.2% (tR = 7.49 min). IR (KBr) 3259, 1670, 1591, 1571 cm−1; 1H-NMR (DMSO-d6) δ: 8.46 (s, 1H), 7.90 (d, 2H, J = 8.0 Hz), 7.69 (d, 2H, J = 8.0 Hz), 7.48 (s, 2H), 7.22–7.25 (m, 1H), 7.12 (d, 1H, J = 6.4 Hz), 6.98 (d, 1H, J = 8.0 Hz), 6.91–6.93 (t, 1H, J = 8.0 Hz), 6.78–6.80 (t, 1H, J = 5.6 Hz), 4.20 (d, 2H, J = 5.6 Hz), 4.10 (t, 2H, J = 5.6 Hz), 3.55–3.57 (t, 4H, J = 4.8 Hz), 2.71–2.73(t, 2H, J = 5.6 Hz), 2.50–2.52 (t, 4H, J = 5.6 Hz), 1.26 (s, 9H); MS (ESI): m/z 579.25 [M + Na]+. Mp 159.5–161.9 °C.
1-(3-(tert-Butyl)-1-phenyl-1H-pyrazol-5-yl)-3-(2-(2-morpholinoethoxy)benzyl)urea (30d). White solid (2.48 g, 52%). HPLC purity 97.2% (tR = 8.11 min). IR (KBr): 3261, 1671, 1593, 1574 cm−1; 1H-NMR (DMSO-d6) δ: 8.29 (s, 1H), 7.48–7.52 (m, 4H), 7.40–7.44 (m, 1H), 7.21–7.25 (m, 1H), 6.98 (d, 1H, J = 8.0 Hz), 6.89–6.92 (t, 1H, J = 7.6 Hz), 6.76–6.79 (t, 1H, J = 5.6 Hz), 6.28 (s, 1H), 4.20–4.22 (d, 2H, J = 5.6 Hz), 4.09–4.11 (t, 2H, J = 5.6 Hz), 3.55–3.57 (t, 4H, J = 4.8 Hz), 2.70–2.72 (t, 2H, J = 5.6 Hz), 2.50–2.52 (t, 4H, J = 5.6 Hz), 1.25 (s, 9H); MS (ESI): m/z 510.27 [M + Na]+. Mp 160.0–162.2 °C.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}