
Molecular Encapsulation of Histamine H2-Receptor Antagonists by Cucurbit[7]Uril: An Experimental and Computational Study


Abstract
:1. Introduction
2. Results and Discussion
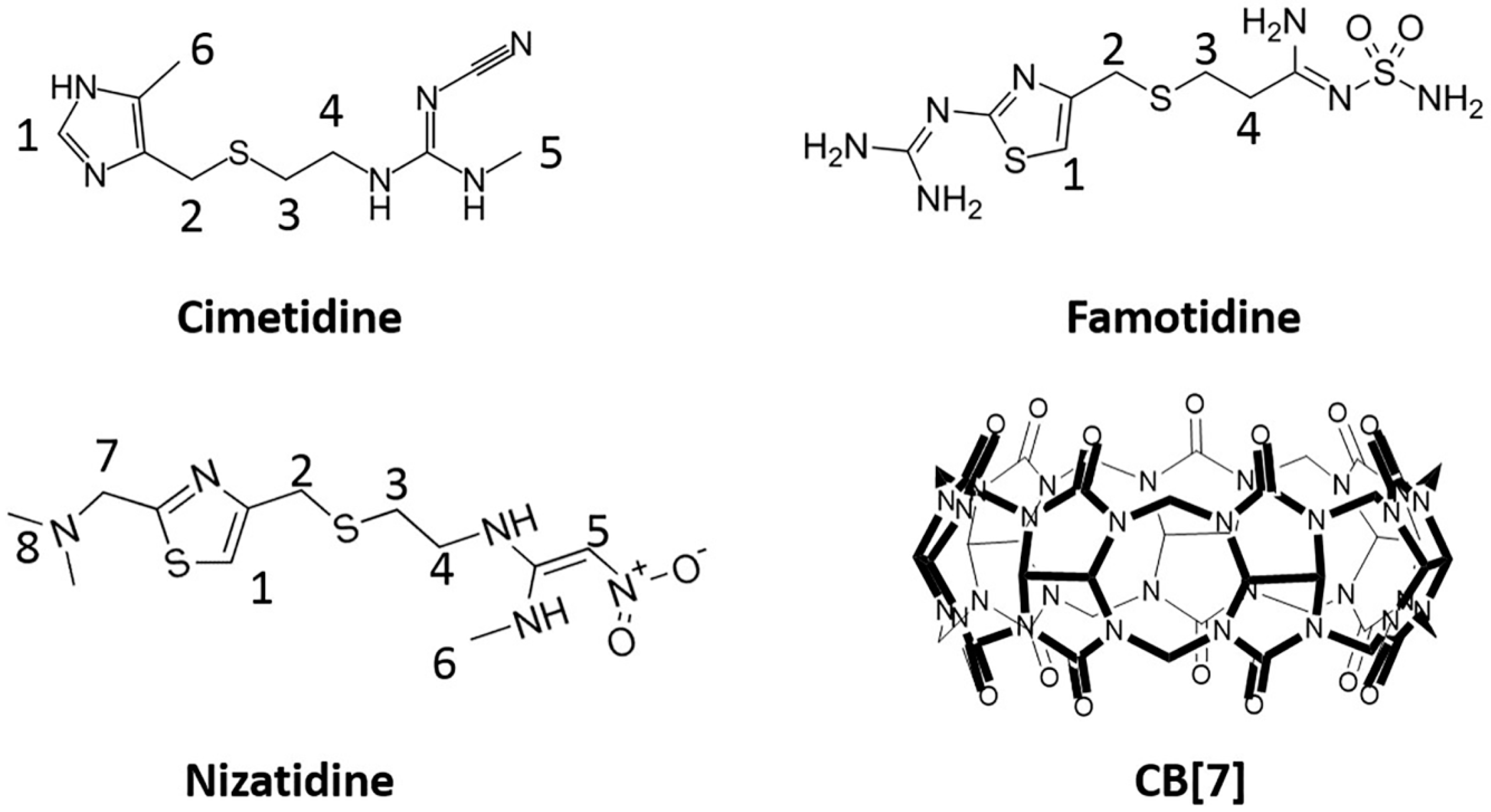
2.1. Experimental Study of Molecular Encapsulations of Histamine H2-Receptor Antagonists
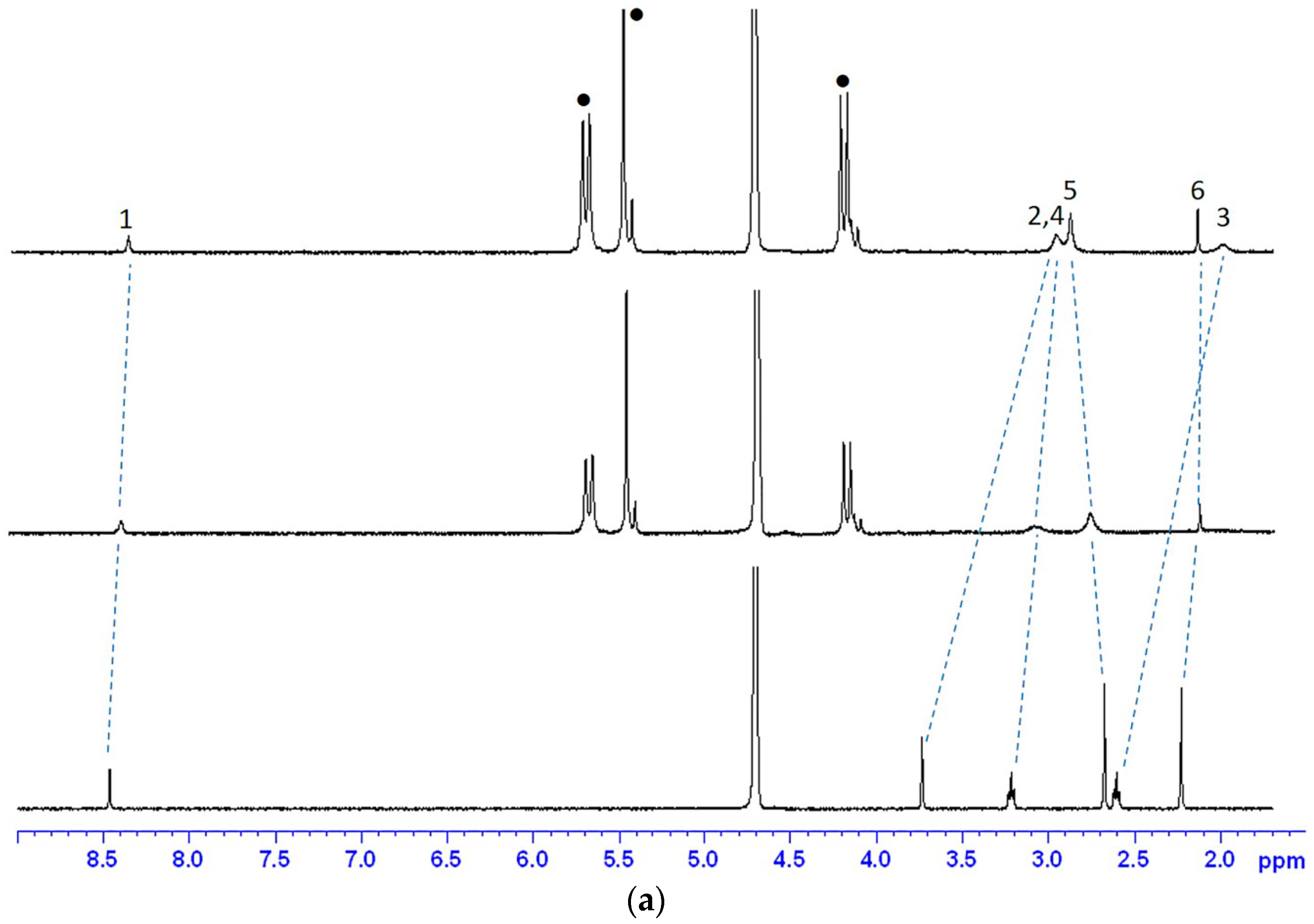
2.1.1. 1H-NMR Studies of the Encapsulation Sites
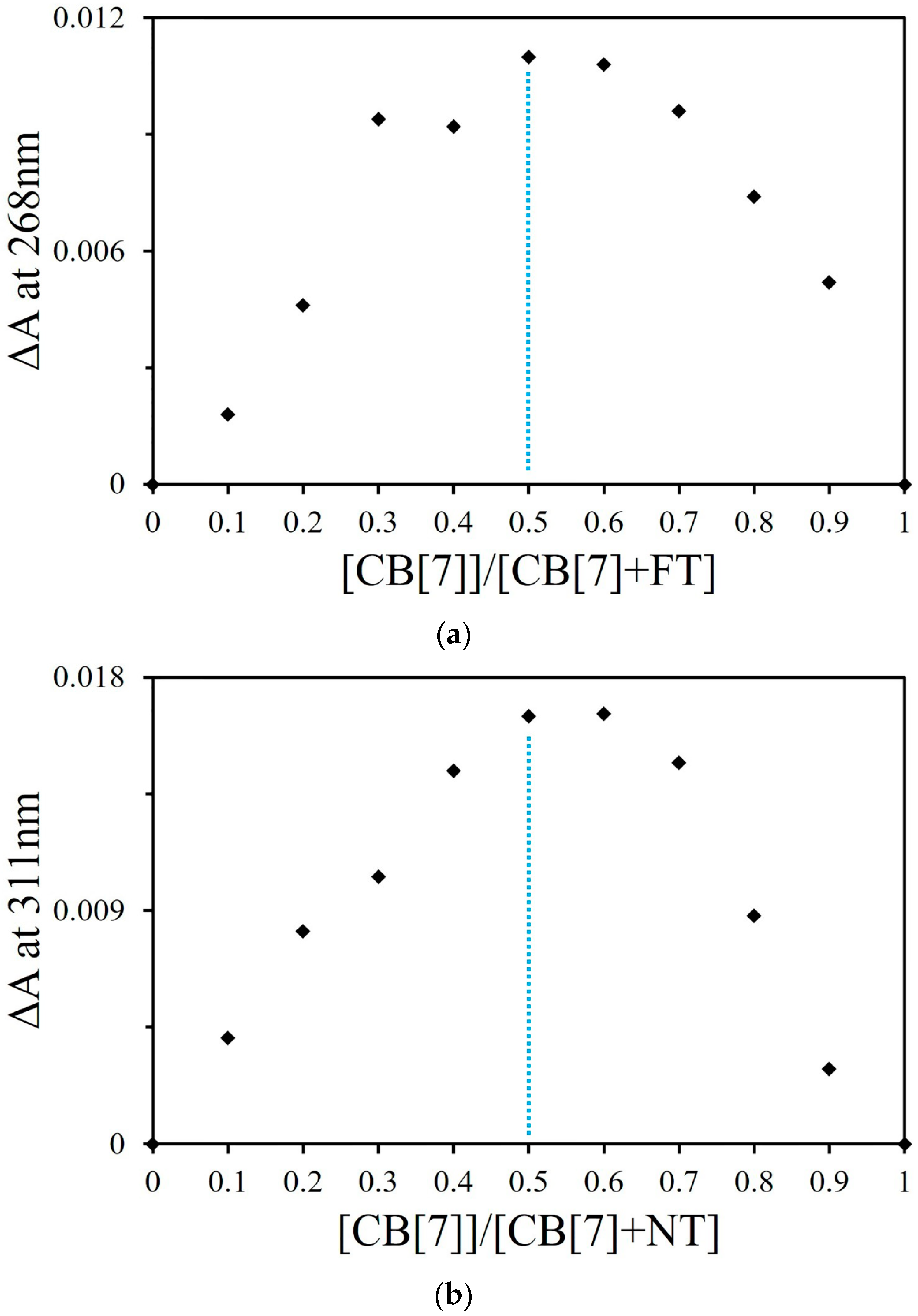
2.1.2. Job Plot and ESI-MS Studies
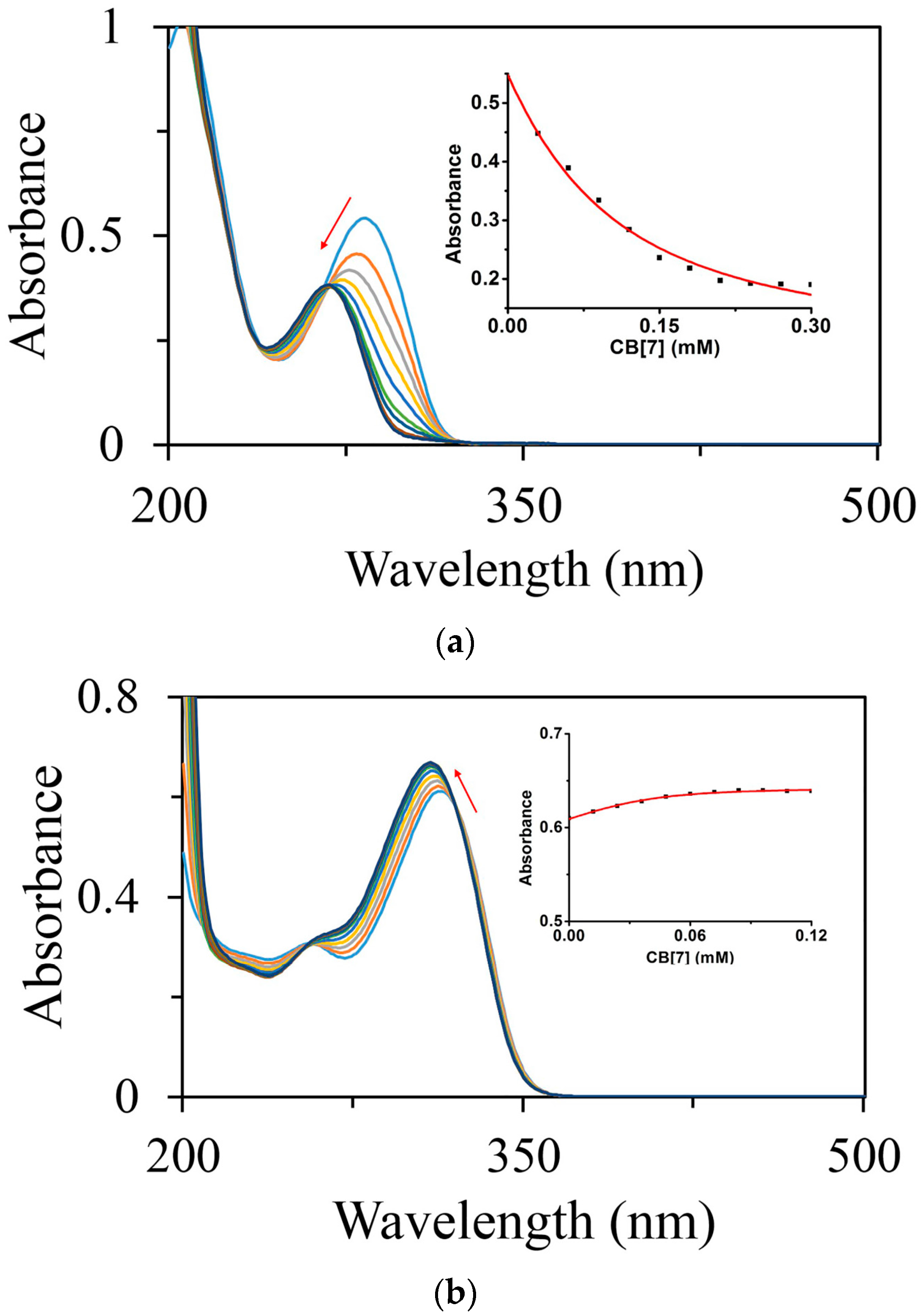
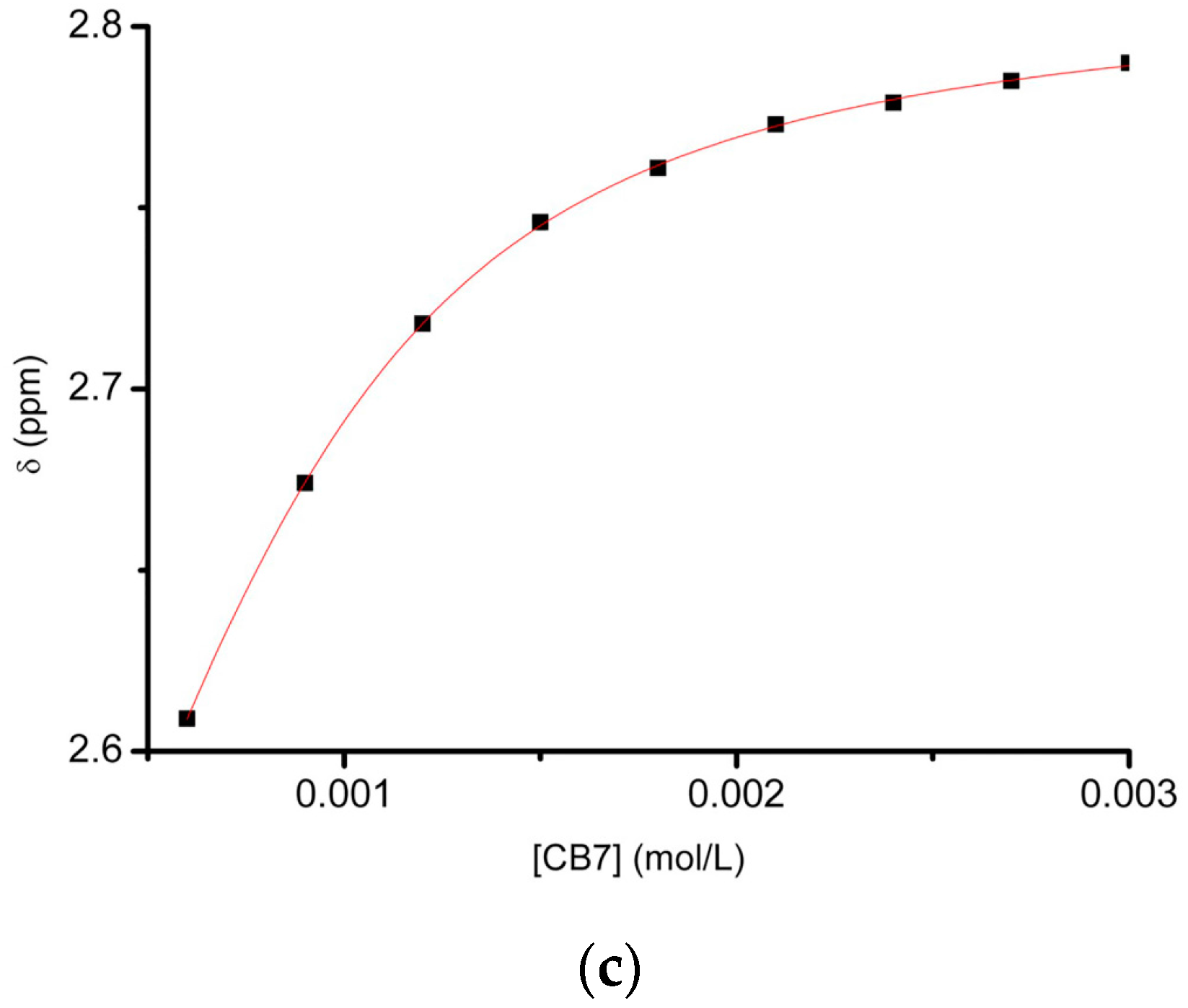
2.1.3. Binding Affinity Studies by UV-Visible and NMR Spectroscopic Titration
2.1.4. ITC Titration Studies
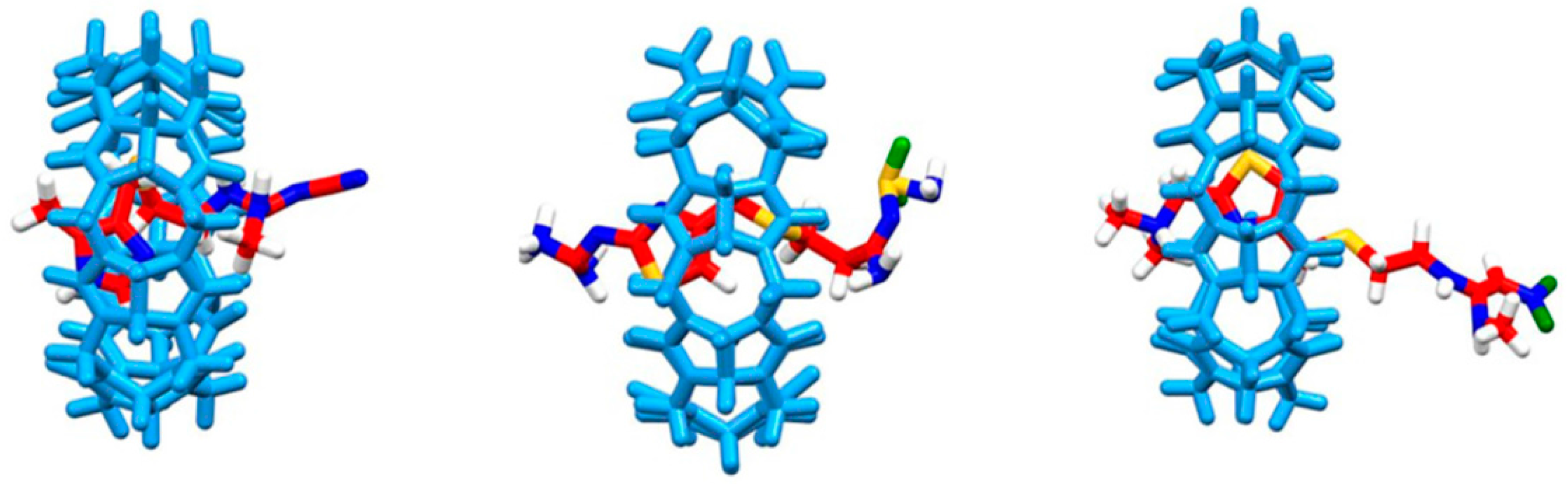
2.2. Computational Study of Host-Guest Complexes
3. Materials and Methods
3.1. Materials
3.2. Instrumentation
3.3. Complexes Preparation and Characterization
3.4. MD Computation
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gavvala, K.; Koninti, R.K.; Sengupta, A.; Hazra, P. Cucurbit[7]uril assisted ultraviolet to visible fluorescence switch of a heart medicine. Phys. Chem. Chem. Phys. 2014, 16, 2823–2826. [Google Scholar] [CrossRef] [PubMed]
- Tonga, G.Y.; Jeong, Y.; Duncan, B.; Mizuhara, T.; Mout, R.; Das, R.; Kim, S.T.; Yeh, Y.-C.; Yan, B.; Hou, S.; et al. Supramolecular regulation of bioorthogonal catalysis in cells using nanoparticle-embedded transition metal catalysts. Nat. Chem. 2015, 7, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Macartney, D.H. Encapsulation of drug molecules by cucurbiturils: Effects on their chemical properties in aqueous solution. Isr. J. Chem. 2011, 51, 600–615. [Google Scholar] [CrossRef]
- Ghosh, I.; Nau, W.M. The strategic use of supramolecular pKa shifts to enhance the bioavailability of drugs. Adv. Drug Deliv. Rev. 2012, 64, 764–783. [Google Scholar] [CrossRef] [PubMed]
- Uzunova, V.D.; Cullinane, C.; Brix, K.; Nau, W.M.; Day, A.I. Toxicity of cucurbit[7]uril and cucurbit[8]uril: An exploratory in vitro and in vivo study. Org. Biomol. Chem. 2010, 8, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- Oun, R.; Floriano, R.S.; Isaacs, L.; Rowan, E.G.; Wheate, N.J. The ex vivo neurotoxic, myotoxic and cardiotoxic activity of cucurbituril-based macrocyclic drug delivery vehicles. Toxicol. Res. 2014, 3, 447–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Chan, J.Y.W.; Yang, X.; Wyman, I.W.; Bardelang, D.; Macartney, D.H.; Lee, S.M.Y.; Wang, R. Developmental and organ-specific toxicity of cucurbit[7]uril: In vivo study on zebrafish models. RSC Adv. 2015, 5, 30067–30074. [Google Scholar] [CrossRef]
- Wheate, N.J.; Day, A.I.; Blanch, R.J.; Arnold, A.P.; Cullinane, C.; Grant Collins, J. Multi-nuclear platinum complexes encapsulated in cucurbit[n]uril as an approach to reduce toxicity in cancer treatment. Chem. Commun. 2004, 1424–1425. [Google Scholar] [CrossRef] [PubMed]
- Jin Jeon, Y.; Kim, S.-Y.; Ho Ko, Y.; Sakamoto, S.; Yamaguchi, K.; Kim, K. Novel molecular drug carrier: Encapsulation of oxaliplatin in cucurbit[7]uril and its effects on stability and reactivity of the drug. Org. Biomol. Chem. 2005, 3, 2122–2125. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Yuan, L.; Macartney, D.H. Inhibition of C(2)-H/D exchange of a bis(imidazolium) dication upon complexation with cucubit[7]uril. Chem. Commun. 2006, 2908–2910. [Google Scholar] [CrossRef]
- Li, S.; Miao, X.; Wyman, I.W.; Li, Y.; Zheng, Y.; Wang, Y.; Macartney, D.H.; Wang, R. High-affinity host-guest complex of cucurbit[7]uril with a bis(thiazolium) salt. RSC Adv. 2015, 5, 56110–56115. [Google Scholar] [CrossRef]
- Wang, R.; Wyman, I.W.; Wang, S.; Macartney, D.H. Encapsulation of a β-carboline in cucurbit[7]uril. J. Incl. Phenom. Macrocycl. Chem. 2009, 64, 233–237. [Google Scholar] [CrossRef]
- Wang, R.; Macartney, D.H. Cucurbit[7]uril host-guest complexes of the histamine H2-receptor antagonist ranitidine. Org. Biomol. Chem. 2008, 6, 1955–1960. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.; Li, Y.; Wyman, I.; Lee, S.M.Y.; Macartney, D.H.; Zheng, Y.; Wang, R. Enhanced in vitro and in vivo uptake of a hydrophobic model drug coumarin-6 in the presence of cucurbit[7]uril. MedChemComm 2015, 6, 1370–1374. [Google Scholar] [CrossRef]
- Wang, R.; MacGillivray, B.C.; Macartney, D.H. Stabilization of the base-off forms of vitamin B12 and coenzyme B12 by encapsulation of the α-axial 5,6-dimethylbenzimidazole ligand with cucurbit[7]uril. Dalton Trans. 2009, 3584–3589. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yin, H.; Wyman, I.W.; Zhang, Q.; Macartney, D.H.; Wang, R. Encapsulation of vitamin B1 and its phosphate derivatives by cucurbit[7]uril: Tunability of the binding site and affinity by the presence of phosphate groups. J. Org. Chem. 2016, 81, 1300–1303. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, S.; Wyman, I.W.; Macartney, D.H.; Zhang, Q.; Zheng, Y.; Wang, R. Supramolecular encapsulation of vitamin B6 by macrocyclic nanocontainer cucurbit[7]uril. J. Nanomater. 2015, 2015. [Google Scholar] [CrossRef]
- Li, S.; Chen, H.; Yang, X.; Bardelang, D.; Wyman, I.W.; Wan, J.; Lee, S.M.Y.; Wang, R. Supramolecular inhibition of neurodegeneration by a synthetic receptor. ACS Med. Chem. Lett. 2015, 6, 1174–1178. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, J.Y.W.; Li, S.; Liu, J.J.; Wyman, I.W.; Lee, S.M.Y.; Macartney, D.H.; Wang, R. In vivo reversal of general anesthesia by cucurbit[7]uril with zebrafish models. RSC Adv. 2015, 5, 63745–63752. [Google Scholar] [CrossRef]
- Li, S.; Yin, H.; Martinz, G.; Wyman, I.W.; Bardelang, D.; Macartney, D.H.; Wang, R. Supramolecular encapsulation of benzocaine and its metabolite para-aminobenzoic acid by cucurbit[7]uril. New J. Chem. 2016, 40, 3484–3490. [Google Scholar] [CrossRef]
- Yang, X.; Wang, Z.; Niu, Y.; Chen, X.; Lee, S.M.Y.; Wang, R. Influence of supramolecular encapsulation of camptothecin by cucurbit[7]uril: Reduced toxicity and preserved anti-cancer activity. MedChemComm 2016, 7, 1392–1397. [Google Scholar] [CrossRef]
- Li, S.; Chan, J.Y.-W.; Li, Y.; Bardelang, D.; Zheng, J.; Yew, W.W.; Chan, D.P.-C.; Lee, S.M.Y.; Wang, R. Complexation of clofazimine by macrocyclic cucurbit[7]uril reduced its cardiotoxicity without affecting the antimycobacterial efficacy. Org. Biomol. Chem. 2016, 14, 7563–7569. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.J.; Apps, M.G.; Wheate, N.J. Chemical factors affecting cucurbit[n]uril formulation into ocular dosage forms: Excipient binding, solubility, corneal permeability and antibiotic encapsulation. Supramol. Chem. 2014, 26, 648–656. [Google Scholar] [CrossRef]
- Saleh, N.i.; Al-Handawi, M.B.; Bufaroosha, M.S.; Assaf, K.I.; Nau, W.M. Tuning protonation states of tripelennamine antihistamines by cucurbit[7]uril. J. Phys. Org. Chem. 2016, 29, 101–106. [Google Scholar] [CrossRef]
- Barrow, S.J.; Kasera, S.; Rowland, M.J.; del Barrio, J.; Scherman, O.A. Cucurbituril-based molecular recognition. Chem. Rev. 2015, 115, 12320–12406. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The mm/pbsa and mm/gbsa methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Day, A.; Arnold, A.P.; Blanch, R.J.; Snushall, B. Controlling factors in the synthesis of cucurbituril and its homologues. J. Org. Chem. 2001, 66, 8094–8100. [Google Scholar] [CrossRef] [PubMed]
- Bardelang, D.; Udachin, K.A.; Leek, D.M.; Margeson, J.C.; Chan, G.; Ratcliffe, C.I.; Ripmeester, J.A. Cucurbit[n]urils (n = 5–8): A comprehensive solid state study. Cryst. Growth Des. 2011, 11, 5598–5614. [Google Scholar] [CrossRef]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; CheathamIII, T.E.; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. Amber 14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Ouyang, D.; Zhang, H.; Herten, D.-P.; Parekh, H.S.; Smith, S.C. Structure, dynamics, and energetics of sirna-cationic vector complexation: A molecular dynamics study. J. Phys. Chem. B 2010, 114, 9220–9230. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, D.; Zhang, H.; Parekh, H.S.; Smith, S.C. The effect of pH on pamam dendrimer-sirna complexation —Endosomal considerations as determined by molecular dynamics simulation. Biophys. Chem. 2011, 158, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhou, H.; Siu, S.W.I.; Gan, Y.; Wang, Y.; Ouyang, D. Comparison of three molecular simulation approaches for cyclodextrin-ibuprofen complexation. J. Nanomater. 2015, 2015. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complexes | Method | N | Ka (M−1) | ΔH (kCal/mol) | ΔG (kCal/mol) | TΔS (kCal/mol) |
|---|---|---|---|---|---|---|
| CT@CB[7] | ITC | 0.99 | 1.44 (± 0.38) × 104 | −13.23 ± 4.01 | −5.69 ± 0.16 | −7.57 ± 0.01 |
| NMR | 6.57 (± 0.19) × 103 | −5.21 ± 0.02 | ||||
| FT@CB[7] | ITC | 1.12 | 4.95 (± 0.36) × 104 | −11.08 ± 0.38 | −6.40 ± 0.04 | −4.68 ± 0.01 |
| UV-vis | 1.30 (± 0.27) × 104 | −5.62 ± 0.12 | ||||
| NT@CB[7] | ITC | 0.95 | 1.34 (± 0.21) × 104 | −15.60 ± 2.18 | −5.64 ± 0.09 | −9.96 ± 0.01 |
| UV-vis | 1.05 (± 0.33) × 105 | −6.85 ± 0.19 |
| kCal/mol | CT@CB[7] | FT@CB[7] | NT@CB[7] |
|---|---|---|---|
| ΔETOT | −37.55 ± 5.24 | −33.12 ± 3.93 | −32.47 ± 3.36 |
| TΔSTOT | −20.10 ± 1.09 | −20.86 ± 1.09 | −19.68 ± 1.63 |
| ΔG | −17.45 ± 4.92 | −12.25 ± 3.77 | −12.79 ± 2.97 |
| Parameter | CT-CB[7] | FT-CB[7] | NT-CB[7] |
|---|---|---|---|
| Water shell (Å) | 10 | 10 | 10 |
| Atom number of CB7 | 126 | 126 | 126 |
| Atom number of drug | 34 | 37 | 44 |
| Atom number of Cl- | 1 | 1 | 1 |
| Number of water | 1413 | 1736 | 1365 |
| Total atom number | 4400 | 5374 | 4265 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, H.; Wang, R.; Wan, J.; Zheng, Y.; Ouyang, D.; Wang, R. Molecular Encapsulation of Histamine H2-Receptor Antagonists by Cucurbit[7]Uril: An Experimental and Computational Study. Molecules 2016, 21, 1178. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21091178
Yin H, Wang R, Wan J, Zheng Y, Ouyang D, Wang R. Molecular Encapsulation of Histamine H2-Receptor Antagonists by Cucurbit[7]Uril: An Experimental and Computational Study. Molecules. 2016; 21(9):1178. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21091178
Chicago/Turabian StyleYin, Hang, Runmiao Wang, Jianbo Wan, Ying Zheng, Defang Ouyang, and Ruibing Wang. 2016. "Molecular Encapsulation of Histamine H2-Receptor Antagonists by Cucurbit[7]Uril: An Experimental and Computational Study" Molecules 21, no. 9: 1178. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21091178