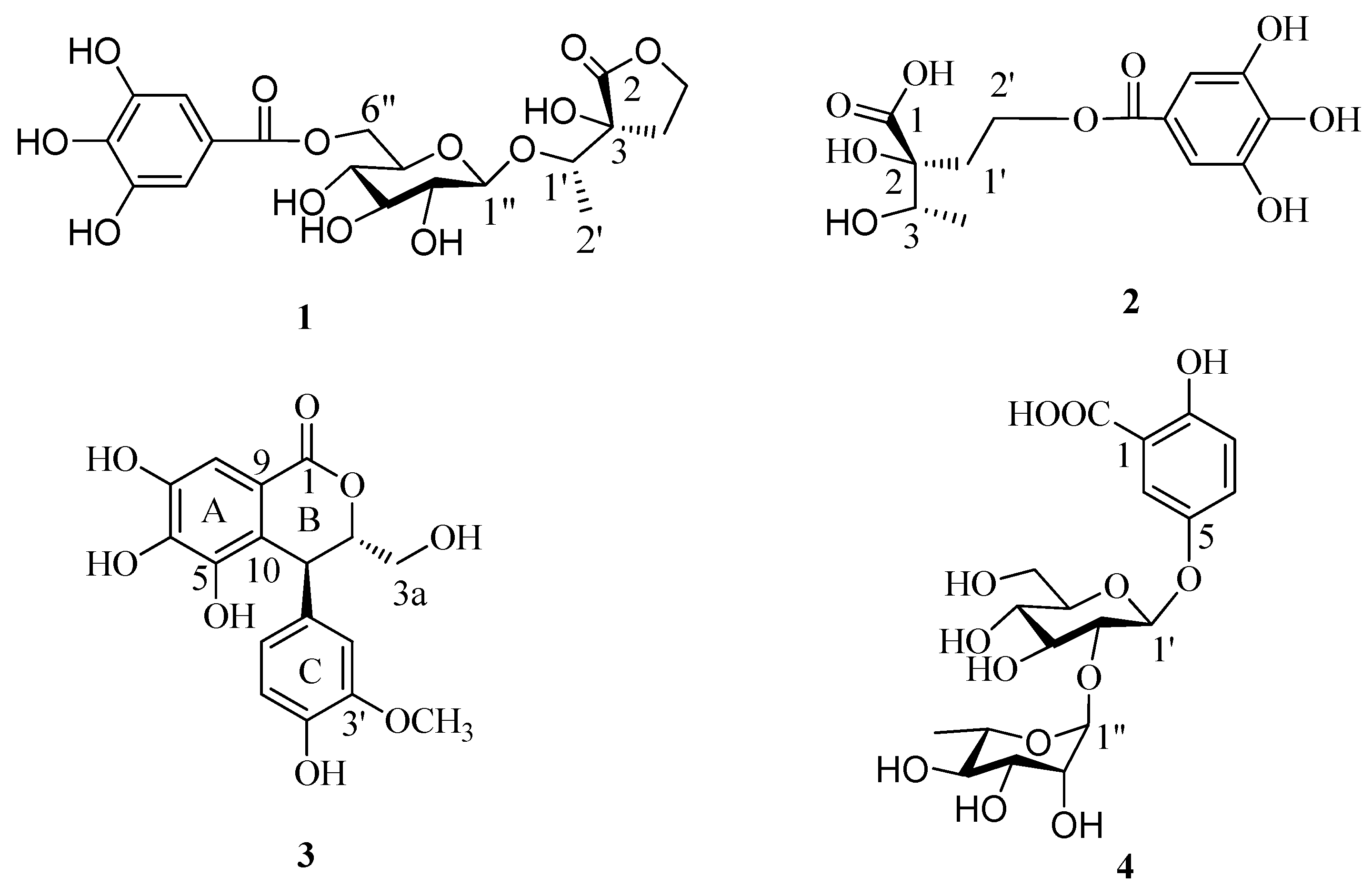
2. Results and Discussion
The fresh leaves of
C. fargesii were extracted with 80% aqueous ethanol, and the extract was partitioned between Et
2O and water. The Et
2O and aqueous fractions were separated by a combination of Sephadex LH-20, MCI gel CHP 20P, Toyopearl Butyl-650C, Chromatorex ODS, and Diaion HP20SS column chromatography and semi-preparative reverse-phase HPLC, to yield three new compounds
1–
3 and one known phenolic compound gentisic acid 5-
O-α-
l-rhamnopyranosyl-(1→2)-β-
d-glucoside [
9] (
4) (
Figure 1).
Compound
1 was isolated as a brown amorphous powder and gave a positive FeCl
3 test (dark blue), which suggested the presence of phenol moieties in the molecule. The molecular formula C
19H
24O
13 was determined based on the liquid chromatography-mass spectrometry IT-TOF (LC-MS/IT-TOF), which showed [M − H]
− and [M + Na]
+ ion peaks at
m/
z 459.1143 (calcd. for C
19H
23O
13, 459.1144) and 483.1118 (calcd. for C
19H
24O
13Na, 483.1109), respectively. In the
1H- and
13C-NMR spectra (
Table 1), two proton singlets at δ
H 7.13 (2H, s) and four aromatic carbon signals at δ
C 109.1 and 145.2, along with an ester carbonyl signal at δ
C 166.4 suggested the presence of a galloyl group [
10]. The hydrolysis of
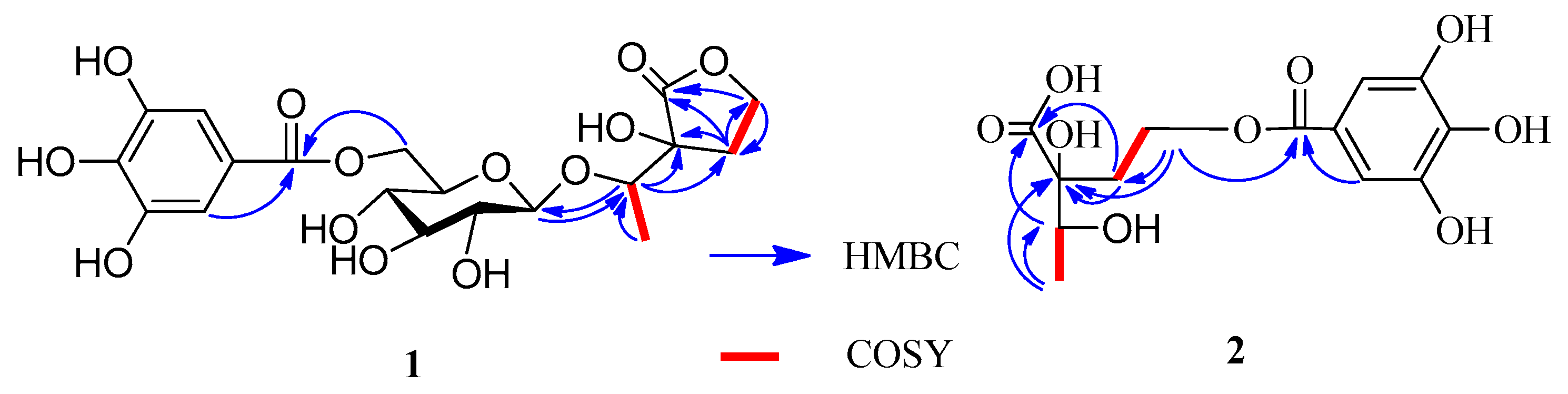
1 produced
d-glucose, which was identified by GC analysis. The coupling constant of the anomeric sugar proton was 7.8 Hz, indicating that the sugar moiety was in the β configuration. The large downfield shift of the glucose H-6″ protons (δ
H 4.32 and 4.58) suggested esterification with the galloyl moiety at this position. This was confirmed by the HMBC correlation between H-6″ and the carboxy carbon (δ
C 166.4) (
Figure 2). A second HMBC correlation between H-1″ and C-1′ suggested that the glycosyl group is linked to C-1′. HSQC experiment showed that the remaining moiety was composed of six carbons: a carboxy carbon (δ
C 178.6, C-2), an oxygenated methine (δ
C 77.8, C-1′), an oxygenated quaternary carbon (δ
C 76.6, C-3), an oxygenated methylene (δ
C 65.8, C-5), a methylene (δ
C 29.9, C-4), and a methyl (δ
C 15.6, C-2′) carbon. The presence of a γ-lactone structure in the remaining moiety was suggested by the lower field shift of the carboxy carbon (δ
C 178.6) and the unsaturation index (eight). This was also confirmed by HMBC correlations shown in
Figure 2, in which the oxygenated methylene protons (δ
H 4.31–4.34, H-5) correlated with C-2. Furthermore, the
1H-
1H COSY correlations of H-4 (δ
H 2.10) with H-5 (δ
H 4.32) and H-1′ with H-2′ and the HMBC correlations of H-1′ with C-3 and C-4 and H-4 with C-2, C-3, and C-5 indicated that the remaining moiety is 3-hydroxy-3-(1′-hydroxyethyl)dihydrofuran-2(3
H)-one. Consequently, the structure of compound
1 was established as 3-[1′-(6″-
O-galloyl-β-
d-glucopyranosyl)oxyethyl]-3-hydroxy-dihydrofuran-2(3
H)-one.
Compound
2 was isolated as a yellow amorphous powder and gave a positive FeCl
3 test (dark blue). The presence of a galloyl group was deduced from the
13C-NMR signals (
Table 1) [
6]. The molecular formula of C
13H
16O
9 was established based on the LC-MS/IT-TOF (
m/
z 339.0672 [M + Na]
+, calcd. for C
13H
16O
9Na, 339.0687) and
13C-NMR data. The
13C-NMR and DEPT spectra showed signals attributable to a methyl (δ
C 16.9), a methylene (δ
C 34.2), an oxymethine (δ
C 71.4), an oxymethylene (δ
C 60.2), an oxy quaternary (δ
C 77.7), and a carboxyl (δ
C 175.4) carbons. The NMR data of
2 (
Table 1) were similar to those of
1, except for the absence of the signals for one glucosyl moiety, which was supported by its MS data. A 2,3-dihydroxy-2-(2′-oxyethyl)-butanoic acid moiety could be constructed by
1H-
1H COSY correlations (
Figure 2) of H-4 with H-3 and H-1′a with H-2′a and the HMBC correlations of H-4 with C-3 (δ
C 71.4), H-3 with C-1, H-1′ with C-1, and H-2′ with C-2 (
Figure 2). The HMBC correlation of H-2′ with the carboxy carbon (δ
C 165.9) indicated that the galloyl group was attached to C-2′. Based on these results, the structure of
2 was determined to be 2-[2′-(galloyl)-oxyethyl]-2,3-dihydroxybutanoic acid.
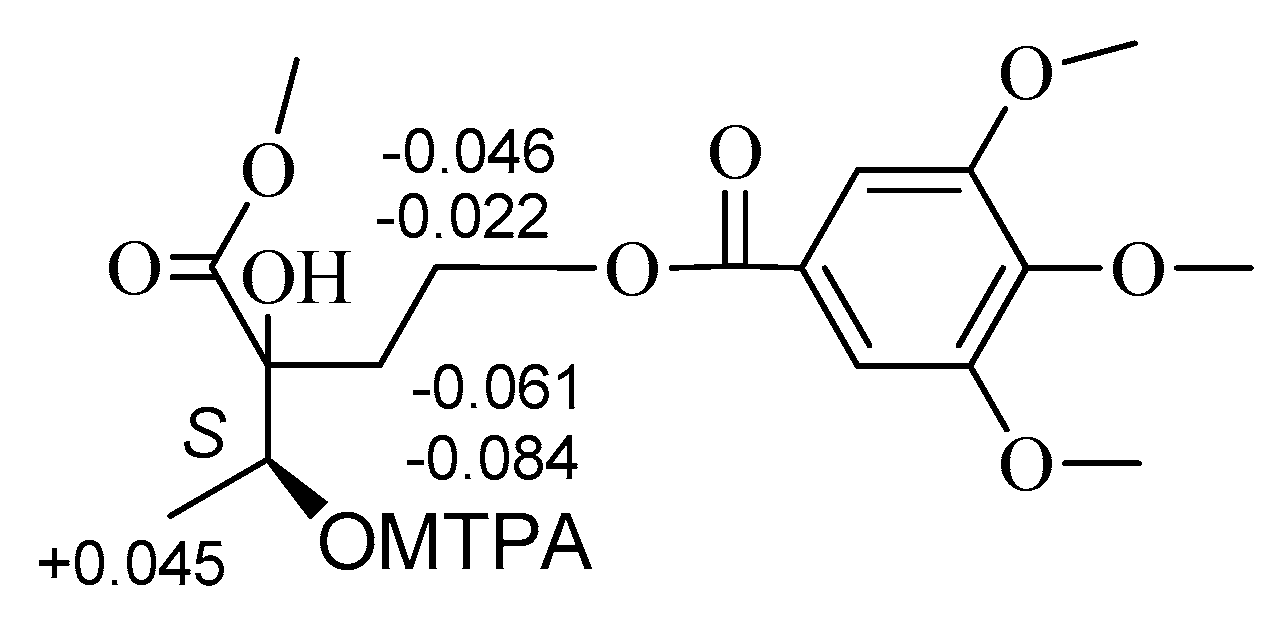
The absolute configurations at C-2 and C-3 of
2 were established using the modified Mosher’s method [
11,
12]. Treatment of
2 with CH
3I, then with (
R)-(−)- and (
S)-(+)-2-methoxy-2-trifluoromethyl-2-phenylacetyl (MTPA) chloride to get the C-3 (
S)- and (
R)-MTPA ester derivatives, respectively. Δδ values obtained from the
1H-NMR data of the C-3 (
R)- and (
S)-MTPA ester derivative indicated that the absolute configuration at C-3 of
2 was
S (
Figure 3). Compound
2 reacted with 2,2-dimethoxypropane (DMP) and pyridinium
p-toluene sulfonate (PPTS) to form the 2,3-
O-isopropylidene derivative. The C-2 and C-3 relative configuration of 2,3-
O-isopropylidene derivative was determined based on the NOE correlation of H-1′ with H
3-4 (
Figure 4). Thus, the absolute configurations at C-2 and C-3 of
2 were assigned as
R and
S, respectively.
Compounds 1 and 2 both contain a 2,3-dihydroxy-2-(2-hydroxyethyl)-butanoic acid moiety. The hydrolysis of 1 in 1 M HCl yielded 3-hydroxy-3-(1-hydroxyethyl)dihydrofuran-2(3H)-one that was identified to have the same absolute configuration as 2 by comparing their and CD data. Hence, the absolute configurations of 1 were assigned as 3R,1′S.
Compound
3 was obtained as a brown amorphous powder, which gave a dark blue color with FeCl
3. The molecular formula C
17H
16O
8 was deduced from the [M − H]
− peak at
m/
z 347.0768 in the LC-MS/IT-TOF (calcd. for C
17H
15O
8, 347.0772). Comparison of the
1H- and
13C-NMR data of
3 (
Table 2) and (3
S,4
S)-3-[(β-
d-glucopyranosyl)oxymethyl]-3,4-dihydro-5,6,7-trihydroxy-4-(4′-hydroxy-3′-methoxyphenyl)-1
H-[2]-benzopyran-1-one [
13] revealed that the methyl at C-3 in the known compound was replaced by a hydroxymethyl in
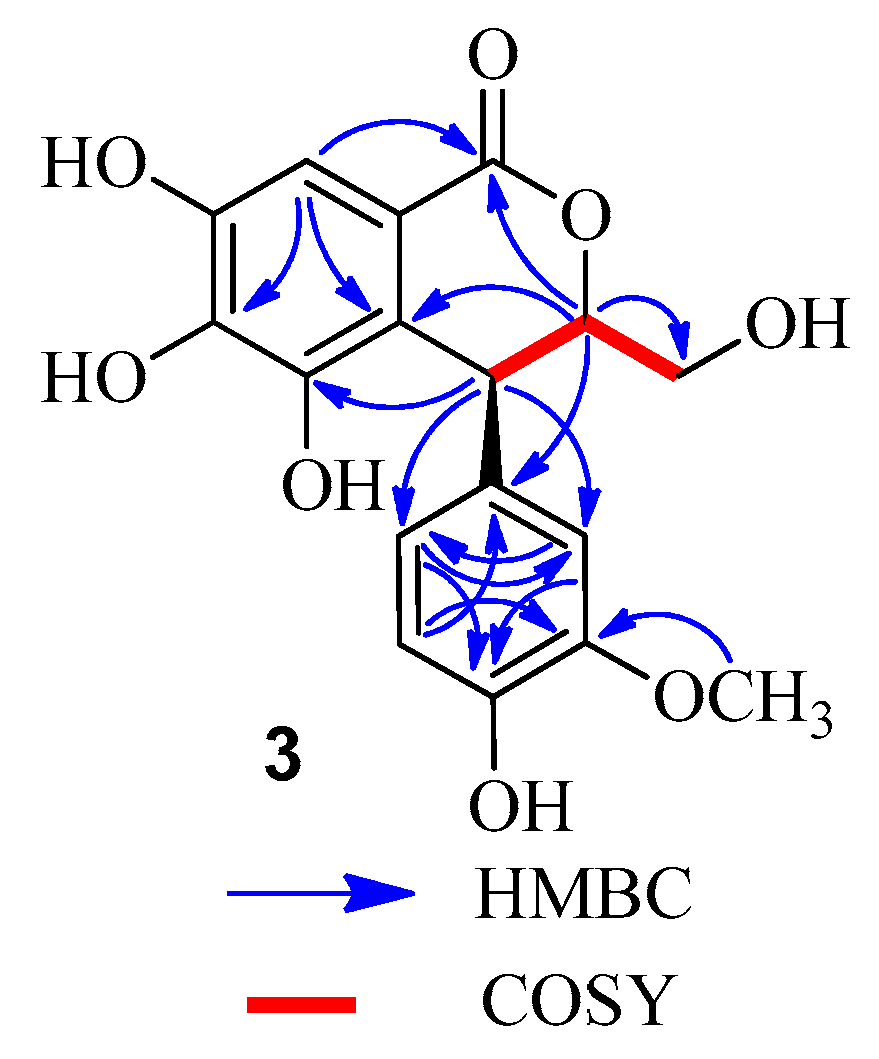
3. This was confirmed by the MS data and the correlations of the methine proton (δ
H 4.67) with the methylene carbon (δ
C 62.4) in the HMBC spectrum (
Figure 5), as well as
1H-
1H COSY correlations of H-3 (δ
H 4.67) with H-3a (δ
H 3.54) and H-4 (δ
H 4.50) (
Figure 5). Comparison of the CD and the optical rotation (
+18.3°) data of
3 with those of similar compounds suggested that the absolute configuration is 3
S,4
S [
14]. Based on the above evidences, the structure of compound
3 was concluded to be (3
S,4
S)-3-hydroxymethyl-3,4-dihydro-5,6,7-trihydroxy-4-(4′-hydroxy-3′-methoxyphenyl)-1
H-[2]-benzopyran-1-one.
All isolates were subjected to a cytotoxicity assay in vitro against human lung epithelial A549, human hepatocellular carcinoma SMMC-7721 cell, human gastric carcinoma MGC-803 cell, liver hepatocellular HepG2 cell, and human breast adenocarcinoma MCF-7 tumour cell. Unfortunately, none of the isolates showed inhibitions of those tumour cells at the highest concentration tested (IC50 value > 10 μM).
3. Experimental Section
3.1. Materials
The leaves of C. fargesii were collected at Guangxi Institute of Botany, Guangxi, China, in August 2014, and were identified by Prof. Shi-Hong Lu. A voucher specimen (20140627) was deposited in the Guangxi Key Laboratory of Functional Phytochemicals Research and Utilization, Guangxi Institute of Botany, China.
3.2. General Experimental Procedures
Optical rotations were measured with a 341 digital polarimeter (Perkin-Elmer Corp., Waltham, MA, USA). 1H- and 13C-NMR spectra were measured in acetone at 27 °C, using an Avance 500 spectrometer (500 MHz for 1H and 125 MHz for 13C, Bruker Biospin AG, Fällanden, Switzerland). Coupling constants and chemical shifts were given in Hz and on a δ (ppm) scale, respectively. GC was performed on a 6890N instrument equipped with a FID detector (Agilent Technologies, Santa Clara, CA, USA) operated at 280 °C (column: 28 m × 0.32 mm i.d. HP-5, column temp. 160 °C). LC-MS/IT-TOF was recorded on a LCMS-IT-TOF spectrometer (Shimadzu, Kyoto, Japan). Semi-preparative HPLC was performed on an Agilent 1200 apparatus equipped with a UV detector and a Zorbax SB-C-18 (9.4 × 250 mm) column (Agilent). Column chromatography (CC) was performed using Sephadex LH-20 (25–100 μm; GE Healthcare Bio-Science AB, Uppsala, Sweden), MCI gel CHP 20P (75–150 μm; Mitsubishi Chemical, Tokyo, Japan), Diaion HP20SS (Mitsubishi Chemical), Chromatorex ODS (100–200 mesh; Fuji Silysia Chemical, Aichi, Japan), and Toyopearl Butyl-650C (TOSOH, Tokyo, Japan) columns. TLC was performed on precoated Kieselgel 60 F254 plates (0.2 mm thick; Merck, Darmstadt, Germany) with toluene–HCO2Et–HCO2H (1:7:1, v/v) as the solvent, and spots were detected by spraying with a 2% ethanolic FeCl3.
3.3. Extraction and Separation
The fresh leaves of C. fargesii (5.20 kg) were cut into small pieces and extracted three times with EtOH/H2O (8:2, v/v, 36 L) by maceration at room temperature for 7 days. The extracts were combined and concentrated under reduced pressure to give an aqueous solution. The solution was partitioned with Et2O four times to give the Et2O fraction (32.4 g). The aqueous layer was subjected to Sephadex LH-20 CC (8 cm i.d. × 40 cm) with 0%–100% MeOH–H2O (20% stepwise elution, each 1.5 L) to give 9 fractions: frs 1 (15.6 g), 2 (84.5 g), 3 (26.6 g), 4 (27.0 g), 5 (130.0 g), 6 (12.9 g), 7 (3.3 g), 8 (2.3 g), and 9 (2.2 g). Fraction fr. 2 (84.5 g) was separated by MCI gel CHP 20PCC (6 cm i.d. × 40 cm) with MeOH–H2O (10% stepwise elution, each 1.0 L) to yield seven fractions, and fraction fr. 2-2 (5.3 g) was further fractionated by Diaion HP20SS CC (4 cm i.d. × 30 cm) with H2O containing increasing proportions of MeOH (0%–100%, 10% stepwise elution, each 0.5 L) to give 4 (105 mg). The Et2O fraction was subjected to MCI gel CHP 20PCC (5 cm i.d. × 50 cm) with 0%–100% MeOH in H2O (10% stepwise elution, each 0.5 L) to yield 10 fractions: frs E-1 (5.3 g), 2 (7.5 g), 3 (1.6 g), 4 (3.2 g), 5 (4.3 g), 6(1.5 g), 7 (2.3 g), 8 (1.0 g), 9 (9.9 g) and 10 (3.5 g). Fr. E-3 was fractionated by Toyopearl Butyl-650C CC (3 cm i.d. × 30 cm) with 0%–100% MeOH–H2O containing 0.1% CF3CO2H (TFA) (10% stepwise elution, each 0.3 L) to give fr. E-31 (1.3 g) and fr. E-32 (122 mg). Fr. E-32 was further purified by Chromatorex ODS CC (3 cm i.d. × 30 cm) with 0%–80% MeOH in H2O (5% stepwise elution, each 0.2 L) to give 3 (12 mg). Fraction E-4 was separated by Sephadex LH-20 CC (4 cm i.d. × 40 cm) with H2O containing increasing amounts of MeOH (0%–100%, 10% stepwise elution, each 0.5 L) to yield Fr. E-41 (250 mg), Fr. E-42 (150 mg), Fr. E-43 (296 mg) and Fr. E-44 (1.7 g). The Fr. E-42 and Fr. E-43 were further purified by semi-preparative HPLC (MeCN/H2O, 20:80, 2.5 mL/min) to give 1 (46 mg, tR 14.5 min) and 2 (68 mg, tR 13.2 min), respectively.
3.4. Spectroscopic Data
(3R,1'S)-[1'-(6"-O-Galloyl-β-d-glucopyranosyl)oxyethyl]-3-hydroxy-dihydrofuran-2(3H)-one (
1): Brown amorphous powder;
+52.1° (
c = 0.12, MeOH); UV (MeOH) λ
max nm (log ε): 272 (4.32); CD (MeOH) λ
max (Δε) 278 (8.4), 254 (4.5), 209 (2.7).
1H- and
13C-NMR data, see
Table 1; LC-MS/IT-TOF
m/
z [M − H]
− 459.1143 (calcd. for C
19H
23O
13, 459.1144) and [M + Na]
+ 483.1118 (calcd. for C
19H
24O
13Na, 483.1109).
(3R,1'S)-3-Hydroxy-1'-hydroxyethyl-dihydrofuran-2(3H)-one (Hydrochloride of 1): −17.0° (c = 0.15, MeOH); UV (MeOH) λmax nm (log ε): 265 (3.16); CD (MeOH) λmax (Δε) 267 (5.7), 251 (3.2), 211 (1.2). 1H-NMR (MeOH-d4, 500 MHz) δ 4.36 (1H, m, H-5a), 4.25 (1H, m, H-5b), 2.11 (1H, m, H-4a), 2.33 (1H, m, H-4b), 1.21 (3H, d, J = 6.5 Hz, H-4), 4.12 (1H, q, J = 6.5 Hz, H-1′), 1.21 (3H, d, J = 6.5 Hz, H-2′); LC-MS/IT-TOF m/z 169.0475 [M + Na]+ (calcd. for C6H10O4Na, 169.0477).
(2R,3S)-2-[2'-(Galloyl)oxyethyl]-dihydroxybutanoic acid (
2): Yellow amorphous powder;
−16.7° (
c = 0.12, MeOH); UV (MeOH) λ
max nm (log ε): 267 (4.26); CD (MeOH) λ
max (Δε) 268 (10.6), 252 (4.6), 212 (1.4).
1H- and
13C-NMR data, see
Table 1; LC-MS/IT-TOF
m/
z 339.0672 [M + Na]
+ (calcd. for C
13H
16O
9Na, 339.0687).
(3S,4S)-3-Hydroxymethyl-3,4-dihydro-5,6,7-trihydroxy-4-(4'-hydroxy-3'-methoxyphenyl)-1H-[2]-benzopyran-1-one (
3): Brown amorphous powder;
+18.3° (
c = 0.11, MeOH); UV (MeOH) λ
max nm (log ε): 220 (4.26), 278 (2.35); CD (MeOH) λ
max (Δε) 288 (11.4), 242 (6.5), 218 (2.7).
1H- and
13C-NMR data, see
Table 2; LC-MS/IT-TOF
m/
z 347.0768 [M − H]
− (calcd. for C
17H
15O
8, 347.0772).
3.5. Preparation of MTPA Esters Derivatives
CH3I (30 mg) and K2CO3 (15 mg) were added to a solution of 2 (10 mg) in DMF (5 mL). After stirring for 24 h at room temperature (r.t.), the reaction mixture was suspended in H2O and extracted with CHCl3. The CHCl3 layer was vacuum dried to afford a residue (6.2 mg). Then, DMAP (3.8 mg), Et3N (4.0 μL), and (R)-(−)-MTPACl (3.0 μL) were added to a solution of the residue (3.1 mg) in CH2Cl2 (1.0 mL) and stirred for 4 h at r.t. The reaction mixture was dried under a stream of N2. Separation of the residue was done by a silica gel column (hexane/EtOAc, 4:1) to afford the (S)-MTPA ester derivative (2.1 mg). The (R)-MTPA ester derivative (2.3 mg) was obtained according to the same procedure using (S)-(+) MTPACl.
(3S)-MTPA Ester derivative of 2: Colorless oil; 1H-NMR (MeOH-d4, 500 MHz) δ 7.0512–7.3901 (7H), 4.8212 (1H, q, J = 6.6 Hz, H-3), 1.3648 (3H, d, J = 6.6 Hz, H-4), 2.2511 (1H, m, H-1′a), 2.3004 (1H, m, H-1′b), 4.2526 (1H, m, H-2′a), 4.2812 (1H, m, H-2′b), 3.3206–3.8516 (-OCH3 × 5). LC-MS/IT-TOF m/z 611.17147 [M + Na]+ (calcd. for C27H31F3O11Na, 611.17162).
(3R)-MTPA Ester derivative of 2: Colorless oil; 1H-NMR (MeOH-d4, 500 MHz) δ 7.0510–7.3902 (7H), 4.8210 (1H, q, J = 6.6 Hz, H-3), 1.3603 (3H, d, J = 6.6 Hz, H-4), 2.2572 (1H, m, H-1′a), 2.3088 (1H, m, H-1′b), 4.2548 (1H, m, H-2′a), 4.2858 (1H, m, H-2′b), 3.3312–3.8716 (-OCH3 × 5). LC-MS/IT-TOF m/z 611.1710 [M + Na]+ (calcd. for C27H31F3O11Na, 611.1716).
3.6. Preparation of Acetonide Derivative of 2
Compound 2 (10.2 mg) was dissolved in acetone (1.0 mL) and treated with DMP (0.2 mL) and PPTS (6.5 mg) at r.t. After 4 h, Et3N (7.5 μL) was added and the mixture was concentrated by N2 blowing. The residue was separated on a silica gel column (CH2Cl2/EtOAc, 4:1–2:1) to afford the 2,3-O-isopropylidene derivative (3.2 mg) of 2.
2,3-O-Isopropylidene Derivative of 2: Colorless oil; 1H-NMR (MeOH-d4, 500 MHz) δ 6.98 (2H, s), 4.36 (1H, q, J = 6.5 Hz, H-3), 1.21 (3H, d, J = 6.5 Hz, H-4), 2.11 (1H, m, H-1′a), 2.23 (1H, m, H-1′b), 4.25 (1H, m, H-2′a), 4.29 (1H, m, H-2′b), 1.27 (3H, s, acetonide-CH3), 1.31 (3H, s, acetonide-CH3); LC-MS/IT-TOF m/z 379.1007 [M + Na]+ (calcd. for C18H20O9Na, 379.1005).
3.7. Acid Hydrolysis and Sugar Analysis by GC
Compound
1 (6 mg) was dissolved in MeOH (4.0 mL) and 1 M H
2SO
4 (2.0 mL) and refluxed for 2 h on a H
2O bath. After the hydrolysate was cool, H
2O (8.0 mL) was added, then extracted with EtOAc (3 × 10.0 mL). The EtOAc layer was vacuum dried and chromatographed on semi-preparative HPLC eluting with a gradient of MeOH–H
2O (5:95–25:75,
v/
v) to afford 3-hydroxy-1′-hydrooxyethyl-dihydrofuran-2(3
H)-one. The aqueous layer was neutralized with aqueous Ba(OH)
2 and evaporated under reduced pressure to give a residue. The residue was dissolved in pyridine (100 μL), subsequent treated with 0.1 mL cysteine methyl ester hydrochloride (150 μL; Sigma, St. Louis, MO, USA) and warmed at 60 °C for 1 h, then the trimethysilylation reagent HMDS/TMCS (hexamethyldisilazane/trimethylchlorosilane/pyridine 2:1:10; Acros Organics, Geel, Belgium) was added and warmed at 60 °C for 30 min. The reaction mixture was partitioned between water and hexane. The hexane extract was analyzed by GC [
15] (detector temperature: 280 °C; injector temperature: 250 °C; temperature gradient: start at 160 °C, hold for 5 min, increase to 280 °C at 5 °C/min, hold for 10 min). The authentic samples were analyzed in the same way. The
tR values of
d-glucose and
l-glucose were 13.25 min and 15.32 min, respectively. The thiazolidine derivatives of the samples were confirmed by comparison with authentic standards.
3.8. Cytotoxicity Assay
All isolates were tested for cytotoxicity in vitro against A549, SMMC-7721, MGC-803, HepG2, and MCF-7 tumour cells via the MTT assay [
16,
17] with hydroxycamptothecine as a positive control.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}