From α-Bromomethylbutenolide to Fused Tri(Tetra) Cyclic Dihydrofurandiones through Barbier Reaction–Heck Arylation Sequence


Abstract
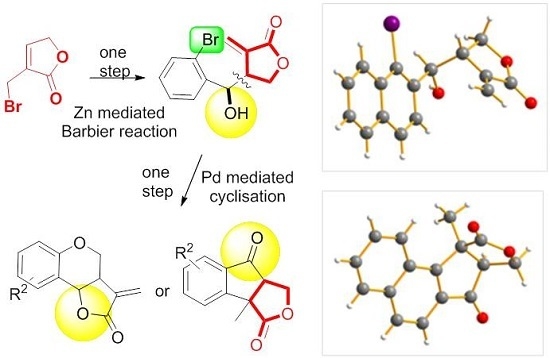
:
1. Introduction
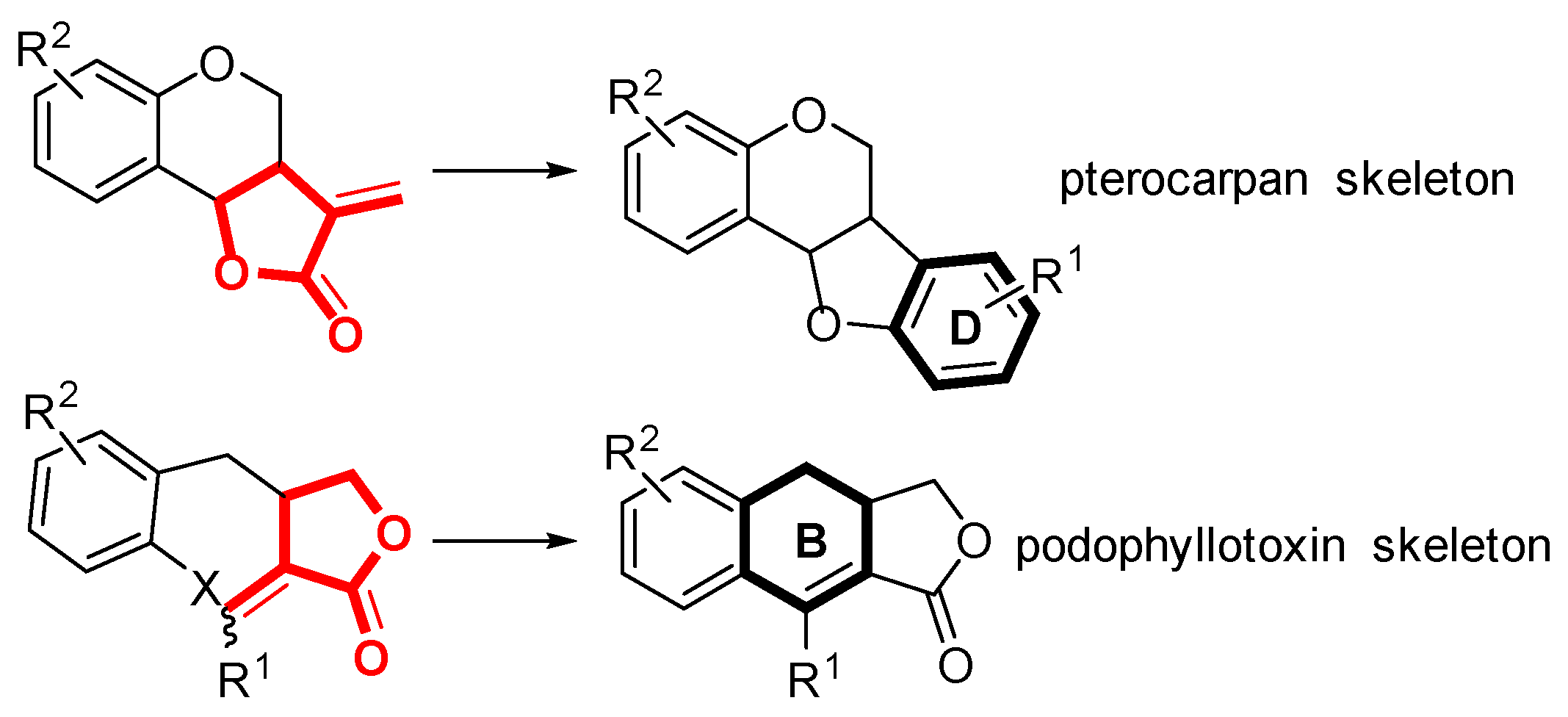
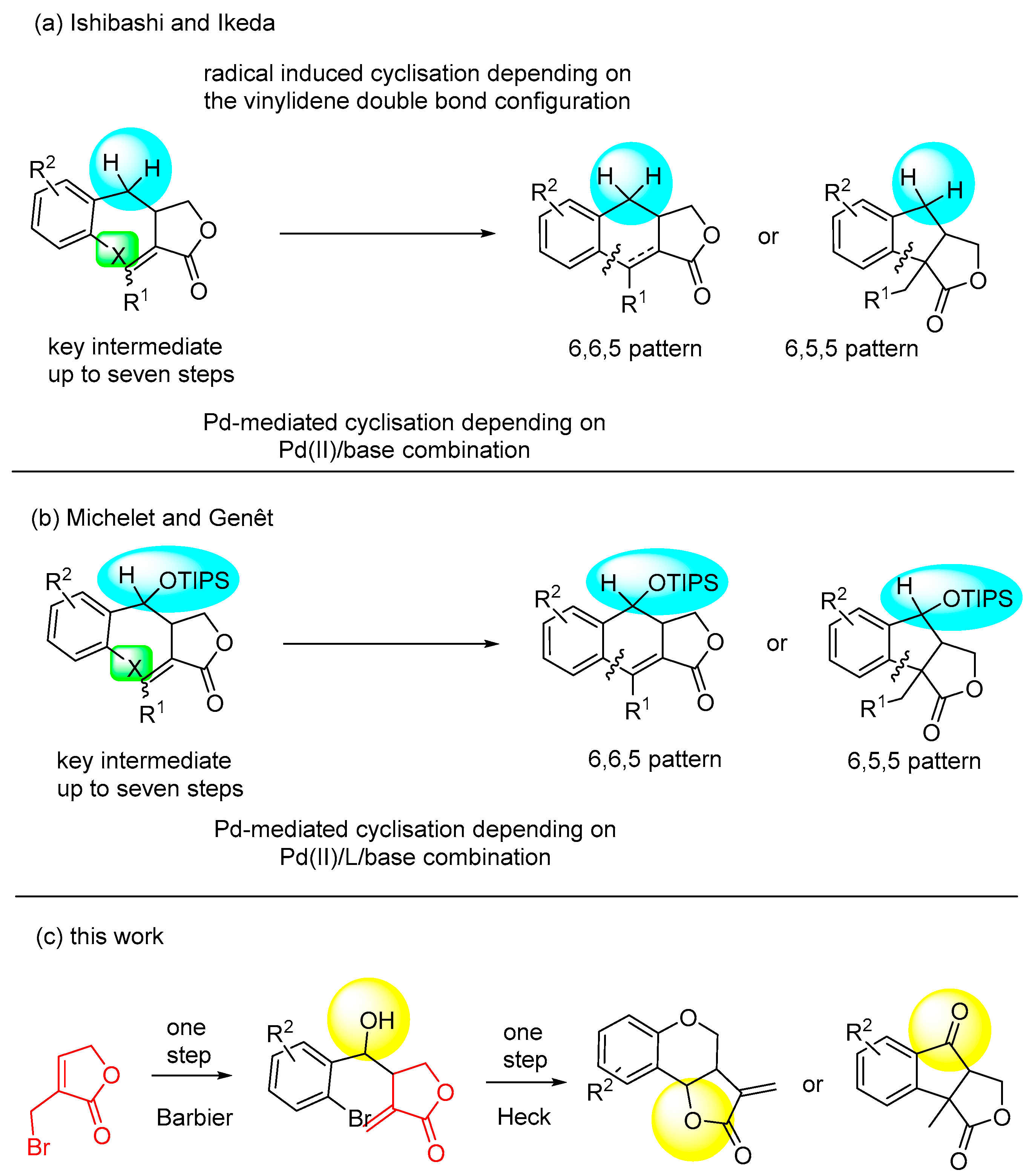
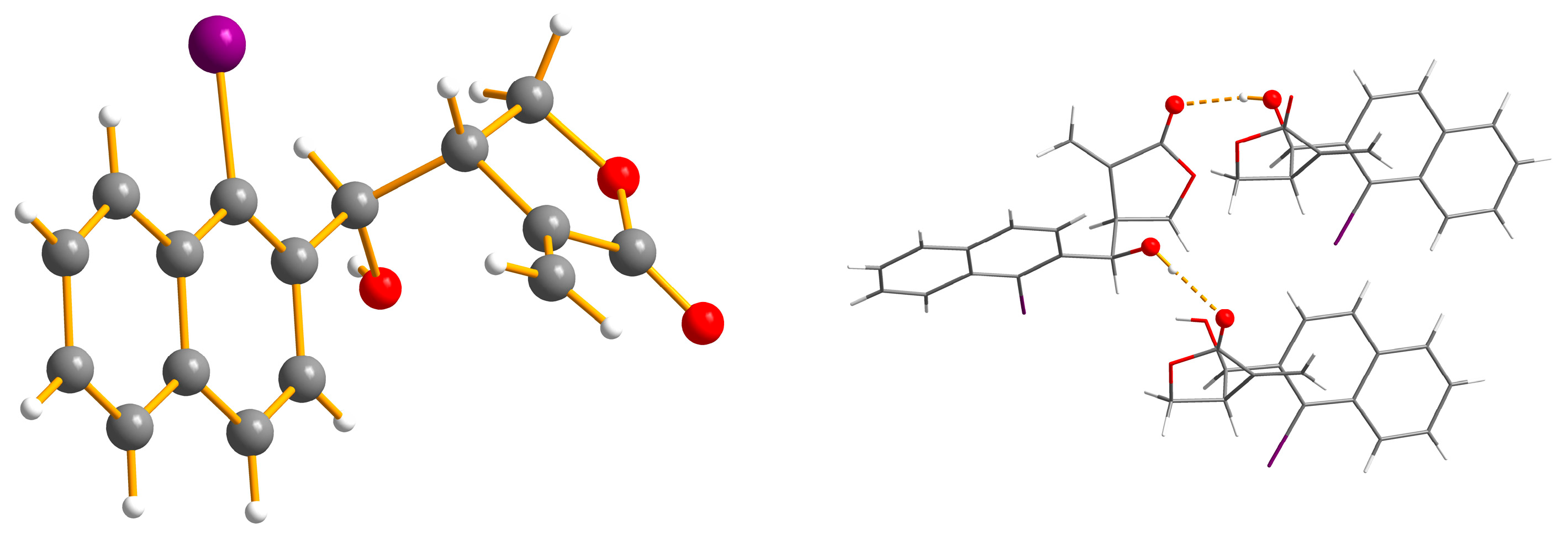
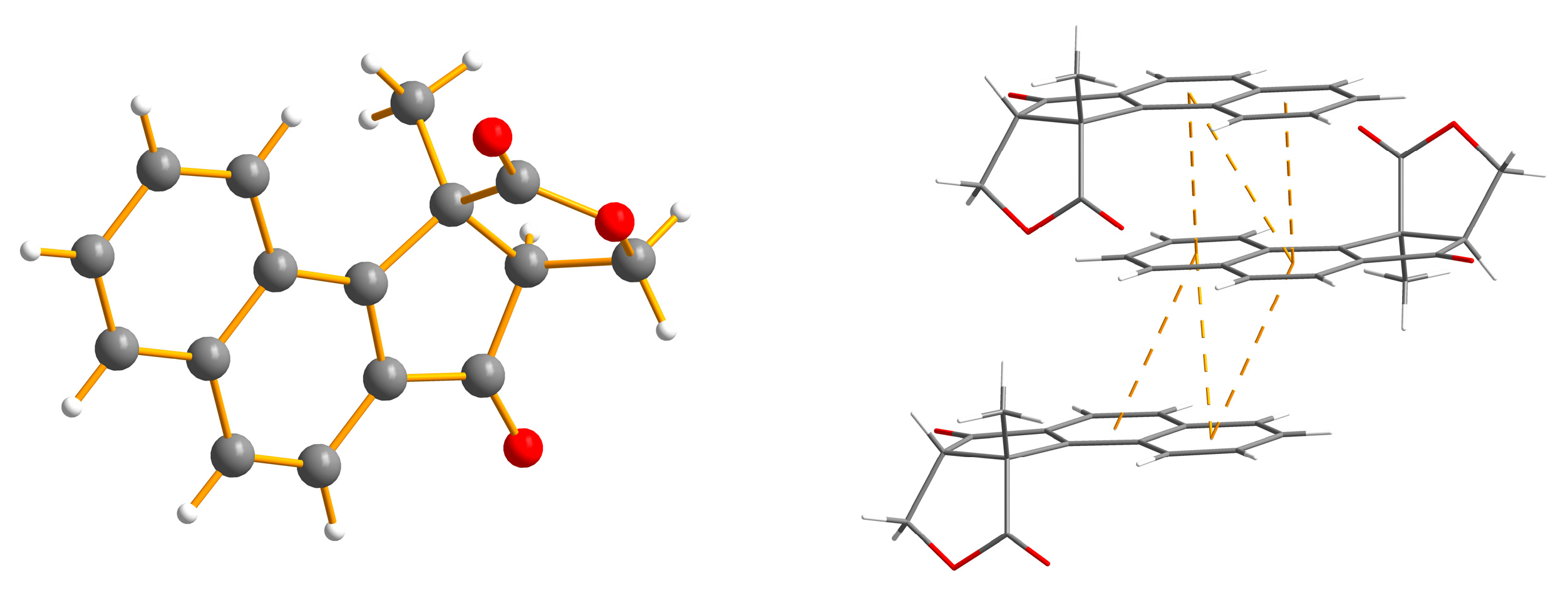
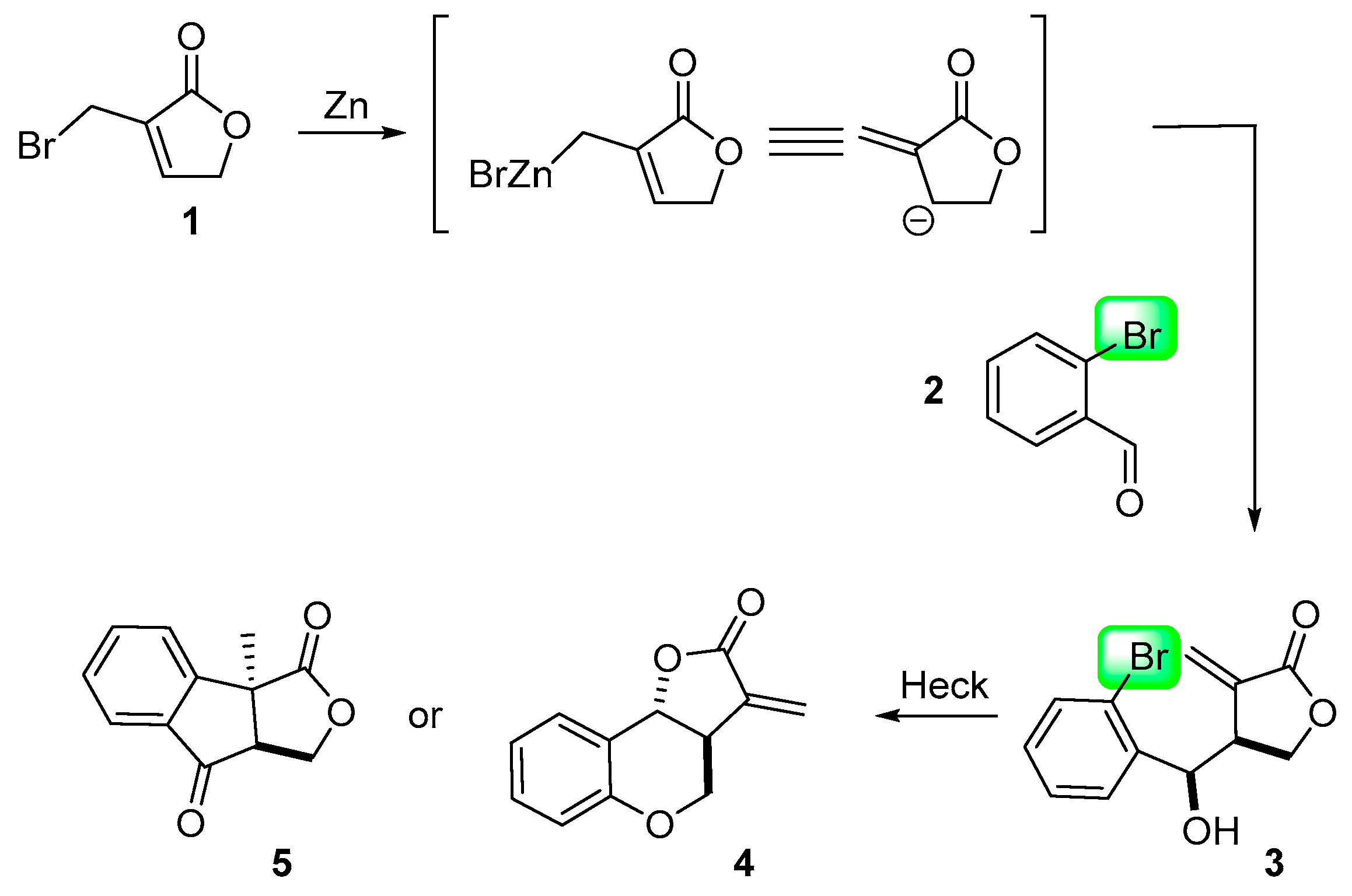
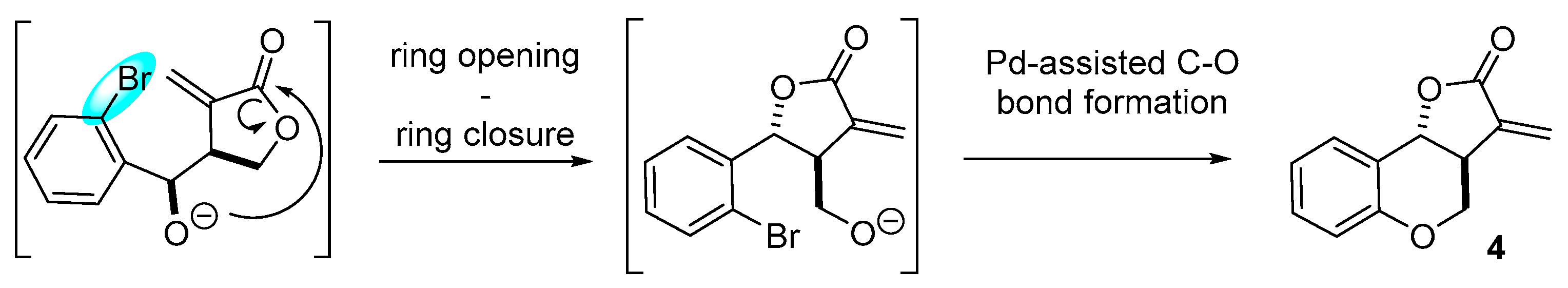
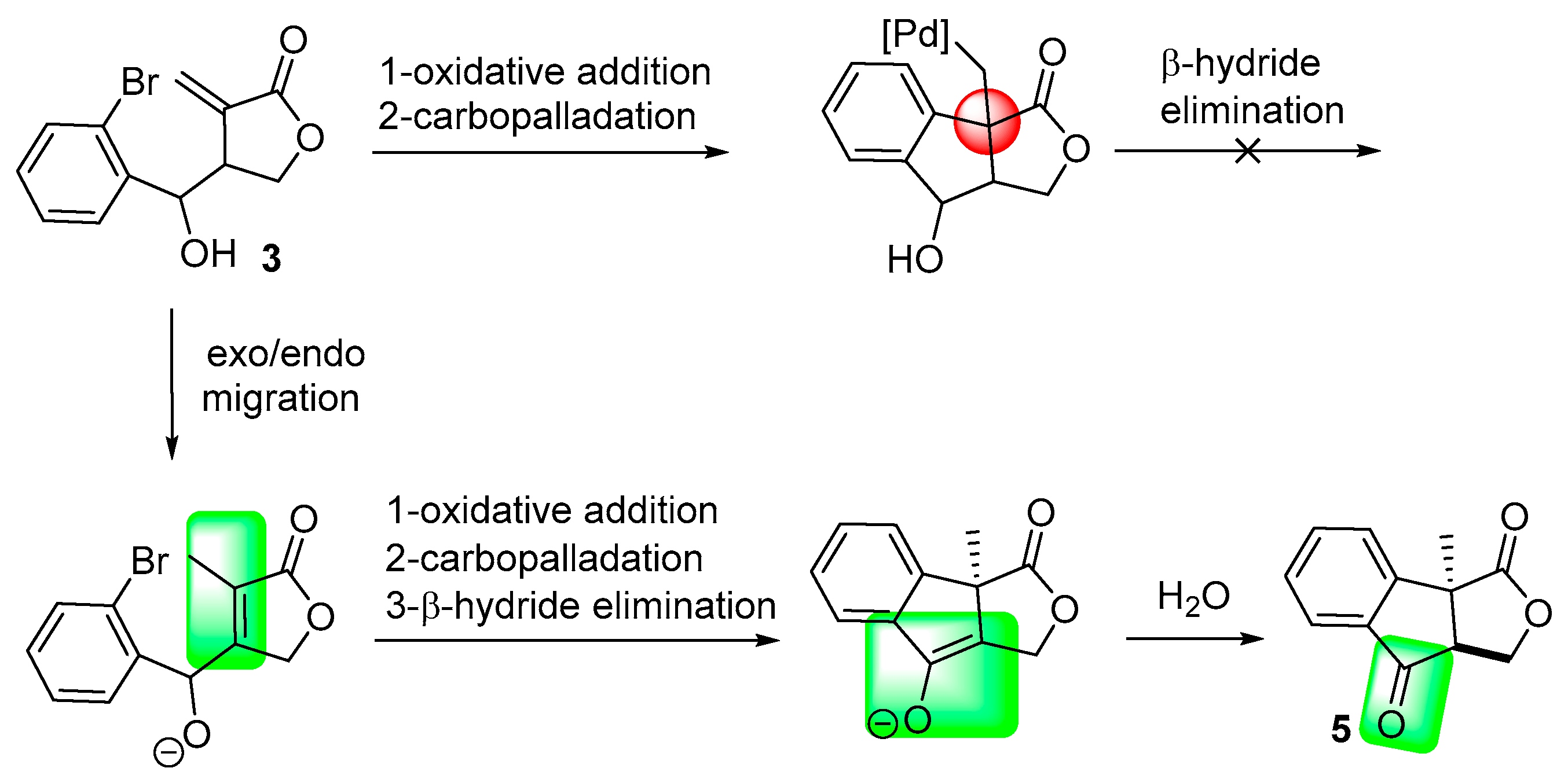
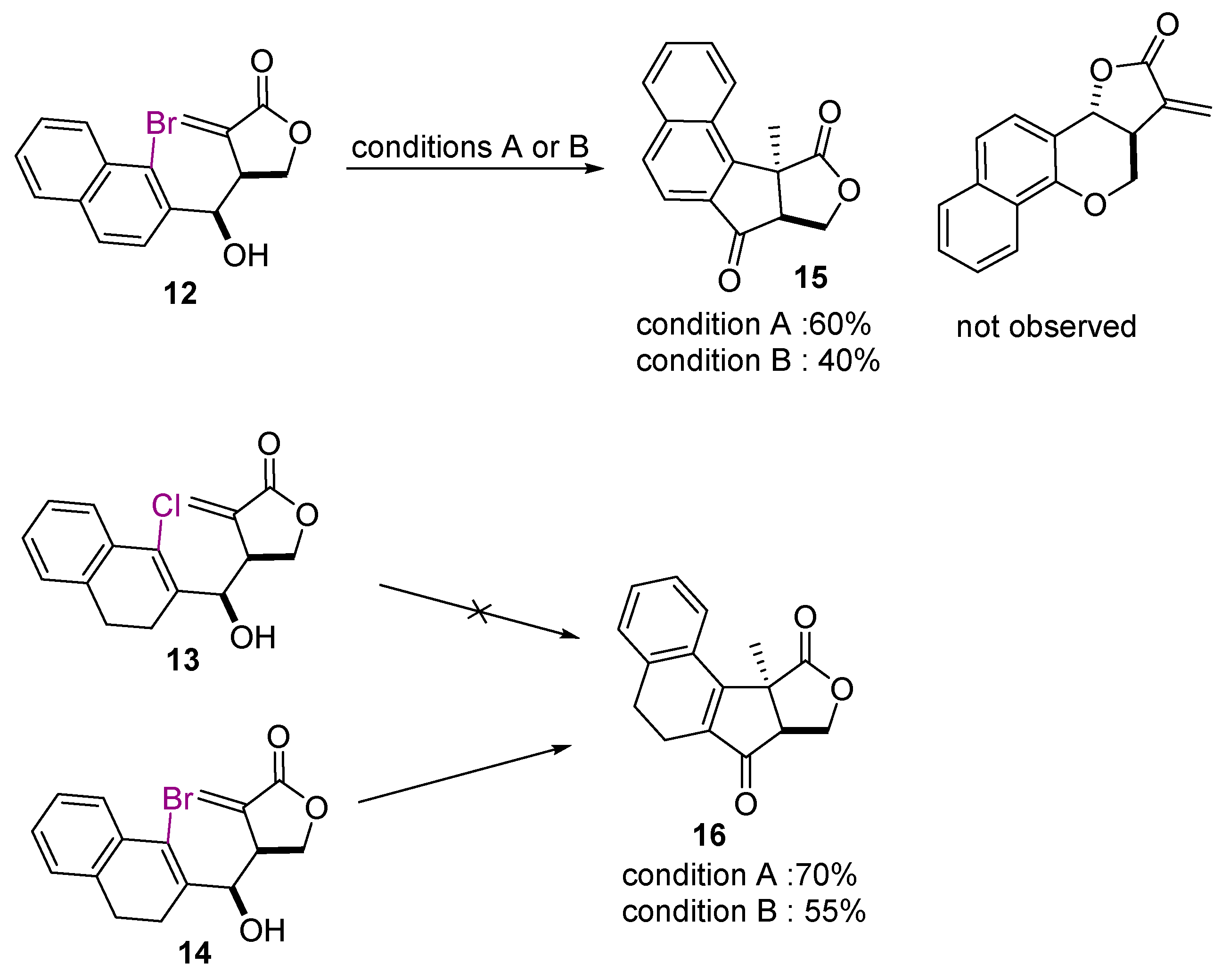
2. Results and Discussion
3. Materials and Methods
3.1. Methods
3.1.1. Representative Procedure for the Barbier Allylation Reaction of 3-Bromomethyl-5H-furan-2-one
3.1.2. Procedure A for Intramolecular Heck Reaction
3.1.3. Procedure B for Intramolecular Heck Reaction
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Westmeier, J.; Kress, S.; Pfaff, C.; Von Zezschwitz, P. Total synthesis of (R)-sarkomycin via asymmetric rhodium-catalyzed conjugate addition. J. Org. Chem. 2013, 78, 10718–10723. [Google Scholar] [CrossRef] [PubMed]
- Usuki, T.; Sato, M.; Hara, S.; Yoshimoto, Y.; Kondo, R.; Zimmermann, S.; Kaiser, M.; Brun, R.; Hamburger, M.; Adams, M. Antitrypanosomal structure–activity-relationship study of synthetic cynaropicrin derivatives. Biorg. Med. Chem. Lett. 2014, 24, 794–798. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, D.M.; Talbot, E.P.A.; Clark, B.P. Stereoselective synthesis of β-(hydroxymethylaryl/alkyl)-α-methylene-γ-butyrolactones. Org. Lett. 2011, 13, 2594–2597. [Google Scholar] [CrossRef] [PubMed]
- Shen, A.; He, Z.-T.; Yu, H.-J.; Fukui, Y.; Tian, P.; Lin, G.-Q. Zinc-mediated asymmetric allylation of chiral N-tert-butanesulfinyl aldimines with 3-bromomethyl-5H-furan-2-one. Synlett 2013, 24, 1649–1656. [Google Scholar] [CrossRef]
- Ferreira, M.; Bisol, T.B.; da Conceiçao, H.P.; Russo, T.V.C.; Bortoluzzi, A.J.; Sa, M.M. One-pot synthesis of α-ylidene δ-lactones from functionalized allylic bromides in a water–isopropanol medium. Synthesis 2017, 49, 667–676. [Google Scholar] [CrossRef]
- Zhang, F.; Yang, Y.; Xie, L.; Xu, X. Pd-catalyzed diastereoselective allylation of aldehydes with 3-bromomethyl-5H-furan-2-one: Stereoselective synthesis of β-(hydroxymethylaryl/alkyl)-α-methylene-γ-butyrolactones with a syn configuration. Chem. Commun. 2013, 49, 4697–4699. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, M.; Schober, M.; Orthaber, A.; Faber, K. Asymmetric synthesis of β-substituted α-methylenebutyro-lactones via TRIP-catalyzed allylation: Mechanistic studies and application to the synthesis of (S)-(−)-hydroxymatairesinol. Adv. Synth. Catal. 2013, 355, 2499–2505. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, Y.; Mochida, K.; Kim, S.-W. Total synthesis of sophorapterocarpan A, maackiain, and anhydropisatin: Application of a 1,3-Michael-Claisen annulations to aromatic synthesis. J. Chem. Soc. Perkin Trans. 1989, 1, 1219–1224. [Google Scholar] [CrossRef]
- Goel, A.; Kumar, A.; Hemberger, Y.; Raghuvanshi, A.; Jeet, R.; Tiwari, G.; Knauer, M.; Kureel, J.; Singh, A.K.; Gautam, A.; et al. Synthesis, optical resolution, absolute configuration, and osteogenic activity of cis-pterocarpans. Org. Biomol. Chem. 2012, 10, 9583–9592. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, H.; Ito, K.; Tabuchi, M.; Ikeda, M. Studies on aconitum species. XIV. Deoxygenation of pseudokobusine to kobusine. Heterocycles 1991, 32, 1297–1300. [Google Scholar] [CrossRef]
- Ishibashi, H.; Ito, K.; Hirano, T.; Tabuchi, M.; Ikeda, M. Synthesis of podophyllotoxin derivatives by means of tributyltin hydride- or palladium-mediated cyclization of α-benzylidene-β-(o-bromobenzyl)-γ-lactones. Tetrahedron 1993, 49, 4173–4182. [Google Scholar] [CrossRef]
- Charruault, L.; Michelet, V.; Genêt, J.-P. Pd-catalyzed route to (±)-podophyllotoxin skeleton. Synthesis of the aryltetralin derivative. Tetrahedron Lett. 2002, 43, 4757–4760. [Google Scholar] [CrossRef]
- Galland, J.-C.; Dias, S.; Savignac, M.; Genêt, J.-P. Cycloisomerization of 1,6-enynes in organoaqueous medium: An efficient and eco-friendly access to furan derivatives. Synthesis of a key intermediate of podophyllotoxin. Tetrahedron 2001, 57, 5137–5148. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, X.; Sun, L.; Xie, L.; Xu, X. Zinc or indium-mediated Barbier-type allylation of aldehydes with 3-bromomethyl-5H-furan-2-one in aqueous media: An efficient synthesis method for α-methylene-γ-butyrolactone. Org. Biomol. Chem. 2012, 10, 3991–3998. [Google Scholar] [CrossRef] [PubMed]
- Santoso, H.; Casana, M.I.; Donner, C.D. Exploring O-stannyl ketyl and acyl radical cyclizations for the synthesis of γ-lactone-fused benzopyrans and benzofurans. Org. Biomol. Chem. 2014, 12, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Pandey, G.; Vaitla, J. Desulfonylative methenylation of β-keto sulfones. Org. Lett. 2015, 17, 4890–4893. [Google Scholar] [CrossRef] [PubMed]
- Donner, C.D.; Casana, M.I. Synthesis of novel pyranoquinones using an acyl radical cyclization strategy. Tetrahedron Lett. 2012, 53, 1105–1107. [Google Scholar] [CrossRef]
- Prim, D.; Campagne, J.-M.; Joseph, D.; Andrioletti, B. Palladium-catalysed reactions of aryl halides with soft, non-organometallic nucleophiles. Tetrahedron 2002, 58, 2041–2075. [Google Scholar] [CrossRef]
- Ashimori, A.; Bachand, B.; Overman, L.E.; Poon, D.J. Catalytic asymmetric synthesis of quaternary carbon centers. Exploratory investigations of intramolecular Heck reactions of (E)-α,β-unsaturated 2-haloanilides and analogues to form enantioenriched spirocyclic products. J. Am. Chem. Soc. 1998, 120, 6477–6487. [Google Scholar] [CrossRef]
- Coquerel, Y.; Bremond, P.; Rodriguez, J. Pd–H from Pd/C and triethylamine: Implications in palladium catalysed reactions involving amines. J. Organomet. Chem. 2007, 692, 4805–4808. [Google Scholar] [CrossRef] [Green Version]
- Jefford, C.W.; Rossier, J.-C.; Boukouvalas, J.; Sledeski, A.W.; Huang, P.-Z. A concise synthesis of siphonodictidine. J. Nat. Prod. 2004, 67, 1383–1386. [Google Scholar] [CrossRef] [PubMed]
- Moise, J.; Arseniyadis, S.; Cossy, J. Cross-metathesis between α-methylene-γ-butyrolactone and olefins: A dramatic additive effect. Org. Lett. 2007, 9, 1695–1698. [Google Scholar] [CrossRef] [PubMed]
- Boufroura, H.; Souibgui, A.; Gaucher, A.; Marrot, J.; Pieters, G.; Aloui, F.; Ben Hassine, B.; Clavier, G.; Prim, D. 3D shapes of aryl(dihydro)naphthothiophenes: A comprehensive and structural study. Org. Biomol. Chem. 2015, 13, 10844–10851. [Google Scholar] [CrossRef] [PubMed]
- Cambridge Crystallographic Data Centre deposit numbers for compounds 12: 1565162 and 16: 1565163. (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: [email protected]). These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html.
- Yang, H.S.; Qiao, X.X.; Cui, Q.; Xu, X.H. First synthesis of cedarmycin B. Chin. Chem. Lett. 2009, 20, 1023–1024. [Google Scholar] [CrossRef]
- Brunner, B.; Stogaitis, N.; Lautens, M. Synthesis of 1,2-dihydropyridines using vinyloxiranes as masked dienolates in imino-aldol reactions. Org. Lett. 2006, 8, 3473–3476. [Google Scholar] [CrossRef] [PubMed]
- Requet, A.; Souibgui, A.; Pieters, G.; Ferhi, S.; Letaieff, A.; Carlin-Sinclair, A.; Marque, S.; Marrot, J.; Ben Hassine, B.; Gaucher, A.; et al. Synthesis of partially hydrogenated oxa[5] and oxa[6]helicenes from β-chlorovinylaldehydes. Tetrahedron Lett. 2013, 54, 4721–4725. [Google Scholar] [CrossRef]
- Shunatona, H.P.; Früh, N.; Wang, Y.-M.; Rauniyar, V.; Toste, F.D. Enantioselective fluoroamination: 1,4-addition to conjugated dienes using anionic phase-transfer catalysis. Angew. Chem. Int. Ed. 2013, 52, 7724–7727. [Google Scholar] [CrossRef] [PubMed]
- Turrini, N.G.; Hall, M.; Faber, K. Enzymatic synthesis of optically active lactones via asymmetric bioreduction using ene-reductases from the old yellow enzyme family. Adv. Synth. Catal. 2015, 357, 1861–1871. [Google Scholar] [CrossRef]
- Smith, C.R. Activated zinc dust. Synlett 2009, 9, 1522–1523. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
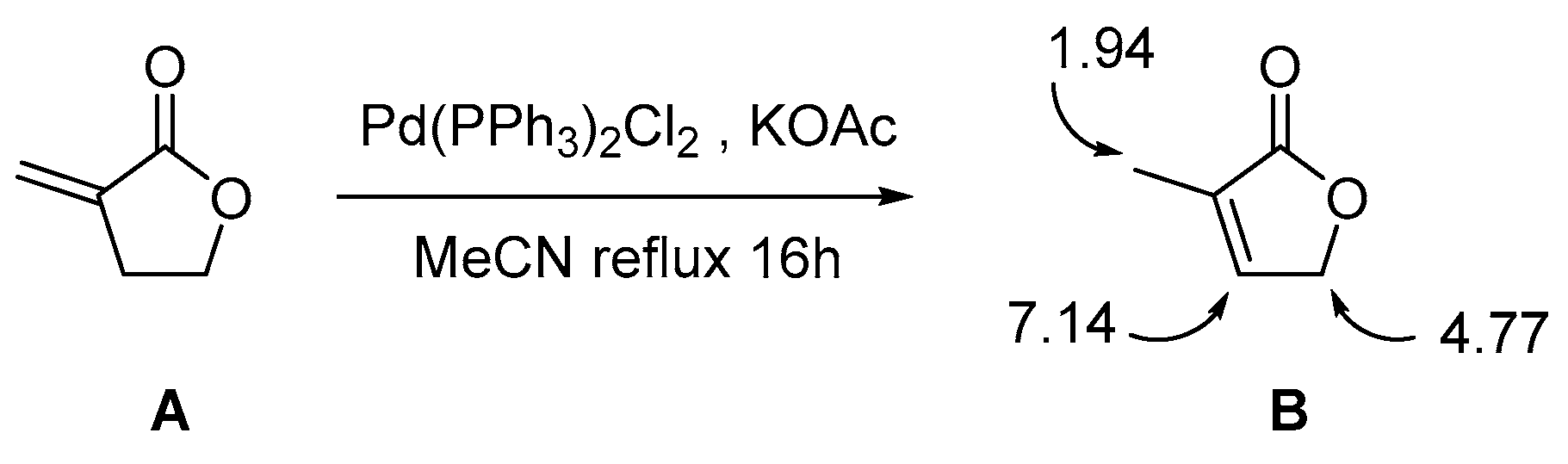{kind=link}
{kind=link}

| Entry | Catalyst | Base | Additive | Solvent | Conditions | 3/4/5 a | Yield (%) |
|---|---|---|---|---|---|---|---|
| 1 | Pd(PPh3)2Cl2 | K2CO3 | THF | 65 °C, 2 h | 1/1/0 | 4(20) | |
| 2 | Pd(PPh3)2Cl2 | K2CO3 | THF | 65 °C, 16 h | 0/1/0 | 4(60) | |
| 3 | Pd(PPh3)2Cl2 | K2CO3 | Ag2CO3 | THF | 65 °C, 16 h | - | - c |
| 4 | Pd(PPh3)2Cl2 | K2CO3 | MeCN | 90 °C, 16 h | 0/1/0 | 4(50) | |
| 5 | Pd(PPh3)2Cl2 | Cs2CO3 | MeCN | 90 °C, 2 h | - | - c | |
| 6 | - | K2CO3 | THF | 65 °C, 16 h | - | - d | |
| 7 | Pd(dppf)Cl2 | K2CO3 | MeCN | 90 °C, 2 h | 0.5/1/0 | 4(30) | |
| 8 | Pd(dppf)Cl2 | K2CO3 | THF | 65 °C, 16 h | 0/1/0 | 4(45) | |
| 9 | Pd(PPh3)2Cl2 | KOAc | THF | 65 °C, 16 h | 1/0/0.1 | nd | |
| 10 | Pd(PPh3)2Cl2 | KOAc | MeCN | 90 °C, 16 h | 0/0/1 | 5(40) | |
| 11 | Pd(PPh3)2Cl2 | KOAc | AgOAc | MeCN | 90 °C, 16 h | 0/0/1 | 5(14) |
| 12 | Pd(PPh3)2Cl2 | KOAc, K2CO3 b | THF | 65 °C, 16 h | 1/1.2/0.1 | - a |
| Entry | Starting Halide | Compound | Condition | α-Methylidene Butyrolactone | Product | Yield (%) | Dr a |
|---|---|---|---|---|---|---|---|
| 1 |  | 6 | THF, 18 h |  | 11 | - | - |
| 2 |  | 7 | THF, 18 h | 11 | - | - | |
| 3 |  | 8 | THF, 18 h |  | 12 | 60 | 87/13 |
| 4 |  | 9, X = Cl | THF, 16 h |  | 13, X = Cl | 80 | 89/11 |
| 5 | 10, X = Br | 14, X = Br | 89 | 94/6 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Talbi, A.; Gaucher, A.; Bourdreux, F.; Marrot, J.; Efrit, M.L.; M’Rabet, H.; Prim, D. From α-Bromomethylbutenolide to Fused Tri(Tetra) Cyclic Dihydrofurandiones through Barbier Reaction–Heck Arylation Sequence. Molecules 2017, 22, 2171. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22122171
Talbi A, Gaucher A, Bourdreux F, Marrot J, Efrit ML, M’Rabet H, Prim D. From α-Bromomethylbutenolide to Fused Tri(Tetra) Cyclic Dihydrofurandiones through Barbier Reaction–Heck Arylation Sequence. Molecules. 2017; 22(12):2171. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22122171
Chicago/Turabian StyleTalbi, Arbia, Anne Gaucher, Flavien Bourdreux, Jérôme Marrot, Mohamed L. Efrit, Hédi M’Rabet, and Damien Prim. 2017. "From α-Bromomethylbutenolide to Fused Tri(Tetra) Cyclic Dihydrofurandiones through Barbier Reaction–Heck Arylation Sequence" Molecules 22, no. 12: 2171. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22122171