
Synthesis and Structural Evaluation of Organo-Ruthenium Complexes with β-Diketonates

Abstract
:1. Introduction
2. Results
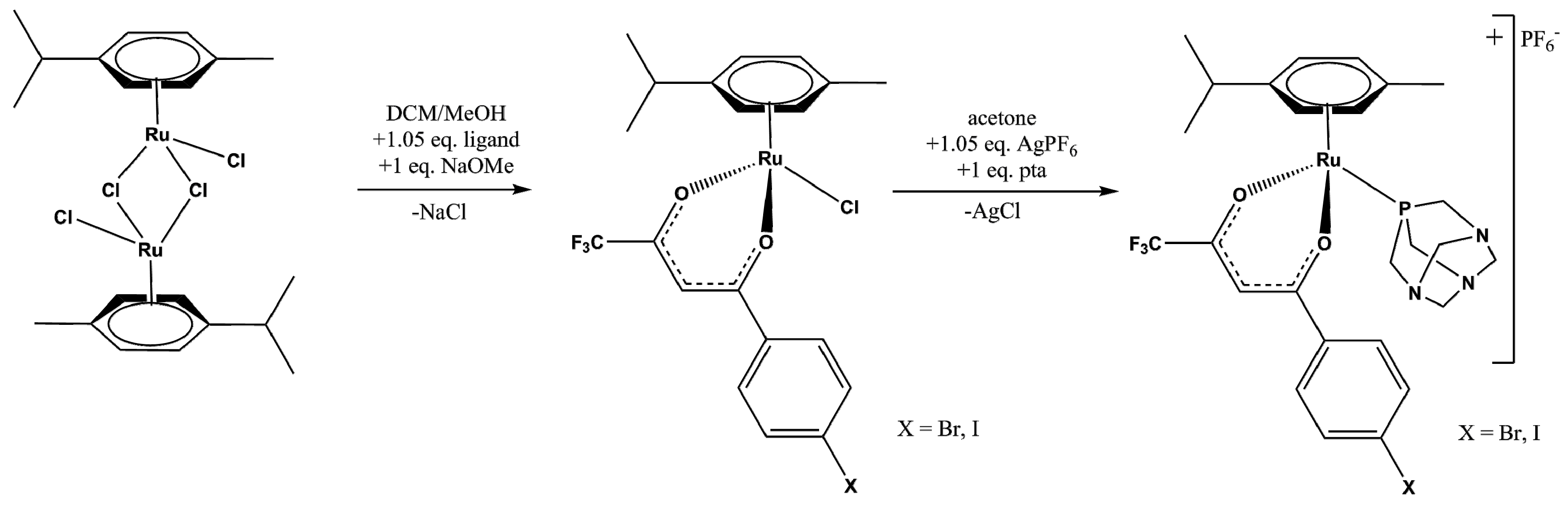
2.1. Synthesis and Spectroscopic Analysis
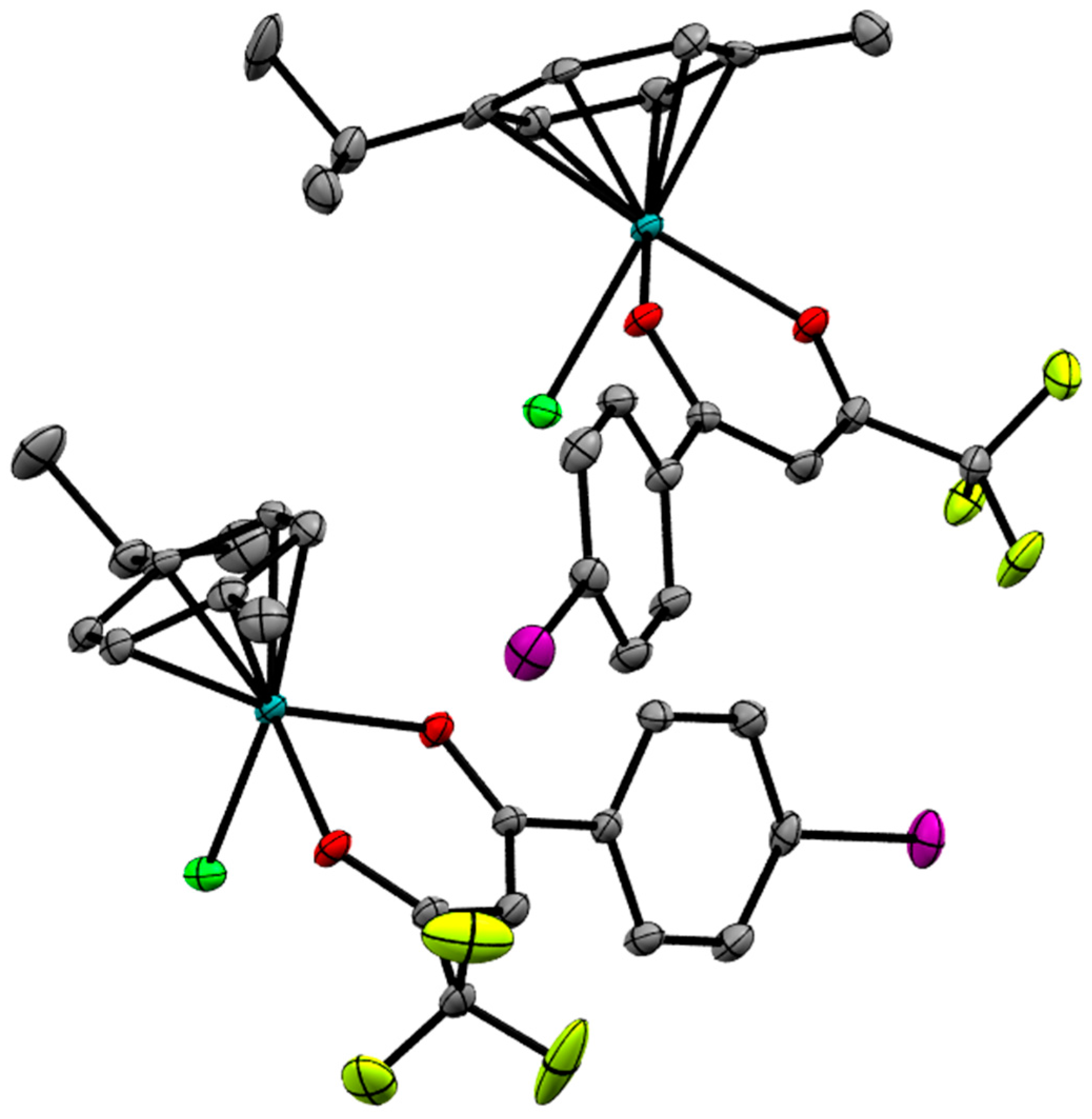
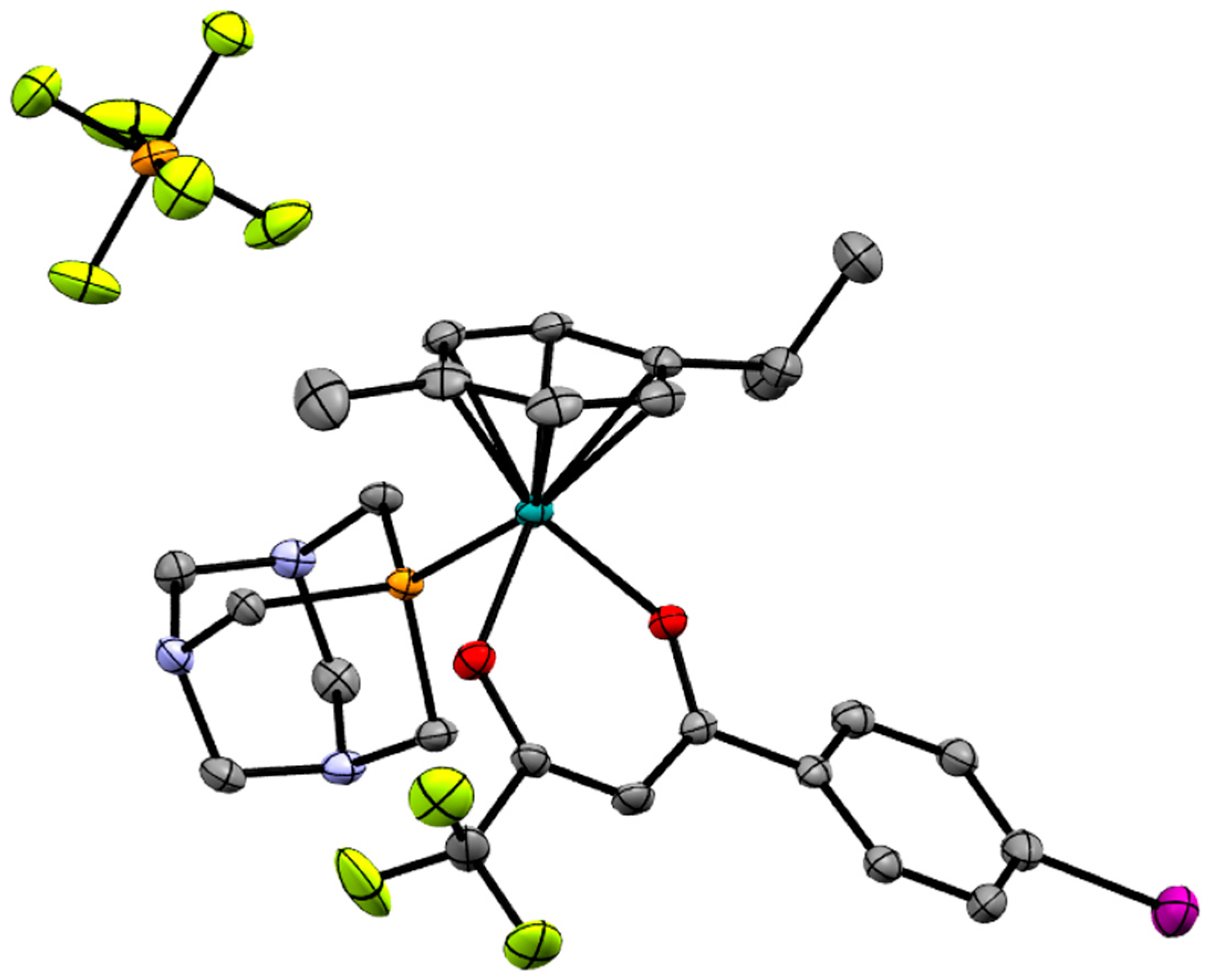
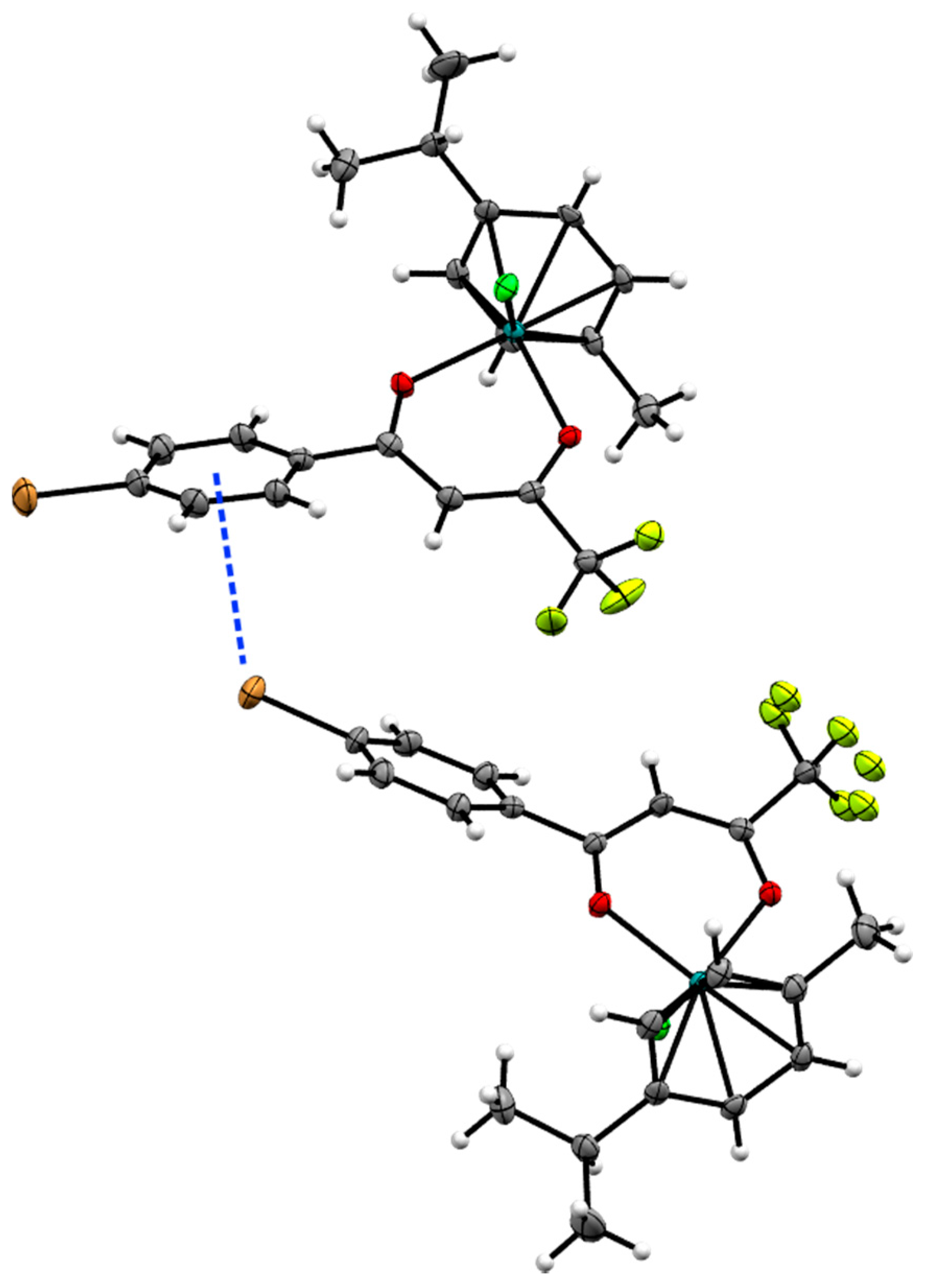
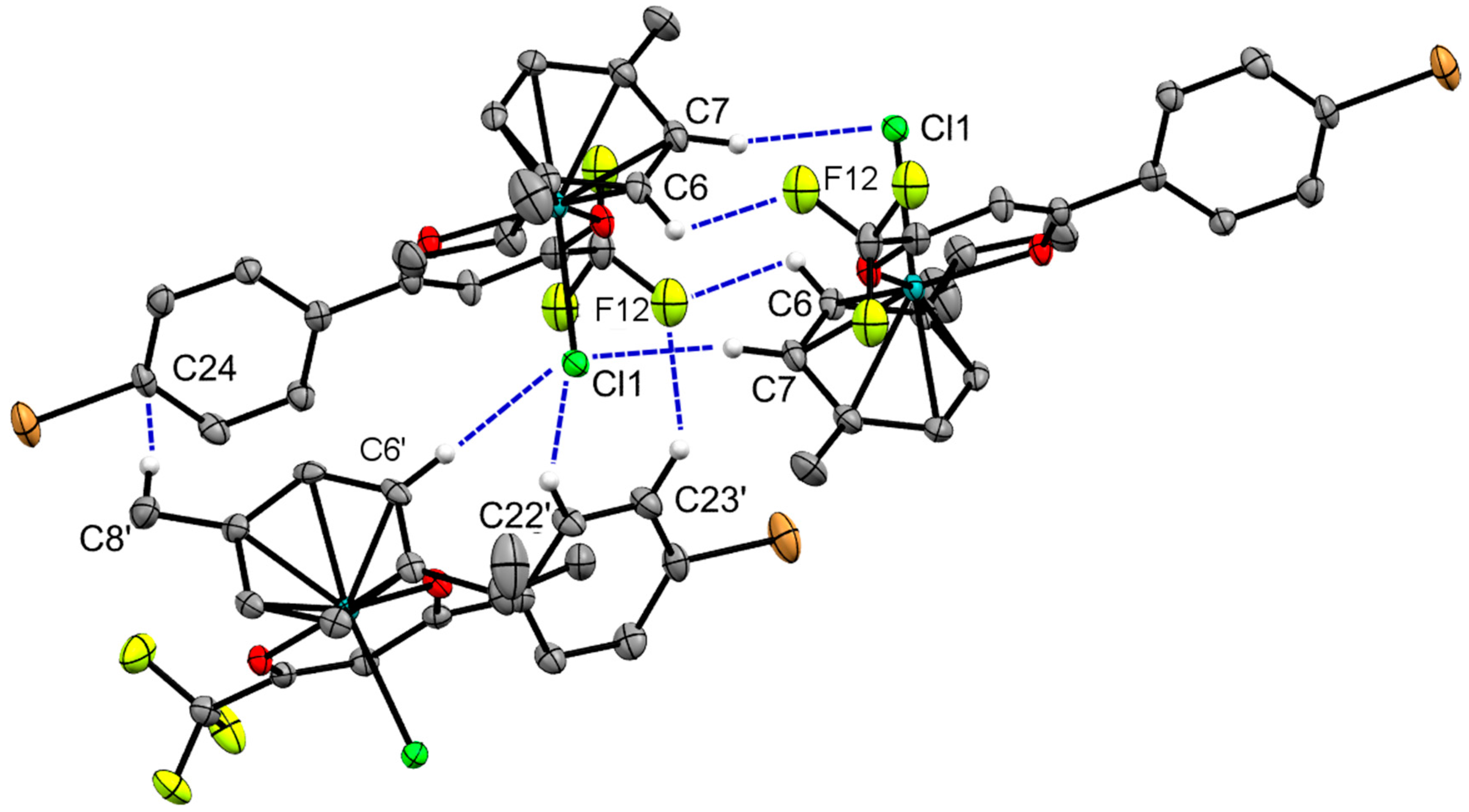
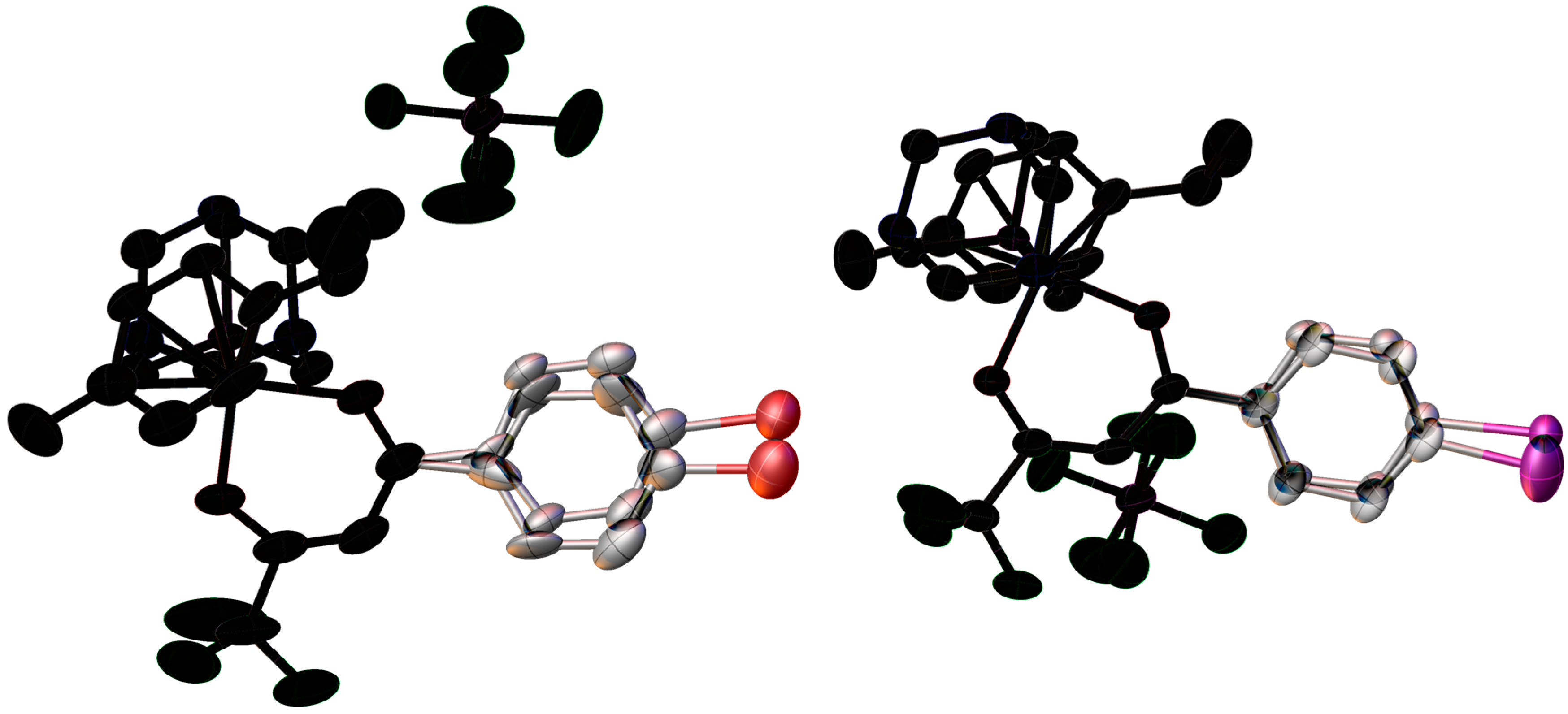
2.2. X-ray Crystal Structures
3. Experimental Section
3.1. Materials and Methods
3.2. Crystal Structure Determination
3.3. Synthesis
3.3.1. Synthesis of 1 and 3
3.3.2. Synthesis of 2 and 4
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Turel, I. Special issue: Practical applications of metal complexes. Molecules 2015, 20, 7951–7956. [Google Scholar] [CrossRef] [PubMed]
- Bergamo, A.; Sava, G. Ruthenium complexes can target determinants of tumour malignancy. Dalton Trans. 2007, 1267–1272. [Google Scholar] [CrossRef] [PubMed]
- Trondl, R.; Heffeter, P.; Kowol, C.R.; Jakupec, M.A.; Berger, W.; Keppler, B.K. Nkp-1339, the first ruthenium-based anticancer drug on the edge to clinical application. Chem. Sci. 2014, 5, 2925–2932. [Google Scholar] [CrossRef]
- Scolaro, C.; Bergamo, A.; Brescacin, L.; Delfino, R.; Cocchietto, M.; Laurenczy, G.; Geldbach, T.J.; Sava, G.; Dyson, P.J. In vitro and in vivo evaluation of ruthenium(II)-arene pta complexes. J. Med. Chem. 2005, 48, 4161–4171. [Google Scholar] [CrossRef] [PubMed]
- Naota, T.; Takaya, H.; Murahashi, S.I. Ruthenium-catalyzed reactions for organic synthesis. Chem. Rev. 1998, 98, 2599–2660. [Google Scholar] [CrossRef] [PubMed]
- Gunanathan, C.; Milstein, D. Bond activation and catalysis by ruthenium pincer complexes. Chem. Rev. 2014, 114, 12024–12087. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Astruc, D. Magnetically recoverable ruthenium catalysts in organic synthesis. Molecules 2014, 19, 4635–4653. [Google Scholar] [CrossRef] [PubMed]
- Suriboot, J.; Bazzi, H.; Bergbreiter, D. Supported catalysts useful in ring-closing metathesis, cross metathesis, and ring-opening metathesis polymerization. Polymers 2016, 8, 140. [Google Scholar] [CrossRef]
- Paradiso, V.; Costabile, C.; Grisi, F. NHC backbone configuration in ruthenium-catalyzed olefin metathesis. Molecules 2016, 21, 117. [Google Scholar] [CrossRef] [PubMed]
- Seršen, S.; Kljun, J.; Požgan, F.; Štefane, B.; Turel, I. Novel organoruthenium(II) β-diketonates as catalysts for ortho-arylation via C–H activation. Organometallics 2013, 32, 609–616. [Google Scholar]
- Seršen, S.; Kljun, J.; Kryeziu, K.; Panchuk, R.; Alte, B.; Körner, W.; Heffeter, P.; Berger, W.; Turel, I. Structure-related mode-of-action differences of anticancer organoruthenium complexes with β-diketonates. J. Med. Chem. 2015, 58, 3984–3996. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, K. Applications in inorganic chemistry. In Infrared and Raman Spectra of Inorganic and Coordination Compounds; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008; pp. 149–354. [Google Scholar]
- Habtemariam, A.; Garino, C.; Ruggiero, E.; Alonso-de Castro, S.; Mareque-Rivas, J.; Salassa, L. Photorelease of pyridyl esters in organometallic ru(II) arene complexes. Molecules 2015, 20, 7276–7291. [Google Scholar] [CrossRef] [PubMed]
- Betanzos-Lara, S.; Salassa, L.; Habtemariam, A.; Novakova, O.; Pizarro, A.M.; Clarkson, G.J.; Liskova, B.; Brabec, V.; Sadler, P.J. Photoactivatable organometallic pyridyl ruthenium(II) arene complexes. Organometallics 2012, 31, 3466–3479. [Google Scholar] [CrossRef]
- Malecki, J.G.; Jaworska, M.; Kruszynski, R.; Klak, J. Reaction of [(C6H6)RuCl2]2 with 7,8-benzoquinoline and 8-hydroxyquinoline. Polyhedron 2005, 24, 3012–3021. [Google Scholar] [CrossRef]
- Casellato, U.; Vigato, P.A.; Vidali, M. Transition metal complexes with binucleating ligands. Coord. Chem. Rev. 1977, 23, 31–117. [Google Scholar] [CrossRef]
- Hynes, M.J. Reactions of β-diketone complexes in solution. Rev. Inorg. Chem. 1990, 11, 21–78. [Google Scholar] [CrossRef]
- Guerriero, P.; Tarnburini, S.; Vigato, P.A. From mononuclear to polynuclear macrocyclic or macroacyclic complexes. Coord. Chem. Rev. 1995, 139, 17–243. [Google Scholar] [CrossRef]
- Bray, D.J.; Clegg, J.K.; Lindoy, L.F.; Schilter, D. Self-assembled metallo-supramolecular systems incorporating β-diketone motifs as structural elements. Adv. Inorg. Chem. 2006, 59, 1–37. [Google Scholar]
- Aromí, G.; Gamez, P.; Reedijk, J. Poly beta-diketones: Prime ligands to generate supramolecular metalloclusters. Coord. Chem. Rev. 2008, 252, 964–989. [Google Scholar] [CrossRef]
- Bruno, I.J.; Cole, J.C.C.; Kessler, M.; Luo, J.; Motherwell, W.D.S.; Purkis, L.H.; Smith, B.R.; Taylor, R.; Cooper, R.I.; Harris, S.E.; et al. Retrieval of crystallographically-derived molecular geometry information. J. Chem. Inf. Comput. Sci. 2004, 44, 2133–2144. [Google Scholar] [CrossRef] [PubMed]
- Melchart, M.; Habtemariam, A.; Parsons, S.; Sadler, P.J. Chlorido-, aqua-, 9-ethylguanine- and 9-ethyladenine-adducts of cytotoxic ruthenium arene complexes containing O,O-chelating ligands. J. Inorg. Biochem. 2007, 101, 1903–1912. [Google Scholar] [CrossRef] [PubMed]
- Vock, C.A.; Renfrew, A.K.; Scopelliti, R.; Juillerat-Jeanneret, L.; Dyson, P.J. Influence of the diketonato ligand on the cytotoxicities of [Ru(η6-p-cymene)(R2acac)(PTA)]+ complexes (PTA = 1,3,5-triaza-7-phosphaadamantane). Eur. J. Inorg. Chem. 2008, 2008, 1661–1671. [Google Scholar] [CrossRef]
- Kljun, J.; Bytzek, A.K.; Kandioller, W.; Bartel, C.; Jakupec, M.A.; Hartinger, C.G.; Keppler, B.K.; Turel, I. Physicochemical studies and anticancer potency of ruthenium η6-p-cymene complexes containing antibacterial quinolones. Organometallics 2011, 30, 2506–2512. [Google Scholar] [CrossRef] [PubMed]
- Habtemariam, A.; Melchart, M.; Fernández, R.; Parsons, S.; Oswald, I.D.H.; Parkin, A.; Fabbiani, F.P.A.; Davidson, J.E.; Dawson, A.; Aird, R.E.; et al. Structure-activity relationships for cytotoxic ruthenium(II) arene complexes containing N,N-, N,O-, and O,O-chelating ligands. J. Med. Chem. 2006, 49, 6858–6868. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van De Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Cryst. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Farrugia, L. ORTEP-3 for windows—A version of ORTEP-III with a graphical user interface (GUI). J. Appl. Crystallogr. 1997, 30, 565. [Google Scholar] [CrossRef]
- Spek, A. Single-crystal structure validation with the program platon. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Sample Availability: Compounds 1–4 are available from authors upon agreement.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond/Angle | 1 | 3 | 5 | 2 | 4 | 6 |
|---|---|---|---|---|---|---|
| Ru1–O17 | 2.096(3) | 2.097(4) | 2.091(2) | 2.081(8) | 2.081(7) | 2.094(4) |
| Ru1’–O17’ | 2.095(3) | 2.099(4) | / | / | / | |
| Ru1–O20 | 2.081(3) | 2.079(3) | 2.092(1) | 2.074(8) | 2.074(6) | 2.072(4) |
| Ru1’–O20’ | 2.076(2) | 2.088(4) | / | / | / | / |
| Ru1–Cl1 | 2.393(1) | 2.396(1) | 2.405(1) | / | / | / |
| Ru1’–Cl1’ | 2.394(1) | 2.397(1) | / | / | / | / |
| Ru1–P30 | / | / | / | 2.318(2) | 2.314(2) | 2.3270(7) |
| O17–Ru1–O20 | 87.7(1) | 88.1(2) | 87.44(3) | 87.6(3) | 87.6(2) | 87.44(7) |
| O17’–Ru1’–O20’ | 87.3(1) | 88.0(2) | / | / | / | / |
| Complex Number | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Empirical formula | C20H19BrClF3O2Ru | C26H31BrF9N3O2P2Ru | C20H19ClF3IO2Ru | C26H31F9IN3O2P2Ru |
| Formula weight | 564.78 | 831.46 | 611.77 | 878.45 |
| Temperature/K | 150.00(10) | 150.00(10) | 150.00(10) | 150.00(10) |
| Crystal system | triclinic | monoclinic | triclinic | monoclinic |
| Space group | P-1 | P21/c | P-1 | P21/c |
| a/Å | 10.0249(4) | 10.6584(5) | 9.9973(3) | 10.6889(4) |
| b/Å | 12.9431(6) | 14.8139(6) | 12.9283(5) | 14.8208(5) |
| c/Å | 18.0374(7) | 19.6695(8) | 18.2806(8) | 19.8820(6) |
| α/° | 72.014(4) | 90 | 71.413(4) | 90 |
| β/° | 80.020(3) | 94.661(4) | 80.368(3) | 93.538(3) |
| γ/° | 76.696(4) | 90 | 76.830(3) | 90 |
| Volume/Å3 | 2153.05(17) | 3095.4(2) | 2169.09(14) | 3143.65(17) |
| Z | 4 | 4 | 4 | 4 |
| ρcalc·g/cm3 | 1.742 | 1.784 | 1.873 | 1.856 |
| μ/mm−1 | 2.746 | 1.986 | 18.506 | 1.666 |
| F (000) | 1112.0 | 1656.0 | 1184.0 | 1728.0 |
| Radiation | Mo Kα (λ = 0.7107) | Mo Kα (λ = 0.7107) | Cu Kα (λ = 1.5418) | Mo Kα (λ = 0.7107) |
| Reflections collected | 16,610 | 17,899 | 15,274 | 20,267 |
| Independent reflections | 9837 (Rint = 0.0328, Rsigma = 0.0668) | 8116 (Rint = 0.0400, Rsigma = 0.0660) | 8508 (Rint = 0.0529, Rsigma = 0.0637) | 8291 (Rint = 0.0345, Rsigma = 0.0529) |
| Data/restraints/parameters | 9837/0/509 | 8116/0/440 | 8508/0/511 | 8291/0/410 |
| Goodness-of-fit on F2 | 1.007 | 1.031 | 1.040 | 1.030 |
| Final R indexes (I ≥ 2σ (I)) | R1 = 0.0445, wR2 = 0.0909 | R1 = 0.0665, wR2 = 0.1699 | R1 = 0.0445, wR2 = 0.1217 | R1 = 0.0532, wR2 = 0.1059 |
| Final R indexes (all data) | R1 = 0.0753, wR2 = 0.1027 | R1 = 0.1132, wR2 = 0.1942 | R1 = 0.0548, wR2 = 0.1325 | R1 = 0.0869, wR2 = 0.1207 |
| Largest diff. peak/hole/e Å−3 | 0.94/−1.03 | 1.64/−0.89 | 1.59/−1.29 | 1.55/−0.73 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uršič, M.; Lipec, T.; Meden, A.; Turel, I. Synthesis and Structural Evaluation of Organo-Ruthenium Complexes with β-Diketonates. Molecules 2017, 22, 326. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22020326
Uršič M, Lipec T, Meden A, Turel I. Synthesis and Structural Evaluation of Organo-Ruthenium Complexes with β-Diketonates. Molecules. 2017; 22(2):326. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22020326
Chicago/Turabian StyleUršič, Matija, Tanja Lipec, Anton Meden, and Iztok Turel. 2017. "Synthesis and Structural Evaluation of Organo-Ruthenium Complexes with β-Diketonates" Molecules 22, no. 2: 326. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22020326