
Green Hydroselenation of Aryl Alkynes: Divinyl Selenides as a Precursor of Resveratrol

 , ,
, ,  ,
, 
Abstract
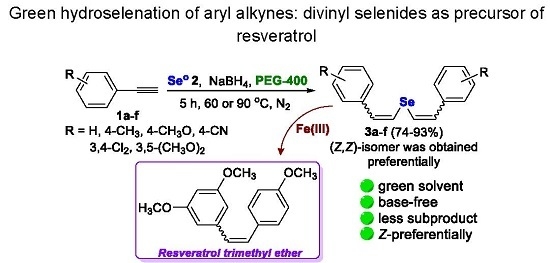
:
1. Introduction
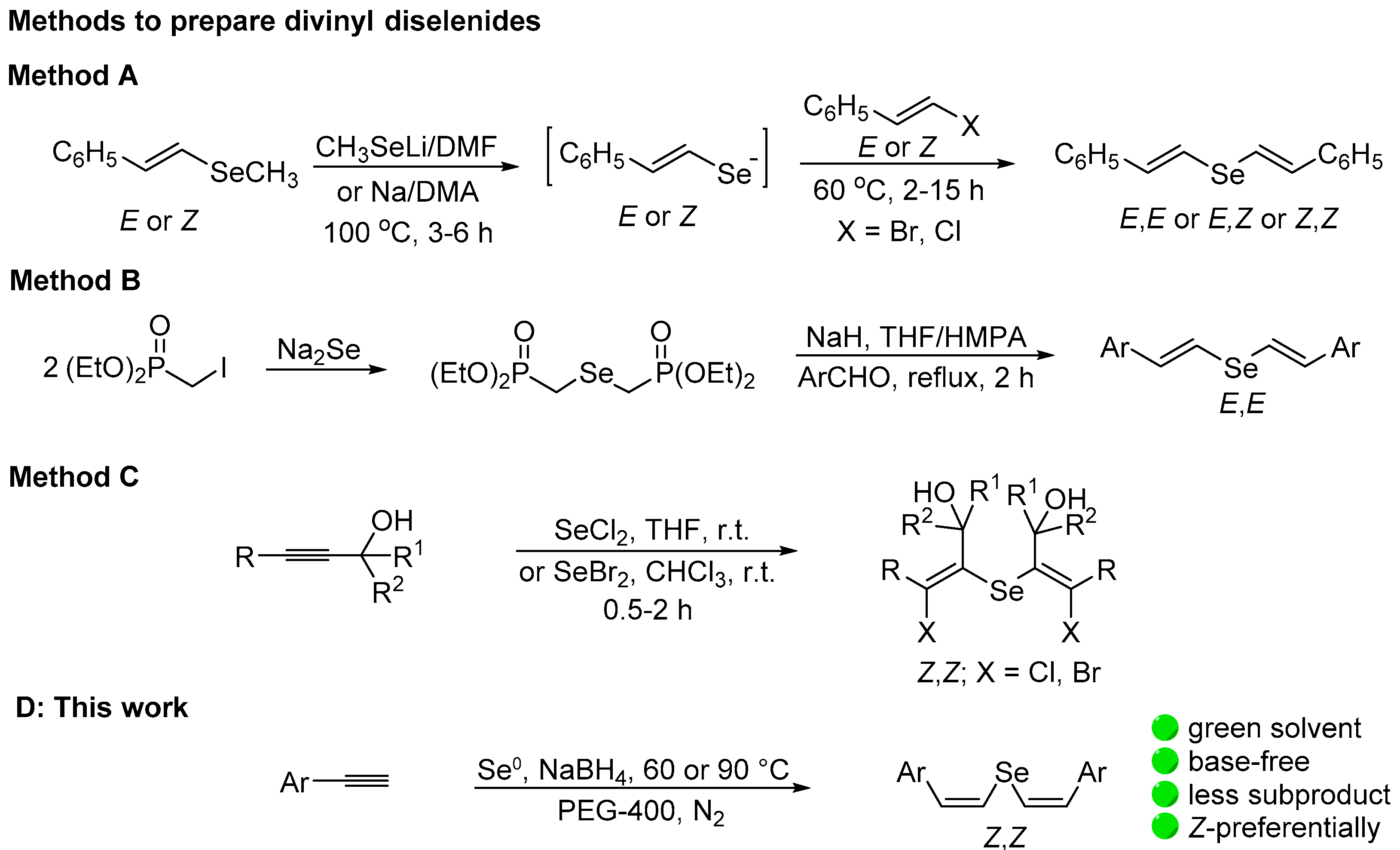
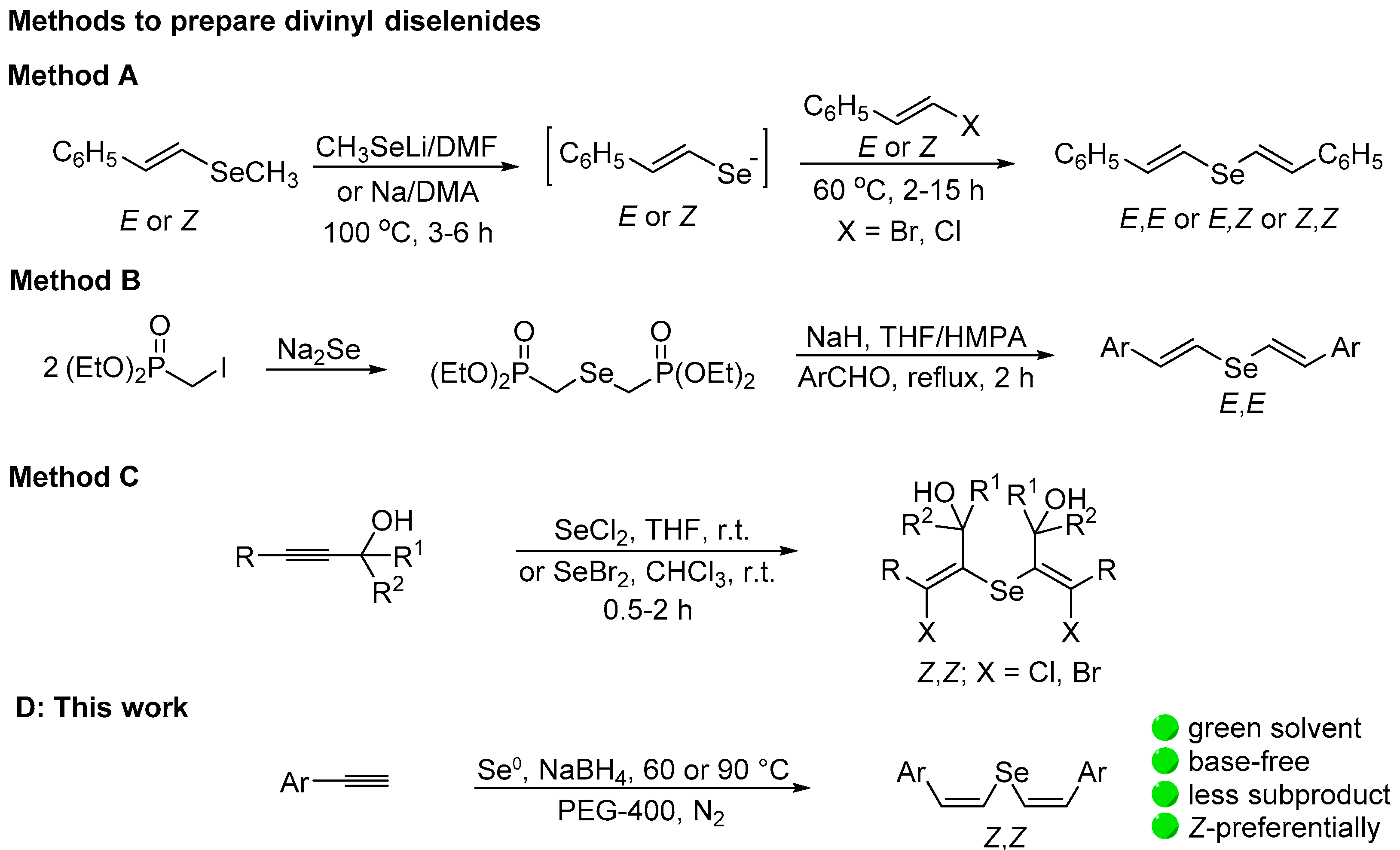
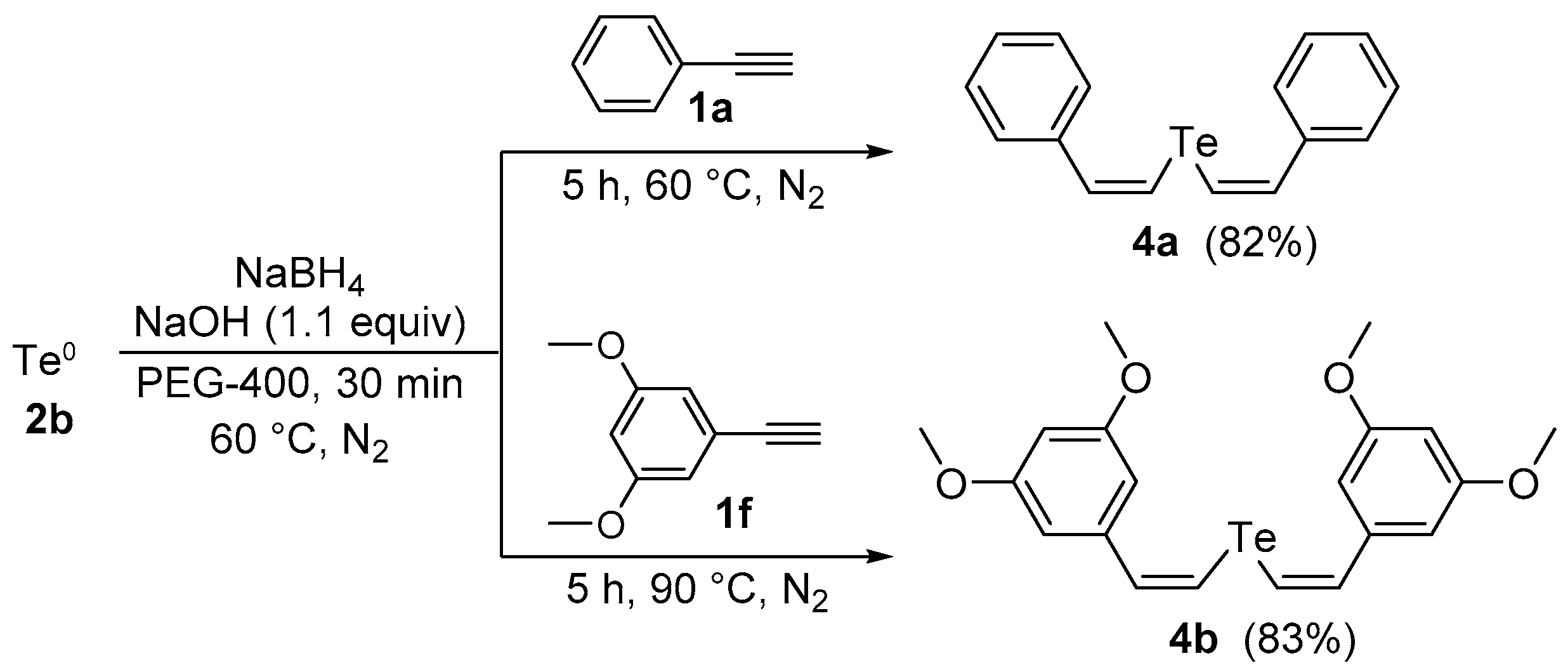
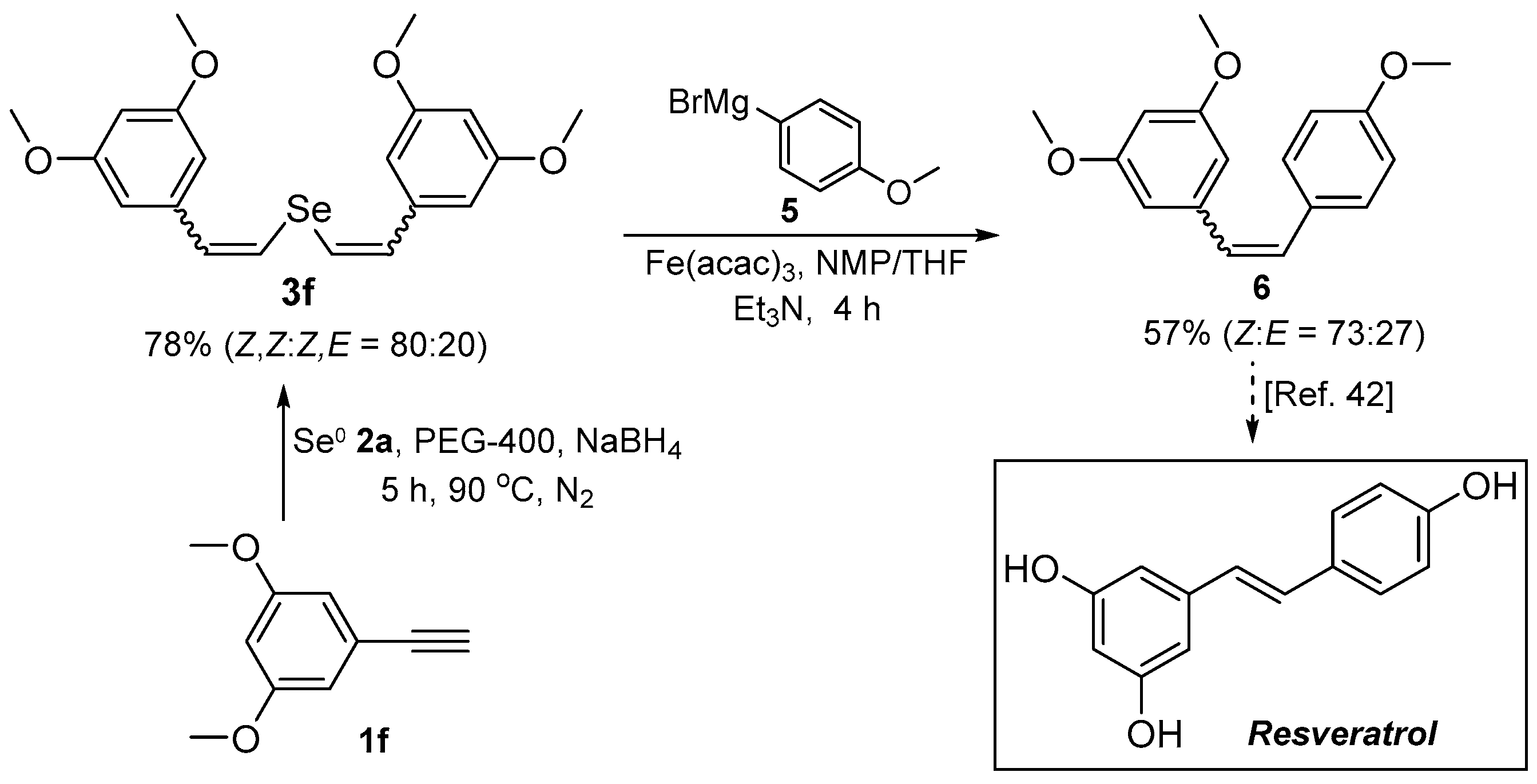
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. General Procedure for the Preparation of Divinyl Selenides 3a–f
3.3. General Procedure for the Preparation of Divinyl Tellurides 4a,b
3.4. Preparation of 3,4’,5-Trimethoxystilbene 6
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zeni, G.; Lüdtke, D.S.; Panatieri, R.B.; Braga, A.L. Vinylic Tellurides: From Preparation to Their Applicability in Organic Synthesis. Chem. Rev. 2006, 106, 1032–1076. [Google Scholar] [CrossRef] [PubMed]
- Perin, G.; Lenardão, E.J.; Jacob, R.G.; Panatieri, R.B. Synthesis of Vinyl Selenides. Chem Rev. 2009, 109, 1277–1301. [Google Scholar] [CrossRef] [PubMed]
- Menezes, P.H.; Zeni, G. Vinyl Selenides in Patai’s Chemistry of Functional Groups; John Wiley & Sons: New York, NY, USA, 2011. [Google Scholar]
- Alonso, F.; Beletskaya, I.P.; Yus, M. Transition-Metal-Catalyzed Addition of Heteroatom-Hydrogen Bonds to Alkynes. Chem. Rev. 2004, 104, 3079–3159. [Google Scholar] [CrossRef] [PubMed]
- Freudendahl, D.M.; Shahzad, S.A.; Wirth, T. Recent Advances in Organoselenium Chemistry. Eur. J. Org. Chem. 2009, 2009, 1649–1664. [Google Scholar] [CrossRef]
- Silveira, C.C.; Mendes, S.R.; Wolf, L. Iron-Catalyzed Coupling Reactions of Vinylic Chalcogenides with Grignard Reagents. J. Braz. Chem. Soc. 2010, 21, 2138–2145. [Google Scholar] [CrossRef]
- Silveira, C.C.; Braga, A.L.; Vieira, A.S.; Zeni, G. Stereoselective Synthesis of Enynes by Nickel-Catalyzed Cross-Coupling of Divinylic Chalcogenides with Alkynes. J. Org. Chem. 2003, 68, 662–665. [Google Scholar] [CrossRef] [PubMed]
- Uemura, S.; Takahashi, H.; Ohe, K. Styryl coupling vs. styryl acetate formation in reactions of styryl tellurides with palladium(II) salts. J. Organomet. Chem. 1992, 423, C9–Cl2. [Google Scholar] [CrossRef]
- Amosova, S.V.; Penzik, M.V.; Albanov, A.I.; Potapov, V.A. The reaction of selenium dichloride with divinyl sulfide. J. Organomet. Chem. 2009, 694, 3369–3372. [Google Scholar] [CrossRef]
- Amosova, S.V.; Penzik, M.V.; Albanov, A.I.; Potapov, V.A. Addition of selenium dibromide to divinyl sulfide: Spontaneous rearrangement of 2,6-dibromo-1,4-thiaselenane to 5-bromo-2-bromomethyl-1,3-thiaselenolane. Tetrahedron Lett. 2009, 50, 306–308. [Google Scholar] [CrossRef]
- Potapov, V.A.; Kurkutov, E.O.; Musalov, M.V.; Amosova, S.V. Reactions of selenium dichloride and dibromide with divinyl sulfone: Synthesis of novel four- and five-membered selenium heterocycles. Tetrahedron Lett. 2010, 51, 5258–5261. [Google Scholar] [CrossRef]
- Potapov, V.A.; Amosova, S.V.; Volkova, K.A.; Penzik, M.V.; Albanov, A.I. Reactions of selenium dichloride and dibromide with divinyl selenide: Synthesis of novel selenium heterocycles and rearrangement of 2,6-dihalo-1,4-diselenanes. Tetrahedron Lett. 2010, 51, 89–92. [Google Scholar] [CrossRef]
- Testaferri, L.; Tiecco, M.; Tingoli, U.; Chianelli, D. Stereospecific synthesis of divinyl selenides nucleophilic substitutions of unactivated vinyl halides by vinyl selenide anions. Tetrahedron 1986, 42, 63–69. [Google Scholar] [CrossRef]
- Comasseto, J.V.; Petragnani, N. The reaction of selenophosphonates with carbonyl compounds. Vinylic selenides. J. Organomet. Chem. 1978, 152, 295–304. [Google Scholar] [CrossRef]
- Silveira, C.C.; Santos, P.C.S.; Braga, A.L. Preparation and nickel-catalyzed coupling reactions of divinylic selenides. Tetrahedron Lett. 2002, 43, 7517–7520. [Google Scholar] [CrossRef]
- Silveira, C.C.; Rinaldi, F.; Guadagnin, R.C. Preparation and Reactivity of Chalcogenyl Phosphonates and Phosphane Oxides. Eur. J. Org. Chem. 2007, 2007, 4935–4939. [Google Scholar] [CrossRef]
- Sheldon, R.A. The E Factor: Fifteen years on. Green Chem. 2007, 9, 1273–1283. [Google Scholar] [CrossRef]
- Braverman, S.; Cherkinsky, M.; Jana, R.; Kalendar, Y.; Sprecher, M. Reaction of selenium and tellurium halides with propargyl alcohols. The regio- and stereoselectivity of addition to the triple bond. J. Phys. Org. Chem. 2010, 23, 1114–1120. [Google Scholar] [CrossRef]
- Potapov, V.A.; Khuriganova, O.I.; Musalov, M.V.; Larina, L.I.; Amosova, S.V. Stereospecific Synthesis of E,E-Bis(2-chlorovinyl)selenide. Russ. J. Gen. Chem. 2010, 80, 541–542. [Google Scholar] [CrossRef]
- Potapov, V.A.; Musalov, M.V.; Khuriganova, O.I.; Larina, L.I.; Amosova, S.V. Reactions of Stereoselective Addition of Selenium Dibromide and Monobromide to Acetylene. Russ. J. Org. Chem. 2010, 46, 753–754. [Google Scholar] [CrossRef]
- Braverman, S.; Pechenick-Azizi, T.; Gottlieb, H.E.; Sprecher, M. Synthesis and Reactivity of Divinylselenium Dichlorides and Dibromides. Synthesis 2011, 2011, 577–584. [Google Scholar] [CrossRef]
- Musalova, M.V.; Potapov, V.A.; Amosova, S.V. Synthesis of Novel E-2-Chlorovinyltellurium Compounds Based on the Stereospecific Anti-addition of Tellurium Tetrachloride to Acetylene. Molecules 2012, 17, 5770–5779. [Google Scholar] [CrossRef] [PubMed]
- Potapov, V.A.; Elokhina, V.N.; Larina, L.I.; Yaroshenko, T.I.; Tatarinova, A.A.; Amosova, S.V. Reactions of sodium selenide with ethynyl and bromoethynyl ketones: Stereo- and regioselective synthesis of functionalized divinyl selenides and 1,3-diselenetanes. J. Organomet. Chem. 2009, 694, 3679–3682. [Google Scholar] [CrossRef]
- Tucci, F.C.; Chieffi, A.; Comasseto, J.V. Tellurium in Organic Synthesis. Preparation of Z-Vinylic Cuprates from Z-Vinylic Tellurides and Their Reaction with Enones and Epoxides. J. Org. Chem. 1996, 61, 4975–4989. [Google Scholar] [CrossRef]
- Barros, S.M.; Comasseto, J.V.; Berriel, J. Vinyllithiums from butyl-vinyl tellurides and bis-vinyl tellurides. Tetrahedron Lett. 1989, 30, 7353–7356. [Google Scholar] [CrossRef]
- Barros, S.M.; Dabdoub, M.J.; Dabdoub, V.M.B.; Comasseto, J.V. Hydrotelluration of Acetylenes: Synthesis of Vinylic Tellurides, Divinyl Tellurides, and l-(Organyltelluro)-l,3-butadienes. Organometallics 1989, 8, 1661–1665. [Google Scholar] [CrossRef]
- Trofimov, B.A.; Amosova, S.V.; Gurasova, N.K.; Musorin, G.K. Reactions of triads Se8-KOH-DMSO, Se8-KOH-DMSO, Te-KOH-HMPA with acetylenes. Tetrahedron 1982, 38, 713–718. [Google Scholar] [CrossRef]
- Kerton, F.M.; Marriott, R. RSC Green Chemistry Book Series—Alternative Solvents for Green Chemistry, 2nd ed.; RSC Publishing: Cambridge, UK, 2013. [Google Scholar]
- Reichardt, C.; Welton, T. Solvents and Solvent Effects in Organic Chemistry, 4th ed.; WILEY-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Perin, G.; Alves, D.; Jacob, R.G.; Barcellos, A.M.; Soares, L.K.; Lenardão, E.J. Synthesis of Organochalcogen Compounds using Non-Conventional Reaction Media. Chem. Sel. 2016, 2, 205–258. [Google Scholar] [CrossRef]
- Perin, G.; Borges, E.L.; Alves, D. Highly stereoselective method to prepare bis-phenylchalcogen alkenes via addition of chalcogenolate to phenylseleno alkynes. Tetrahedron Lett. 2012, 53, 2066–2069. [Google Scholar] [CrossRef]
- Perin, G.; Borges, E.L.; Rosa, P.C.; Carvalho, P.N.; Lenardão, E.J. Simple cleavage of diorganyl diselenides with NaBH4/PEG-400 and direct Michael addition to electron-deficient alkenes. Tetrahedron Lett. 2013, 54, 1718–1721. [Google Scholar] [CrossRef]
- Perin, G.; Borges, E.L.; Peglow, T.J.; Lenardão, E.J. Direct Michael addition to electron-deficient alkenes using diorganyl dichalcogenides (Te/S) and NaBH4/PEG-400. Tetrahedron Lett. 2014, 55, 5652–5655. [Google Scholar] [CrossRef]
- Silva, P.C.; Borges, E.L.; Lima, D.B.; Jacob, R.G.; Lenardão, E.J.; Perin, G.; Silva, M.S. A simple and non-conventional method for the synthesis of selected β-arylalkylchalcogeno substituted alcohols, amines and carboxylic acids. ARKIVOC 2016, V, 376–389. [Google Scholar]
- Neves, A.R.; Lúcio, M.; Lima, J.L.C.; Reis, S. Resveratrol in Medicinal Chemistry: A Critical Review of its Pharmacokinetics, Drug-Delivery, and Membrane Interactions. Curr. Med. Chem. 2012, 19, 1663–1681. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.-L.; Chong, I.-W.; Lee, Y.-C.; Tsai, J.-R.; Wang, H.-M.; Hsieh, C.-C.; Kuo, H.-F.; Liu, W.-L.; Chen, Y.-H.; Chen, H.-L. Anti-inflammatory Effects of Resveratrol on Hypoxia/Reoxygenation-Induced Alveolar Epithelial Cell Dysfunction. J. Agric. Food Chem. 2015, 63, 9480–9487. [Google Scholar] [CrossRef] [PubMed]
- Hong, T.; Jiang, W.; Dong, H.-M.; Qiu, S.-X.; Lu, Y. Synthesis and cytotoxic activities of E-resveratrol derivatives. Chin. J. Nat. Med. 2015, 13, 375–382. [Google Scholar] [CrossRef]
- Park, S.; Cha, S.-H.; Cho, I.; Park, S.; Park, Y.; Cho, S.; Park, Y. Antibacterial nanocarriers of resveratrol with gold and silver nanoparticles. Mater. Sci. Eng. C 2016, 58, 1160–1169. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.-H.; Lee, H.; Lee, S.-R. Protective effect of resveratrol against neuronal damage following transient global cerebral ischemia in mice. J. Nutr. Biochem. 2016, 27, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Guiso, M.; Marra, C.; Farina, A. A new efficient resveratrol synthesis. Tetrahedron Lett. 2002, 43, 597–598. [Google Scholar] [CrossRef]
- Botella, L.; Nájera, C. Synthesis of methylated resveratrol and analogues by Heck reactions in organic and aqueous solvents. Tetrahedron 2004, 60, 5563–5570. [Google Scholar] [CrossRef]
- Lara-Ochoa, F.; Sandoval-Minero, L.C.; Espinosa-Pérez, G. A new synthesis of resveratrol. Tetrahedron Lett. 2015, 56, 5977–5979. [Google Scholar] [CrossRef]
- Brown, J.W.; Jarenwattananon, N.N.; Otto, T.; Wang, J.L.; Glöggler, S.; Bouchard, L.-S. Heterogeneous Heck coupling in multivariate metal–organic frameworks for enhanced selectivity. Catal. Commun. 2015, 65, 105–107. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 3, 4 and 6 are available from the authors.



| Entry | Temperature | Time (h) | Yield (%) b |
|---|---|---|---|
| 1 | r.t. | 12 | 12 |
| 2 | 40 °C | 12 | 42 |
| 3 | 60 °C | 2 | 40 |
| 4 | 60 °C | 3 | 72 |
| 5 | 60 °C | 5 | 92 |
| 6 | 80 °C | 5 | 30 |

| Entry | Alkynes 1 | Product 3 | Yield (%) b | Ratio (Z,Z/Z,E/E,E) |
|---|---|---|---|---|
| 1 |  |  | 92 | 80/20/00 c |
| 2 |  |  | 82 | 71/15/14 d |
| 3 |  |  | 77 | 77/23/00 d,e |
| 4 |  |  | 74 | 53/43/04 c |
| 5 |  |  | 93 | 100/00/00 c |
| 6 |  |  | 78 | 80/20/00 c,e |
| 7 |  |  | nr | - |
| 8 |  |  | nr | - |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perin, G.; Barcellos, A.M.; Luz, E.Q.; Borges, E.L.; Jacob, R.G.; Lenardão, E.J.; Sancineto, L.; Santi, C. Green Hydroselenation of Aryl Alkynes: Divinyl Selenides as a Precursor of Resveratrol. Molecules 2017, 22, 327. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22020327
Perin G, Barcellos AM, Luz EQ, Borges EL, Jacob RG, Lenardão EJ, Sancineto L, Santi C. Green Hydroselenation of Aryl Alkynes: Divinyl Selenides as a Precursor of Resveratrol. Molecules. 2017; 22(2):327. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22020327
Chicago/Turabian StylePerin, Gelson, Angelita M. Barcellos, Eduardo Q. Luz, Elton L. Borges, Raquel G. Jacob, Eder J. Lenardão, Luca Sancineto, and Claudio Santi. 2017. "Green Hydroselenation of Aryl Alkynes: Divinyl Selenides as a Precursor of Resveratrol" Molecules 22, no. 2: 327. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22020327