Molecular Quantum Similarity, Chemical Reactivity and Database Screening of 3D Pharmacophores of the Protein Kinases A, B and G from Mycobacterium tuberculosis


Abstract
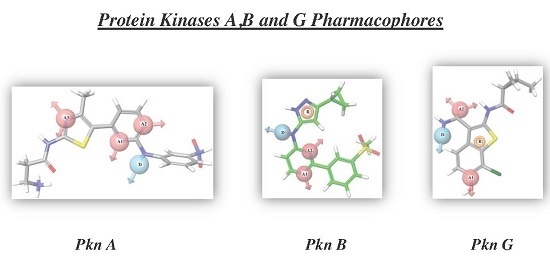
:
1. Introduction
2. Results
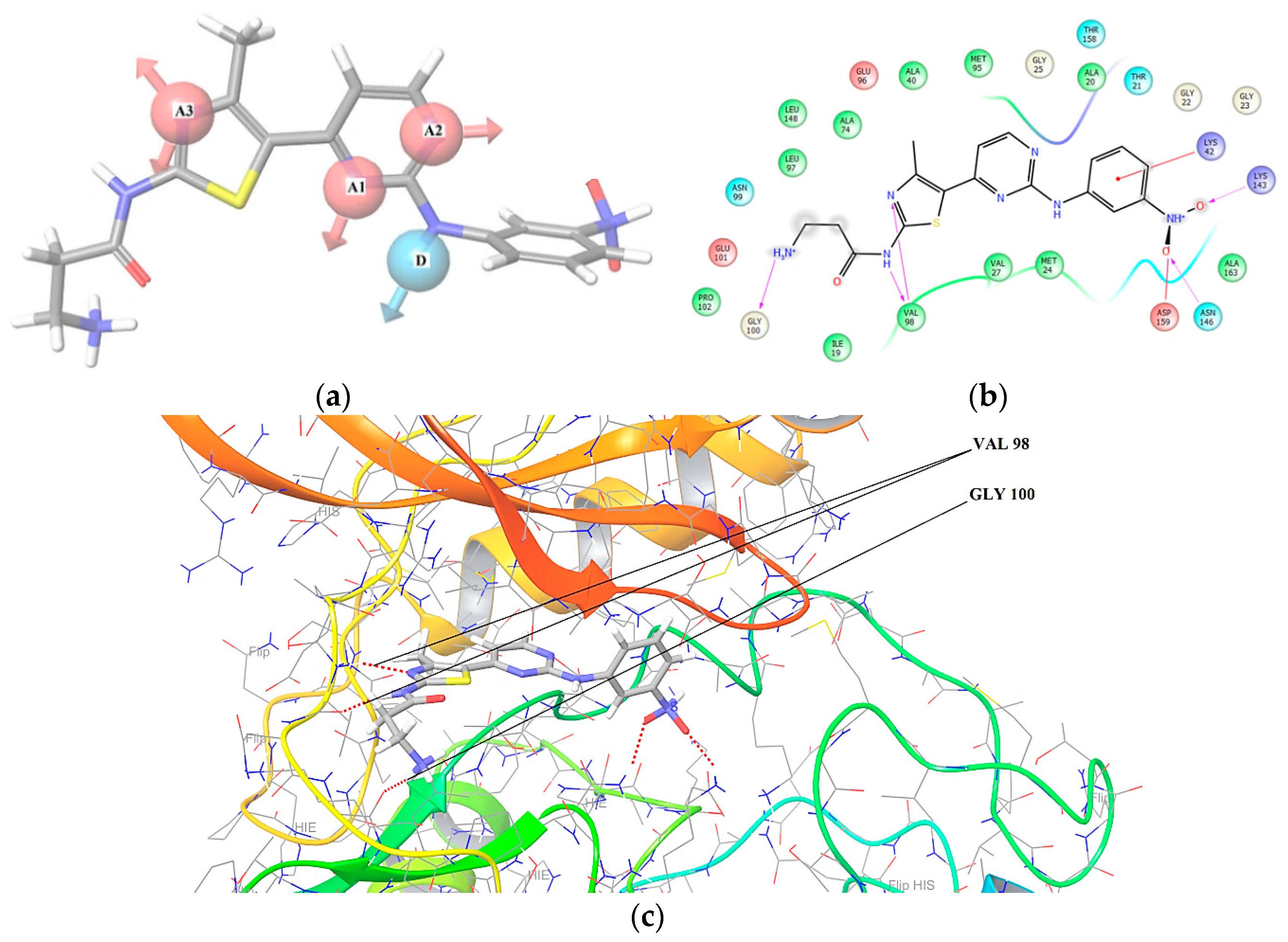
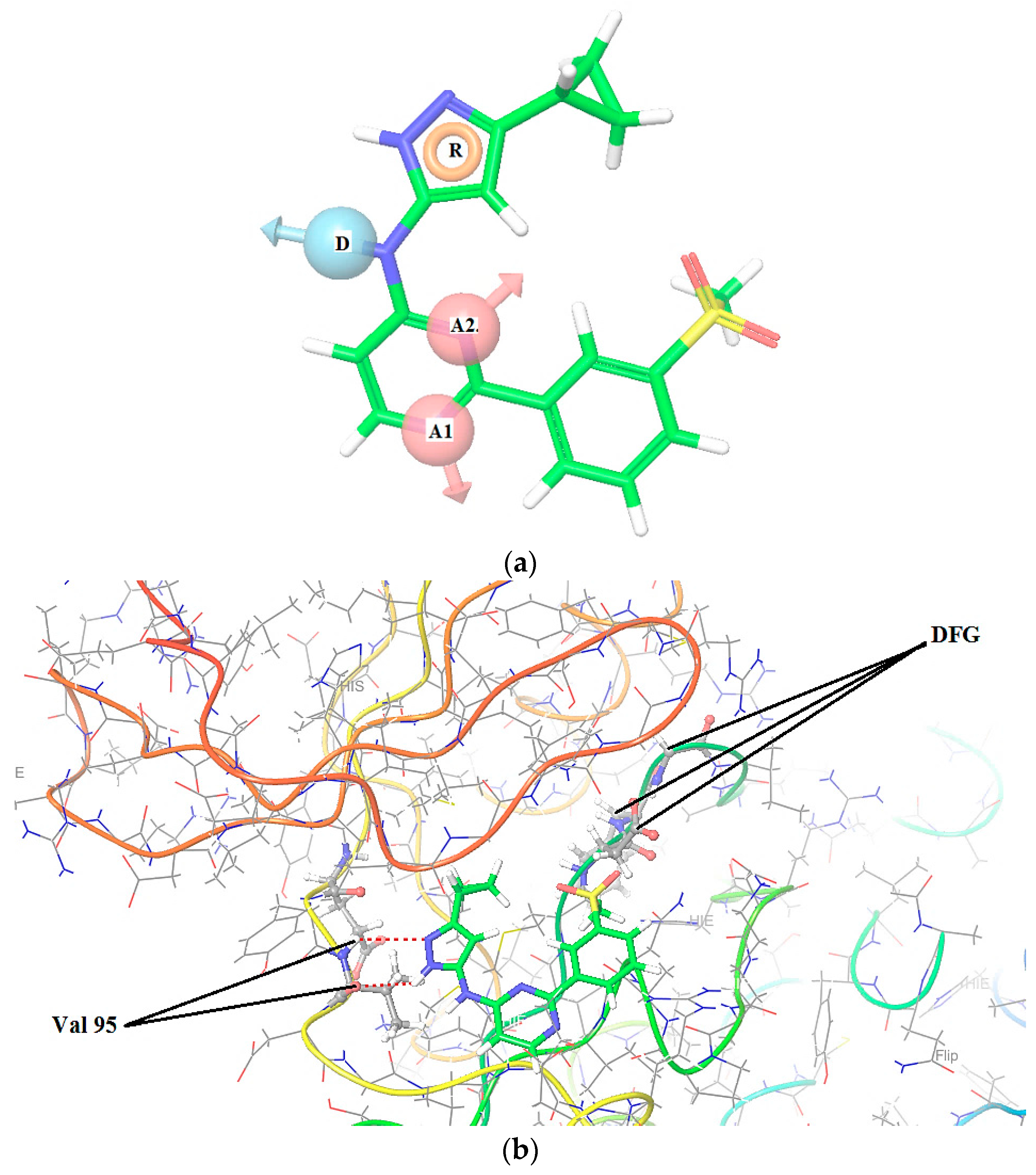
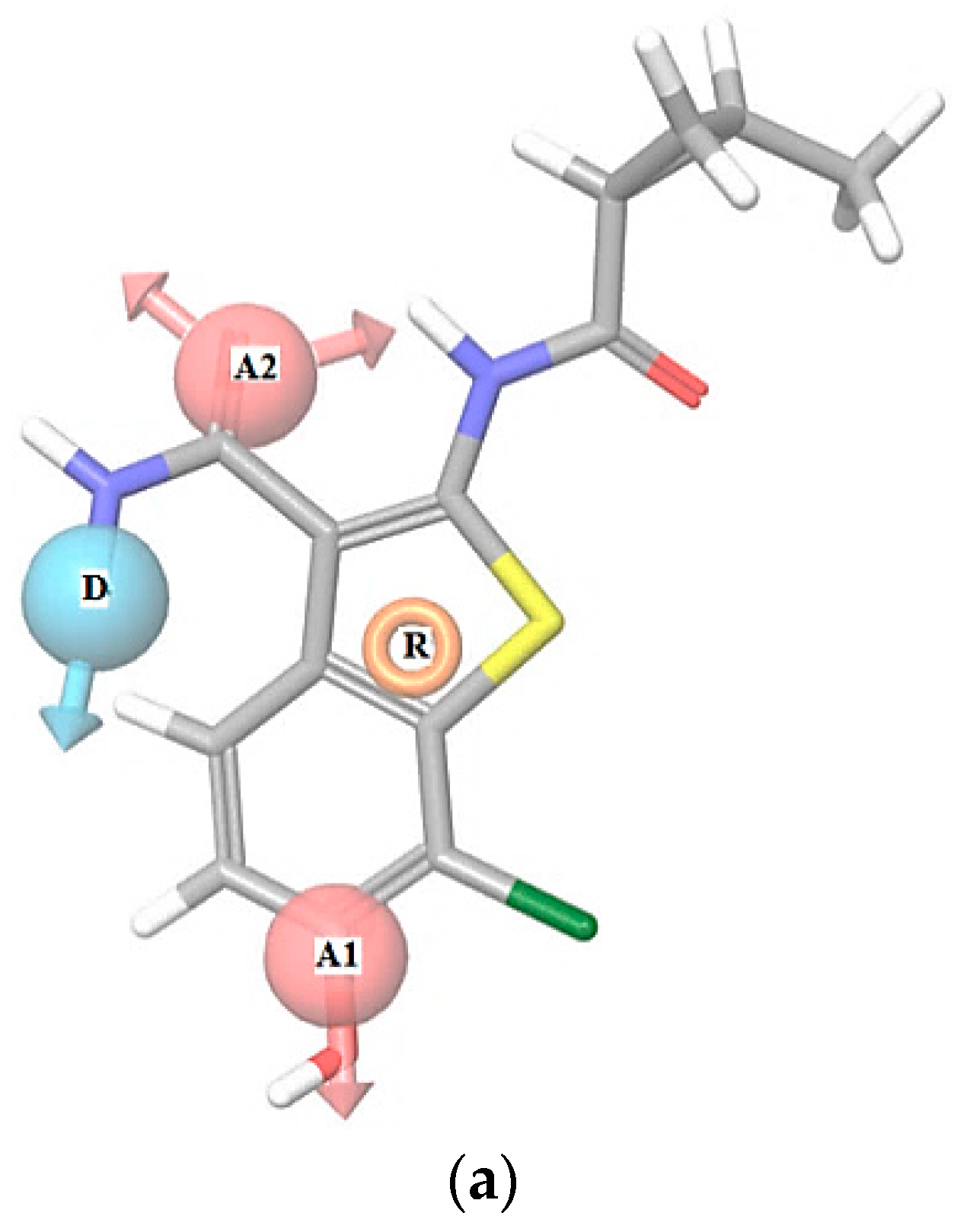
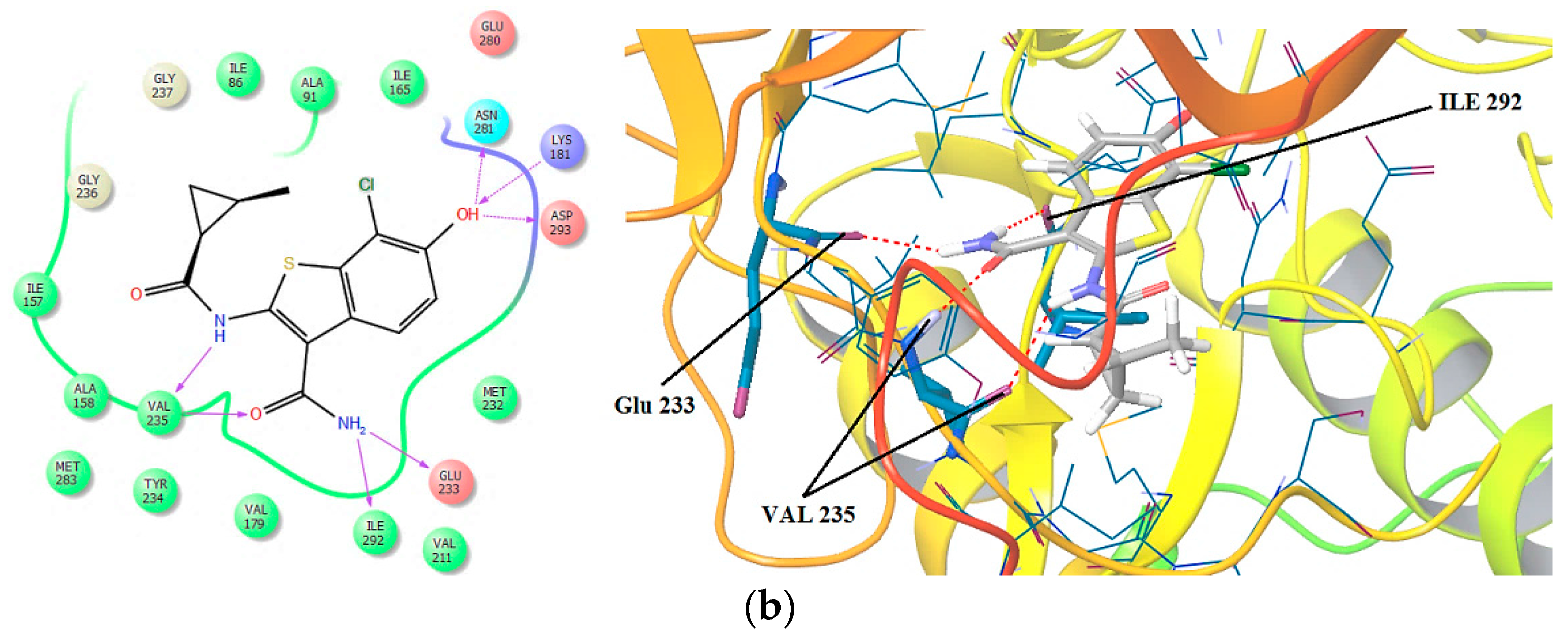
2.1. 3D Pharmacophore Searching: Mechanic Molecular Approach
2.2. Selectivity Analysis: Quantum Chemistry Approach
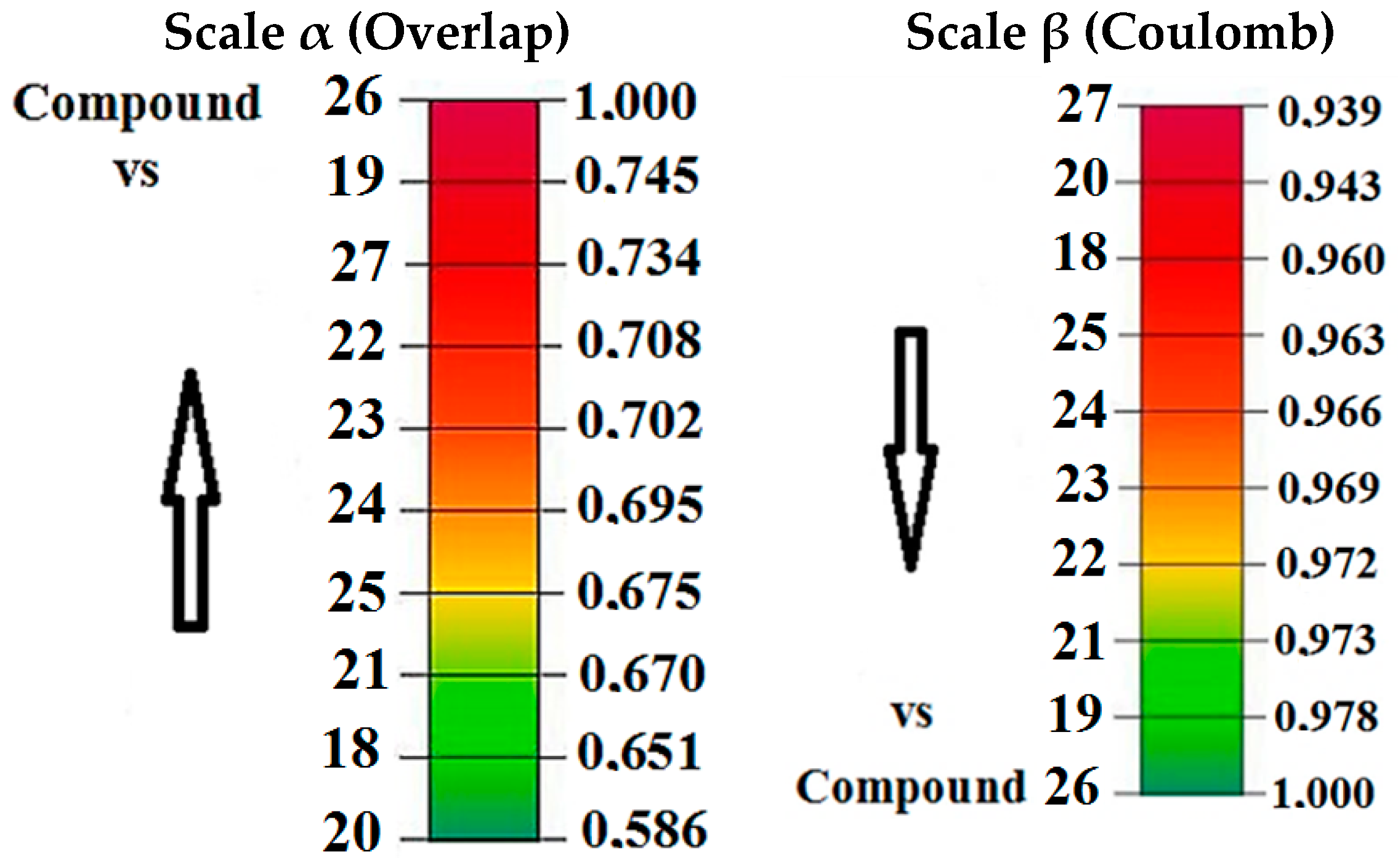
2.2.1. Molecular Quantum Similarity Study
2.2.2. Chemistry Reactivity Study
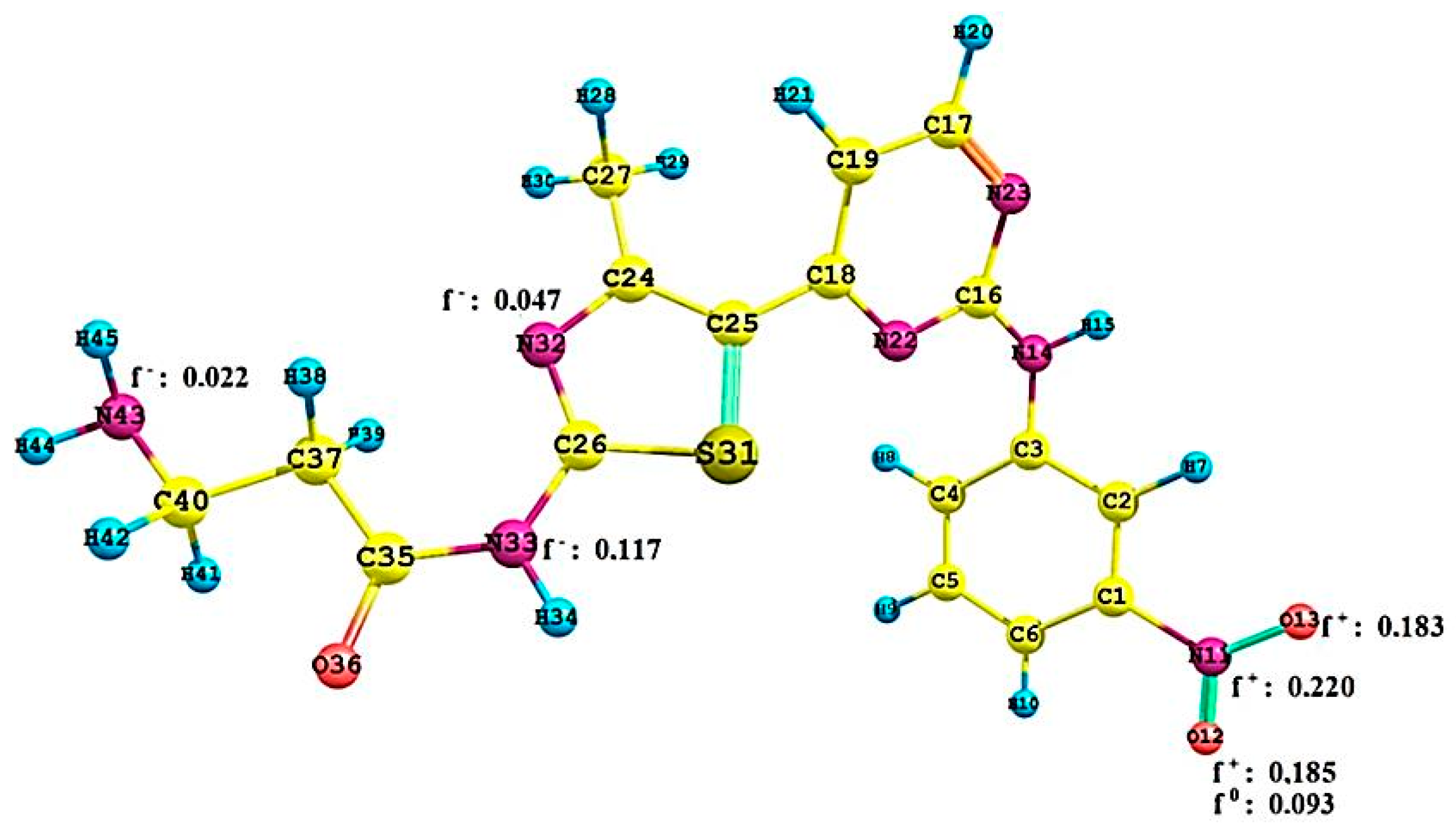
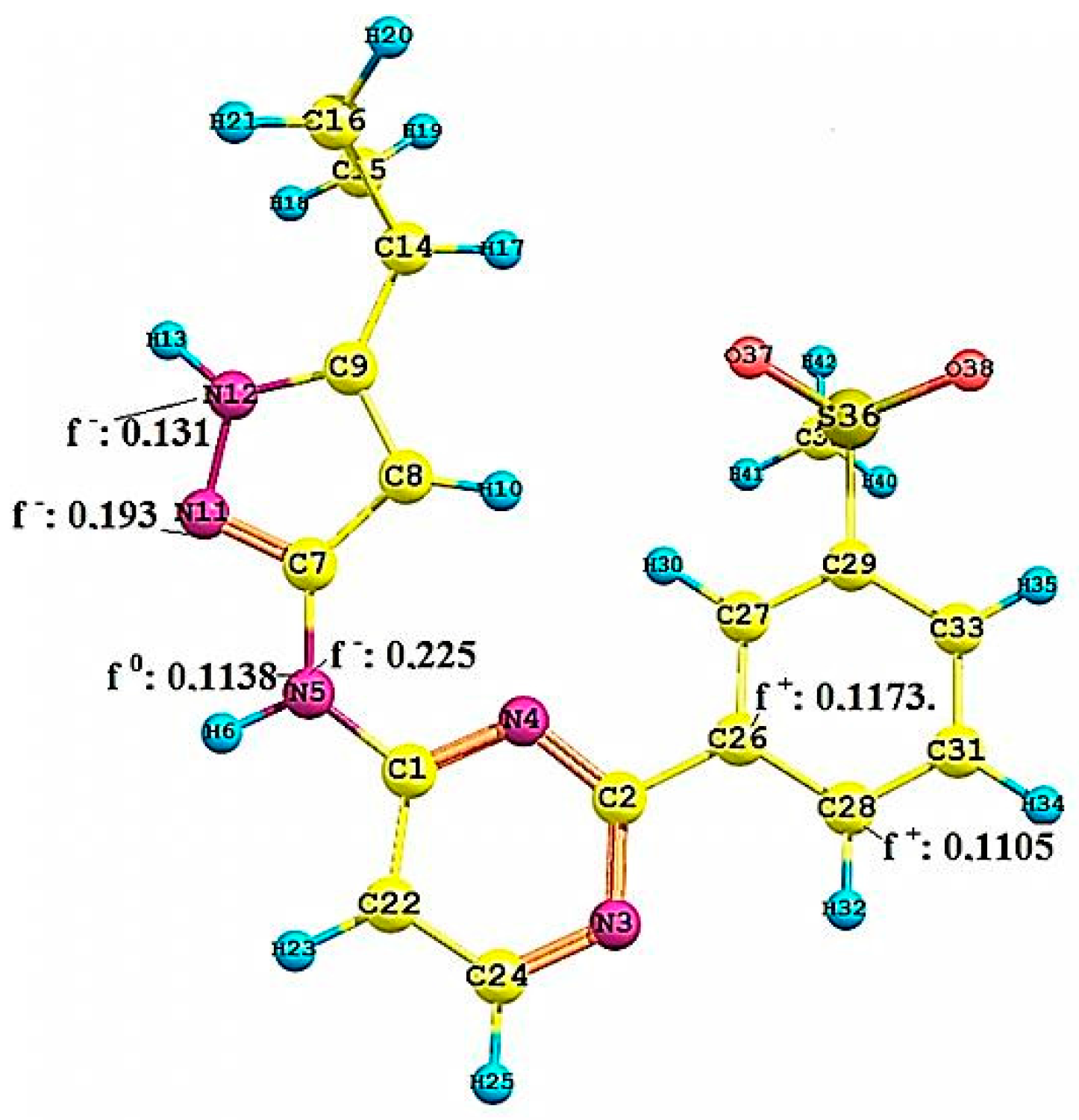
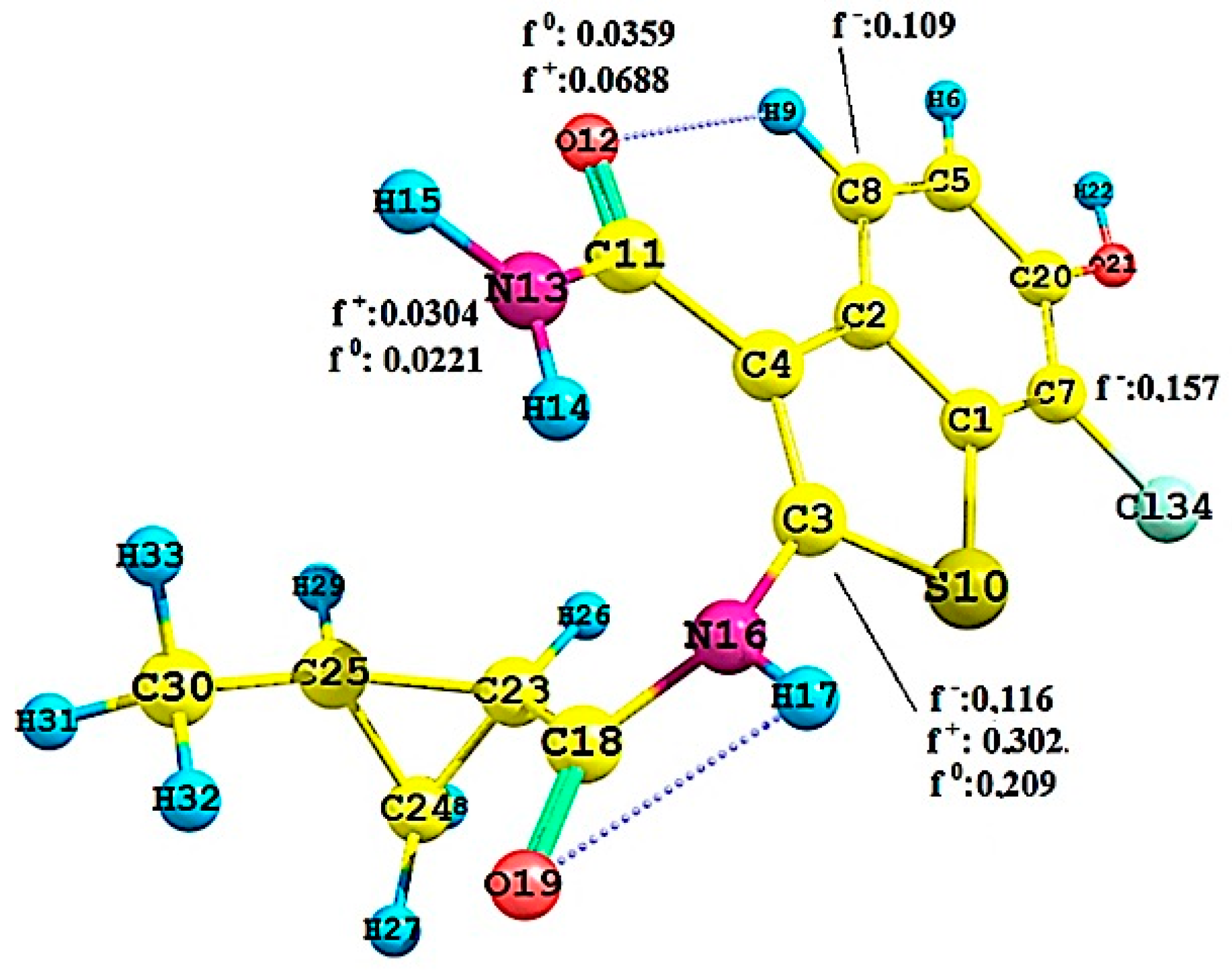
2.2.3. Local Reactivity Study
2.3. 3D Pharmacophore-Based Database Screening on the Chibale’s Database (CD)
3. Materials and Methods
3.1. System Preparation to the Molecular Mechanic (MM) Approach
3.2. Docking Studies and Pharmacophore Research
4. Theoretical and Computational Details to the Quantum Chemistry Approach
4.1. Molecular Quantum Similarity: Steric and Electronic Effects Study
4.2. Molecular Alignment
4.3. Chemical Reactivity Descriptors: Selectivity Study
4.4. Creating the 3D Chibale’s Database: Database Screening
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization (WHO). Health Topics (Tuberculosis). Available online: http://www.who.int/campaigns/tb-day/2017/event/en/ (accessed on 24 March 2017).
- Cully, M. Redesigned antibiotic combats drug-resistant tuberculosis. Nat. Rev. Drug Discov. 2014, 13, 256. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.E.; Hurdle, J.G.; Liu, J.; Bruhn, D.F.; Matt, T.; Scherman, M.S.; Vaddady, P.K.; Zheng, Z.; Qi, J.; Akbergenov, R.; et al. Spectinamides: A new class of semisynthetic antituberculosis agents that overcome native drug efflux. Nat. Med. 2014, 20, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Nahid, P.; Cole, S.T. Advances in the development of new tuberculosis drugs and treatment regimens. Nat. Rev. Drug Discov. 2013, 12, 388–404. [Google Scholar] [CrossRef] [PubMed]
- Smaill, F.; Jeyanathan, M.; Smieja, M.; Medina, M.F.; Zganiacz, N.T.A.; Yin, C.; Heriazon, A.; Damjanovic, D.; Puri, L.; Hamid, J.; et al. A Human Type 5 Adenovirus–Based Tuberculosis Vaccine Induces Robust T Cell Responses in Humans Despite Preexisting Anti-Adenovirus Immunity. Sci. Transl. Med. 2013, 5, 205ra134. [Google Scholar] [CrossRef] [PubMed]
- Raviglione, M.C.; Smith, I.M. XDR Tuberculosis—Implications for Global Public Health. N. Engl. J. Med. 2007, 356, 656–659. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.P.S.; Lakshminarayana, S.B.; Kondreddi, R.R.; Herve, M.; Camacho, L.R.; Bifani, P.; Kalapala, S.K.; Jiricek, J.; Ma, N.L.; Tan, B.H.; et al. Indolcarboxamide is a preclinical candidate for treating multidrug-resistant tuberculosis. Sci. Transl. Med. 2013, 5, 214ra168. [Google Scholar] [CrossRef] [PubMed]
- Mills, H.L.; Cohen, T.; Colijn, C. Community-wide isoniazid preventive therapy drives drug-resistant tuberculosis: A model-based analysis. Sci. Transl. Med. 2013, 5, 180ra49. [Google Scholar]
- Migliori, G.B.; Lange, C.; Centis, R.; Sotgiu, G.; Hoffmann, R.M.H.; Kliiman, K.; de laco, G.; Lauria, F.N.; Richardson, M.D.; Spanevello, A.; et al. Resistance to second-line injectables and treatment outcomes in multidrug-resistant and extensively drug-resistant tuberculosis cases. Eur. Resp. J. 2008, 31, 1155–1159. [Google Scholar] [CrossRef] [PubMed]
- Dye, C.; Williams, B.G.; Espinal, M.A.; Raviglione, M.C. Erasing the world’s slow stain: Strategies to beat multidrug-resistant tuberculosis. Science 2002, 295, 2042–2046. [Google Scholar] [CrossRef] [PubMed]
- Hallows, K.R.; Alzamora, R.; Li, H. AMP-activated protein kinase inhibits alkaline pH- and PKA-induced apical vacuolar H+-ATPase accumulation in epididymal clear cells. Am. J. Physiol. Cell Physiol. 2009, 296, C672–C681. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Haapasalo, A.; Soininen, H.; Hiltunen, M. AMP-activated protein kinase: A potential player in Alzheimer’s disease. J. Neurochem. 2011, 118, 460–474. [Google Scholar] [CrossRef] [PubMed]
- Kotlyarov, A.; Neininger, A.; Schubert, C.; Eckert, R.; Birchmeier, C.; Volk, H.D.; Gaestel, M. MAPKAP kinase 2 is essential for LPS-induced TNF-αbiosynthesis. Nat. Cell. Biol. 1999, 1, 94–97. [Google Scholar] [PubMed]
- Sipos, A.; Pató, J.; Székely, R.; Hartkoorn, R.C.; Kékesi, L.; Őrfi, L.; Szántai-Kis, C.; Mikušová, K.; Svetlíková, Z.; Korduláková, J.; et al. Lead selection and characterization of antitubercular compounds using the Nested Chemical Library. Tuberculosis 2015, 95, S200–S206. [Google Scholar] [CrossRef] [PubMed]
- Székely, R.; Wáczek, F.; Szabadkai, I.; Németh, G.; Hegymegi-Barakonyi, B.; Erős, D.; Szokol, B.; Pató, J.; Hafenbradl, D.; Satchell, J.; et al. A novel drug discovery concept for tuberculosis: Inhibition of bacterial and host cell signalling. Immunol. Lett. 2008, 116, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Lougheed, K.E.A.; Osborne, S.A.; Saxty, B.; Whalley, D.; Chapman, T.; Bouloc, N.; Chugh, J.; Nott, T.J.; Patel, D.; Spivey, V.L.; et al. Effective inhibitors of the essential kinase PknB and their potential as anti-mycobacterial agents. Tuberculosis 2011, 91, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Chapman, T.M.; Bouloc, N.; Buxton, R.S.; Chugh, J.; Lougheed, K.E.A.; Osborne, S.A.; Saxty, B.; Smerdon, S.J.; Taylor, D.L.; Whalley, D. Substituted aminopyrimidine protein kinase B (PknB) inhibitors show activity against Mycobacterium tuberculosis. Bioorg. Med. Chem. Lett. 2012, 22, 3349–3353. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, A.; Malasoni, R.; Srivastava, A.; Pandey, R.R.; Dwivedi, A.K. Design, synthesis and molecular docking of substituted 3-hydrazinyl-3-oxo-propanamides as anti-tubercular agents. Bioorg. Med. Chem. Lett. 2014, 24, 5181–5184. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Goldman, B.B.; Proteins, W.T.W. QSD quadratic shape descriptors. 2. Molecular docking using quadratic shape descriptors (QSDock). Proteins Struct. Funct. Bioinform. 2000, 38, 79–94. [Google Scholar] [CrossRef]
- Meng, E.C.; Shoichet, B.K.; Kuntz, I.D. Automated docking with grid-based energy evalution. J. Comp. Chem. 2004, 13, 505–524. [Google Scholar] [CrossRef]
- Carbó-Dorca, R. Collective Euclidian distances and quantum similarity. J. Math. Chem. 2013, 51, 338–353. [Google Scholar] [CrossRef]
- Lipkowitz, K.B.; Larter, R.; Cundari, T.R. Biomolecular applications of Poisson-Boltzmann methods. Revis. Comput. Chem. 2005, 21, 1–125. [Google Scholar]
- Gironés, X.; Carbó-Dorca, R. Modelling Toxicity using Molecular Quantum Similarity Measures. QSAR Comb. Sci. 2006, 25, 579–589. [Google Scholar] [CrossRef]
- Carbó-Dorca, R.; Gironés, X. Foundation of Quantum Similarity Measures and Their Relationship to QSPR: Density Function Structure, Approximations, and Application Examples. Int. J. Quant. Chem. 2005, 101, 8–20. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Compounds; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Morales-Bayuelo, A. Analyzing the substitution effect on the CoMFA results within the framework of density functional theory (DFT). J. Mol. Mod. 2016, 22, 164. [Google Scholar] [CrossRef] [PubMed]
- Hans, R.H.; Wiid, I.J.F.; van Helden, P.D.; Wanc, B.; Franzblau, S.G.; Gut, J.; Rosenthal, P.J.; Chibale, K. Novel thiolactone–isatin hybrids as potential antimalarial and antitubercular agents. Bioorg. Med. Chem. Lett. 2011, 21, 2055–2058. [Google Scholar] [CrossRef] [PubMed]
- Hans, R.H.; Guantai, E.M.; Lategan, C.; Smith, P.J.; Wanc, B.; Franzblau, S.G.; Gut, J.; Rosenthal, P.J.; Chibale, K. Synthesis, antimalarial and antitubercular activity of acetylenic chalcones. Bioorg. Med. Chem. Lett. 2010, 20, 942–944. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Kumar, M.; Pavadai, E.; Naran, K.; Warner, D.F.; Ruminski, P.G.; Chibale, K. Synthesis of new verapamil analogues and their evaluation in combination with rifampicin against Mycobacterium tuberculosis and molecular docking studies in the binding site of efflux protein Rv1258c. Bioorg. Med. Chem. Lett. 2014, 24, 2985–2990. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Singh, K.; Wan, B.; Franzblau, S.; Chibale, K.; Balzarini, J. Facile transformation of Biginelli pyrimidin-2(1H)-ones to pyrimidines. In vitro evaluation as inhibitors of Mycobacterium tuberculosis and modulators of cytostatic activity. Eur. J. Med. Chem. 2011, 46, 2290–2294. [Google Scholar] [CrossRef] [PubMed]
- Khanye, S.D.; Wan, B.; Franzblau, S.G.; Gut, J.; Rosenthal, P.J.; Smith, G.S.; Chibale, K. Synthesis and in vitro antimalarial and antitubercular activity of gold(III) complexes containing thiosemicarbazone ligands. J. Organomet. Chem. 2011, 696, 3392–3396. [Google Scholar] [CrossRef]
- Zuccotto, F.; Ardini, E.; Casale, E.; Angiolini, M. Through the “Gatekeeper Door”: Exploiting the Active Kinase Conformation. J. Med. Chem. 2010, 53, 2681–2694. [Google Scholar] [CrossRef] [PubMed]
- Scherr, N.; Honnappa, S.; Kunz, G.; Mueller, P.; Jayachandran, R.; Winkler, F.; Pieters, J.; Steinmetz, M.O. Structural basis for the specific inhibition of protein kinase G, a virulence factor of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2014, 104, 12151–12156. [Google Scholar] [CrossRef] [PubMed]
- Lionta, E.; Spyrou, G.; Vassilatis, D. K.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Duffy, E.M. Prediction of Drug Solubility from Structure. Adv. Drug Deliv. Rev. 2002, 54, 355–366. [Google Scholar] [CrossRef]
- Duffy, E.M.; Jorgensen, W.L. Prediction of Properties from Simulations: Free Energies of Solvation in Hexadecane, Octanol, and Water. J. Am. Chem. Soc. 2000, 122, 2878–2888. [Google Scholar] [CrossRef]
- Colmenarejo, G.; Alvarez-Pedraglio, A.; Lavandera, J.L. Cheminformatic Models to Predict Binding Affinities to Human Serum Albumin. J. Med. Chem. 2001, 44, 4370–4378. [Google Scholar] [CrossRef] [PubMed]
- Luco, J.M. Prediction of the Brain–Blood Distribution of a Large Set of Drugs from Structurally Derived Descriptors Using Partial Least-Squares (PLS) Modeling. J. Chem. Inf. Comput. Sci. 1999, 39, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Kelder, J.; Grootenhuis, P.D.; Bayada, D.M.; Delbresine, L.P.; Ploemen, J.P. Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm. Res. 1999, 16, 1514–1519. [Google Scholar] [CrossRef] [PubMed]
- Ajay; Bermis, G.W.; Murkco, M.A. Designing libraries with CNS activity. J. Med. Chem. 1999, 42, 4942–4951. [Google Scholar] [CrossRef] [PubMed]
- Yazdanian, M.; Glynn, S.L.; Wright, J.L.; Hawi, A. Correlating partitioning and caco-2 cell permeability of structurally diverse small molecular weight compounds. Pharm. Res. 1998, 15, 1490–1494. [Google Scholar] [CrossRef] [PubMed]
- Irvine, J.D.; Takahashi, L.; Lockhart, K.; Cheong, J.; Tolan, J.W.; Selick, H.E.; Grove, J.R. MDCK (Madin-Darby canine kidney) cells: A tool for membrane permeability screening. Pharm. Sci. 1999, 88, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Stenberg, P.; Norinder, U.; Luthman, K.; Artursson, P. Experimental and computational screening models for the prediction of intestinal drug absorption. J. Med. Chem. 2001, 44, 1927–1937. [Google Scholar] [CrossRef] [PubMed]
- Potts, R.O.; Guy, R.H. Predicting Skin Permeability. Pharm. Res. 1992, 9, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Potts, R.O.; Guy, R.H. A Predictive Algorithm for Skin Permeability: The Effects of Molecular Size and Hydrogen Bond Activity. Pharm. Res. 1995, 12, 1628–1633. [Google Scholar] [CrossRef] [PubMed]
- Ravala, S.K.; Singh, S.; Yadav, G.S.; Kumar, S.; Karthikeyan, S.; Chakraborti, P.K. Evidence that phosphorylation of threonine in the GT motif triggers activation of Pkn A, a eukaryotic-type serine/threonine kinase from Mycobacterium tuberculosis. FEBS J. 2015, 282, 1419–1431. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Lombardía, M.; Pompeo, F.; Boitel, B.; Alzari, P.M. Crystal Structure of the Catalytic Domain of the PknB Serine/Threonine Kinase from Mycobacterium tuberculosis. J. Biol. Chem. 2003, 278, 13094–13100. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Suite 2014-1 Protein Preparation Wizard, Epik, version 2.7; Schrödinger, LLC: New York, NY, USA, 2013.
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theor. Comp. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W.J. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef] [PubMed]
- Maestro, version 9.7; Schrödinger, LLC: New York, NY, USA, 2014.
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1998, 37, 785–789. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Epik, version 2.7; Schrödinger, LLC: New York, NY, USA, 2014.
- Glide, version 6.2; Schrödinger, LLC: New York, NY, USA, 2014.
- Schrödinger Suite 2014-2 Induced Fit Docking Protocol, Glide, Version 6.3; Schrödinger, LLC: New York, NY, USA, 2014.
- Prime, version 3.6; Schrödinger, LLC: New York, NY, USA, 2014.
- GROMACS Tutorials. Available online: https://www.pdc.kth.se/software/software/GROMACS/centos7/5.1.2/index.html (accessed on 1 March 2017).
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2001, 9, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Phase 3.7 User Manual. Available online: http://helixweb.nih.gov/schrodinger-2013.3-docs/phase/phase_user_manual.pdf (accessed on 1 April 2014).
- Carbó-Dorca, R.; Besalú, E. A general survey of molecular quantum similarity. J. Mol. Struct. THEOCHEM 1998, 451, 11–23. [Google Scholar] [CrossRef]
- Gallegos, A.; Carbó-Dorca, R.; Ponec, R.; Waisser, K. Similarity approach to QSAR: Application to antimycobacterial benzoxazines. Int. J. Pharm. 2004, 269, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Carbó-Dorca, R.; Damme, S.V. A new insight on the quantum quantitative structure-properties relationships. Int. J. Quant. Chem. 2008, 108, 1721–1734. [Google Scholar] [CrossRef]
- Ferro, N.; Gallegos, A.; Bultinck, P.; Jacobsen, H.; Carbó-Dorca, R.; Reinard, T. Coulomb and Overlap Self-Similarities: A Comparative Selectivity Analysis of Structure–Function Relationships for Auxin–like Molecules. J. Chem. Inf. Mod. 2006, 46, 1751–1762. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.; Burt, C.; Graham, R. A Linear Molecular Similarity Index. Quant. Struct. Act. Relat. 1992, 11, 34–35. [Google Scholar] [CrossRef]
- Carbó-Dorca, R. Scaled Euclidian distances: A general dissimilarity index with a suitably defined geometrical foundation. J. Math. Chem. 2012, 50, 734–740. [Google Scholar] [CrossRef]
- Gironés, X.; Robert, D.; Carbó-Dorca, R. TGSA: A molecular superposition program based on topo-geometrical considerations. J. Comput. Chem. 2001, 22, 255–263. [Google Scholar] [CrossRef]
- Ayers, P.W.; Anderson, J.S.M.; Bartolotti, L.J. Perturbative perspectives on the chemical reaction prediction problem. Int J. Quantum Chem. 2005, 101, 520–534. [Google Scholar] [CrossRef]
- Geerlings, P.; de Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Pearson, R.G. Absolute Hardness: Companion Parameter to Absolute Electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Melin, J.; Aparicio, F.; Subramanian, V.; Galván, M.; Chattaraj, P.K. Is the Fukui Function a Right Descriptor of Hard–Hard Interactions? J. Phys. Chem. A 2004, 108, 2487–2491. [Google Scholar] [CrossRef]
- Chermette, H. Chemical reactivity indexes in density functional theory. J. Comp. Chem. 1999, 20, 129–154. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Maiti, B.; Sarkar, U. Philicity: A Unified Treatment of Chemical Reactivity and Selectivity. J. Phys. Chem. A 2003, 107, 4973–4975. [Google Scholar] [CrossRef]
- Liu, S.B. Conceptual density functional theory and some recent developments. Acta Physico-Chim. Sin. 2009, 25, 590–600. [Google Scholar]
- Harbola, M.K.; Chattaraj, P.K.; Parr, R.G. Aspects of the Softness and Hardness Concepts of Density-Functional Theory. Isr. J. Chem. 1991, 31, 395–402. [Google Scholar] [CrossRef]
- Pearson, R.G. Chemical Hardness, Applications from Molecules to Solids; John Wiley and Sons: Weinheim, Germany, 1997. [Google Scholar]
- Ayers, P.W. The physical basis of the hard/soft acid/base principle. Faraday Discuss. 2007, 135, 161–190. [Google Scholar] [CrossRef] [PubMed]
- Mineva, T.; Sicilia, E.; Russo, N. Density-Functional Approach to Hardness Evaluation and Its Use in the Study of the Maximum Hardness Principle. J. Am. Chem. Soc. 1998, 120, 9053–9058. [Google Scholar] [CrossRef]
- DeLuca, G.; Sicilia, E.; Russo, N.; Mineva, T. On the hardness evaluation in solvent for neutral and charged systems. J. Am. Chem. Soc. 2002, 124, 1494–1499. [Google Scholar] [CrossRef]
- Mineva, T.; Parvanov, V.; Petro, I.; Neshev, N.; Russo, N. Fukui Indices from Perturbed Kohn–Sham Orbitals and Regional Softness from Mayer Atomic Valences. J. Phys Chem. A 2001, 105, 1959–1967. [Google Scholar] [CrossRef]
- Parr, R.G.; von Szentpaly, L.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density Functional Approach to the Frontier-Electron Theory of Chemical Reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar] [CrossRef]
- Perdew, J.P.; Parr, R.G.; Levy, M.; Balduz, J.L., Jr. Density-Functional Theory for Fractional Particle Number: Derivative Discontinuities of the Energy. Phys. Rev. Lett. 1982, 49, 1691–1694. [Google Scholar] [CrossRef]
- Yang, W.T.; Zhang, Y.K.; Ayers, P.W. Degenerate Ground States and a Fractional Number of Electrons in Density and Reduced Density Matrix Functional Theory. Phys. Rev. Lett. 2000, 84, 5172–5175. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.T.; Parr, R.G.; Pucci, R. Electron density, Kohn–Sham frontier orbitals, and Fukui functions. J. Chem. Phys. 1984, 81, 2862–2863. [Google Scholar]
- Ayers, P.W.; Yang, W.T.; Bartolotti, L.J. Fukui function. In Chemical Reactivity Theory: A Density Functional View; Chattaraj, P.K., Ed.; CRC Press: Boca Raton, FL, USA, 2009; pp. 255–267. [Google Scholar]
- Ayers, P.W.; Levy, M. Perspective on “Density functional approach to the frontier-electron theory of chemical reactivity”. Theor. Chem. Acc. 2000, 103, 353–360. [Google Scholar] [CrossRef]
- Fuentealba, P.; Pérez, P.; Contreras, R. On the condensed Fukui function. J. Chem. Phys. 2000, 113, 2544–2551. [Google Scholar] [CrossRef]
- Bultnick, P.; Carbó-Dorca, R.; Langenaeker, W. Negative Fukui functions: New insights based on electronegativity equalization. J. Chem. Phys. 2003, 118, 4349–4356. [Google Scholar] [CrossRef]
- Roy, R.K.; Tajima, N.; Hirao, K.J. A Simple Model to Predict Preferable Aldol Products from Unsymmetrical Ketones Using Local Hard–Soft Acid–Base Concept. J. Phys. Chem. A 2001, 105, 2117–2124. [Google Scholar] [CrossRef]
- Ayers, P.W.; Morrison, R.C.; Roy, R.K. Variational principles for describing chemical reactions: Condensed reactivity indices. J. Chem. Phys. 2002, 116, 8731–8734. [Google Scholar] [CrossRef]
- Petersen, G.O.; Saxena, S.; Renuka, J.; Soni, V.; Yogeeswari, P.; Santos, D.S.; Bizarro, C.V.; Sriram, D. Structure-based virtual screening as a tool for the identification of novel inhibitors against Mycobacterium tuberculosis 3-dehydroquinate dehydratase. J. Mol. Graph. Model. 2015, 60, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.-X.; Qian, H.-L.; Huang, Y.-X.; Zhu, N.-Y.; Si, S.-Y.; Xie, Y. Virtual screening for the identification of novel inhibitors of Mycobacterium tuberculosis cell wall synthesis: Inhibitors targeting RmlB and RmlC. Comp. Biol. Med. 2015, 58, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hess, T.N.; Jones, V.; Zhou, J.Z.; McNeil, M.R.; McCammon, J.A. Novel inhibitors of Mycobacterium tuberculosis dTDP-6-deoxy-l-lyxo-4-hexulose reductase (RmlD) identified by virtual screening. Bioorg. Med. Chem. Lett. 2011, 21, 7064–7067. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, H.; Kumar, A.; Chandra, N.; Imran, B.M.; Arora, S.A. Effect of lipophilicity modulation on inhibition of human rhinovirus capsid binders. Bioorg. Med. Chem. Lett. 2011, 21, 6021–6035. [Google Scholar]
- Poyraz, Ö.; Saxena, S.; Schnell, R.; Yogeeswari, P.; Schneider, G.; Sriram, D. Discovery of novel inhibitors targeting the Mycobacterium tuberculosis O-acetylserine sulfhydrylase (CysK1) using virtual high-throughput screening. Bioorg. Med. Chem. Lett. 2013, 23, 1182–1186. [Google Scholar]
- Schrödinger Suite 2014-1 Command Line Tutorial: Creating and Searching 3D Databases, Epik Version 3.5; Schrödinger, LLC: New York, NY, USA, 2013.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PKn A | Survival | Site | Vector | Volume | Matches |
|---|---|---|---|---|---|
| AAAD * | 3.145 | 0.85 | 0.956 | 0.484 | 4 of 4 compounds |
| ADRR | 3.680 | 0.458 | 0.854 | 0.421 | 3 compounds |
| AADR | 3.845 | 0.569 | 0.548 | 0.401 | 3 compounds |
| PKn B | Survival | Site | Vector | Volume | Matches |
| AADR * | 4.316 | 0.93 | 0.992 | 0.627 | 9 of 13 compounds |
| AADR | 4.308 | 0.91 | 0.988 | 0.640 | 7 compounds |
| DHRR | 4.253 | 0.80 | 0.962 | 0.547 | 8 compounds |
| PKn G | Survival | Site | Vector | Volume | Matches |
| AADR * | 3.056 | 0.47 | 0.888 | 0.696 | 10 of 10 compounds |
| AADR | 2.466 | 0.46 | 0.753 | 0.257 | 10 compounds |
| AADR | 2.180 | 0.16 | 0.710 | 0.311 | 10 compounds |
| Compound | C. Potential (μ) | C. Hardness (η) | Softness (S) | Electrophilicity (ω) |
|---|---|---|---|---|
| 1 | −4.124 | 3.839 | 0.261 | 2.215 |
| 2 | −3.485 | 3.543 | 0.282 | 1.714 |
| 3 | −3.418 | 3.959 | 0.253 | 1.476 |
| 4 | −3.491 | 3.154 | 0.317 | 1.932 |
| Compound | C. Potential (μ) | C. Hardness (η) | Softness (S) | Electrophilicity (ω) |
|---|---|---|---|---|
| 5 | −3.575 | 5.655 | 0.177 | 1.129 |
| 6 | −2.965 | 4.118 | 0.243 | 1.067 |
| 7 | −3.575 | 4.089 | 0.245 | 1.563 |
| 8 | −2.621 | 5.030 | 0.199 | 0.683 |
| 9 | −2.981 | 4.351 | 0.230 | 1.021 |
| 10 | −3.531 | 4.937 | 0.202 | 1.268 |
| 11 | −3.313 | 3.764 | 0.266 | 1.458 |
| 12 | −2.769 | 5.888 | 0.170 | 0.651 |
| 13 | −2.887 | 4.630 | 0.216 | 0.900 |
| 14 | −3.494 | 4.399 | 0.227 | 1.386 |
| 15 | −3.111 | 4.705 | 0.213 | 1.028 |
| 16 | −3.549 | 3.642 | 0.275 | 1.721 |
| 17 | −3.252 | 3.428 | 0.292 | 1.543 |
| Compound | C. Potential (μ) | C. Hardness (η) | Softness (S) | Electrophilicity (ω) |
|---|---|---|---|---|
| 18 | −3.730 | 5.103 | 0.196 | 1.363 |
| 19 | −3.386 | 3.940 | 0.254 | 1.455 |
| 20 | −3.905 | 5.127 | 0.195 | 1.487 |
| 21 | −3.622 | 4.471 | 0.224 | 1.467 |
| 22 | −3.731 | 4.473 | 0.224 | 1.556 |
| 23 | −3.745 | 4.478 | 0.223 | 1.566 |
| 24 | −4.131 | 4.651 | 0.215 | 1.835 |
| 25 | −3.241 | 4.407 | 0.227 | 1.198 |
| 26 | −3.623 | 4.375 | 0.229 | 1.500 |
| 27 | −3.611 | 4.490 | 0.223 | 1.459 |
| Compound with Pkn A Affinity | R | n | QPpolz d | SASA e | FOSA f | FISA g | PISA h | WPSA i | Clog P |
|---|---|---|---|---|---|---|---|---|---|
| CB a-11 b RR c′ | H | 3 | 33.435 | 519.287 | 246.394 | 108.866 | 134.095 | 29.932 | 2.12 |
| CB-11RS ′ | H | 3 | 33.444 | 519.272 | 263.242 | 77.672 | 149.322 | 29.035 | 2.12 |
| CB-11SR * | H | 3 | 32.996 | 509.874 | 250.606 | 107.808 | 124.503 | 26.957 | 2.12 |
| CB-11SS * | H | 3 | 33.785 | 524.334 | 265.154 | 85.176 | 149.124 | 24.881 | 2.12 |
| CB-12RR ′ | Br | 3 | 35.103 | 548.141 | 246.203 | 108.886 | 86.116 | 106.937 | 3.12 |
| CB-12RS ′ | Br | 3 | 35.102 | 548.265 | 263.252 | 77.672 | 101.105 | 106.237 | 3.12 |
| CB-12SR ′ | Br | 3 | 34.852 | 533.186 | 249.293 | 105.638 | 76.888 | 101.367 | 3.12 |
| CB-12SS * | Br | 3 | 35.515 | 554.864 | 265.193 | 85.184 | 102.419 | 102.068 | 3.12 |
| CB-13RR ′ | I | 3 | 35.500 | 553.805 | 246.169 | 108.885 | 85.600 | 113.151 | 3.38 |
| CB-13RS * | I | 3 | 35.055 | 542.675 | 250.252 | 106.173 | 78.136 | 108.114 | 3.38 |
| CB-13SR ′ | I | 3 | 35.498 | 553.944 | 263.253 | 77.671 | 100.562 | 112.458 | 3.38 |
| CB-13SS * | I | 3 | 35.911 | 560.530 | 265.180 | 85.178 | 101.877 | 108.294 | 3.38 |
| CB-14RR ′ | F | 3 | 33.723 | 528.294 | 246.429 | 108.875 | 96.135 | 76.855 | 2.40 |
| CB-14RS * | F | 3 | 33.510 | 515.890 | 249.540 | 107.362 | 85.703 | 73.284 | 2.40 |
| CB-14SR ′ | F | 3 | 33.727 | 528.286 | 263.248 | 77.672 | 111.462 | 75.905 | 2.40 |
| CB-14SS * | F | 3 | 34.140 | 534.883 | 265.189 | 85.187 | 85.187 | 71.739 | 2.40 |
| CB-15RR ′ | Cl | 3 | 34.752 | 543.124 | 246.230 | 108.883 | 86.801 | 101.210 | 2.97 |
| CB-15RS * | Cl | 3 | 34.511 | 528.669 | 249.320 | 105.938 | 77.560 | 95.851 | 2.97 |
| CB-15SR ′ | Cl | 3 | 34.752 | 543.237 | 263.253 | 77.672 | 101.828 | 100.483 | 2.97 |
| CB-15SS * | Cl | 3 | 35.165 | 549.828 | 265.186 | 85.186 | 103.137 | 96.319 | 2.97 |
| CB-16RR ′ | CH3 | 3 | 35.277 | 549.512 | 331.983 | 108.869 | 78.728 | 29.933 | 2.62 |
| CB-16RS* | CH3 | 3 | 34.849 | 538.256 | 333.659 | 105.147 | 72.444 | 27.006 | 2.62 |
| CB-16SR ′ | CH3 | 3 | 35.306 | 551.436 | 351.239 | 77.670 | 93.498 | 29.029 | 2.62 |
| CB-16SS* | CH3 | 3 | 35.715 | 557.934 | 353.127 | 85.178 | 112.768 | 24.885 | 2.62 |
| CB-17RR ′ | NO2 | 3 | 35.116 | 556.139 | 245.355 | 204.840 | 76.018 | 29.927 | 2.16 |
| CB-17RS * | NO2 | 3 | 34.684 | 544.945 | 250.325 | 198.137 | 69.525 | 26.958 | 2.16 |
| CB-17SR ′ | NO2 | 3 | 35.157 | 557.839 | 263.310 | 174.709 | 90.792 | 29.029 | 2.16 |
| CB-17SS ′ | NO2 | 3 | 34.555 | 548.924 | 242.772 | 205.494 | 70.921 | 29.737 | 2.16 |
| CB-22RR ′ | H | 5 | 36.703 | 547.504 | 292.222 | 101.786 | 124.211 | 29.285 | 3.24 |
| CB-22RS* | H | 5 | 37.311 | 561.538 | 269.138 | 131.258 | 151.763 | 9.379 | 3.24 |
| CB-22SR ′ | H | 5 | 36.593 | 545.737 | 297.895 | 97.651 | 122.486 | 27.705 | 3.24 |
| CB-22SS* | H | 5 | 37.288 | 558.397 | 290.577 | 105.866 | 137.393 | 24.561 | 3.24 |
| CB-23RR ′ | Cl | 5 | 38.013 | 571.319 | 292.026 | 101.818 | 76.893 | 100.582 | 4.09 |
| CB-23RS ′ | Cl | 5 | 38.624 | 585.586 | 269.148 | 131.276 | 104.292 | 104.292 | 4.09 |
| CB-23SR * | Cl | 5 | 38.638 | 587.477 | 314.807 | 77.673 | 95.413 | 99.584 | 4.09 |
| CB-23SS * | Cl | 5 | 38.182 | 578.661 | 287.071 | 124.580 | 95.665 | 71.345 | 4.09 |
| CB-24RR ′ | F | 5 | 36.985 | 556.516 | 292.243 | 101.807 | 86.274 | 76.192 | 3.52 |
| CB-24RS ′ | F | 5 | 37.595 | 570.571 | 269.155 | 131.263 | 113.869 | 56.283 | 3.52 |
| CB-24SR ′ | F | 5 | 37.691 | 574.185 | 314.788 | 77.664 | 106.723 | 75.009 | 3.52 |
| CB-24SS * | F | 5 | 37.061 | 548.024 | 313.187 | 78.434 | 97.486 | 58.918 | 3.52 |
| CB-25RR ′ | Br | 5 | 38.384 | 576.756 | 291.976 | 101.823 | 101.823 | 106.298 | 4.24 |
| CB-25RS * | Br | 5 | 38.799 | 586.992 | 267.539 | 128.972 | 104.130 | 86.352 | 4.24 |
| CB-25SR ′ | Br | 5 | 38.987 | 592.508 | 314.806 | 77.674 | 94.692 | 105.336 | 4.24 |
| CB-25SS * | Br | 5 | 38.522 | 583.254 | 287.106 | 124.245 | 94.983 | 76.920 | 4.24 |
| CB-26RR ′ | I | 5 | 38.781 | 582.399 | 291.923 | 101.820 | 76.160 | 112.495 | 4.50 |
| CB-26RS * | I | 5 | 39.003 | 590.994 | 269.419 | 128.332 | 100.816 | 92.427 | 4.50 |
| CB-26SR ′ | I | 5 | 39.384 | 598.197 | 314.810 | 77.679 | 94.151 | 111.557 | 4.50 |
| CB-26SS * | I | 5 | 39.003 | 582.006 | 283.206 | 121.666 | 94.068 | 83.066 | 4.50 |
 |  |  | |||
| CB-61 | CB-83-94 | CB-90-98 | |||
| Compound with Pkn A Affinity | R1 | R2 | X | R3 | pIC50 |
| CB a-61 b R c ′ | CH(CH3)2 | 1,2-Dimethoxy-phenylethylamine | - | - | −2.699 |
| CB-61S ′ | CH(CH3)2 | 1,2-Dimethoxy-phenylethylamine | - | - | −2.699 |
| CB-83 | H | C2H5 | NH | H | - |
| CB-84 | C6H5 | C2H5 | NH | H | - |
| CB-85 | H | C2H5 | NH | C6H5CH2 | - |
| CB-86 | C6H5 | C2H5 | NH | C6H5CH2 | −1.805 |
| CB-88 ′ | C6H5 | C2H5 | NH | (CH2)3OH | >−2.107 |
| CB-90 | C6H5 | C2H5 | NH | n-C4H9 | −2.071 |
| CB-92 ″ | C6H5 | C2H5 | NH | 2-OHC6H4 | −2.086 |
| CB-93 ′ | C6H5 | C2H5 | NH | 4-OHC6H4 | - |
| CB-94 ″ | C6H5 | C2H5 | NH | 3-NH2C6H4 | −1.494 |
| CB-98 ′ | 3-Methoxy-4-hydroxyphenyl | H | - | - | >−2.107 |
| Compound | QPpolz d | SASA e | FOSA f | FISA g | PISA h | WPSA i |
|---|---|---|---|---|---|---|
| CB a-61 b R c ′ | 46.733 | 809.665 | 554.715 | 61.543 | 193.408 | 0.000 |
| CB-61S ′ | 46.917 | 824.012 | 572.502 | 56.988 | 194.522 | 0.000 |
| CB-83 | 18.594 | 409.242 | 196.278 | 155.093 | 57.871 | 0.000 |
| CB-84 | 27.711 | 506.534 | 191.029 | 126.786 | 188.719 | 0.000 |
| CB-85 | 31.732 | 572.272 | 279.927 | 91.991 | 200.354 | 0.000 |
| CB-86 | 41.980 | 693.239 | 280.021 | 62.464 | 350.753 | 0.000 |
| CB-88 ′ | 34.639 | 641.288 | 314.312 | 123.633 | 203.343 | 0.000 |
| CB-90 | 36.787 | 661.635 | 391.158 | 67.097 | 203.379 | 0.000 |
| CB-92 ″ | 39.959 | 668.341 | 190.827 | 95.529 | 381.985 | 0.000 |
| CB-93 ′ | 39.974 | 673.321 | 191.826 | 117.150 | 364.345 | 0.000 |
| CB-94 ″ | 40.209 | 675.980 | 191.376 | 123.448 | 361.156 | 0.000 |
| CB-98 ′ | 21.225 | 452.473 | 106.409 | 133.125 | 139.416 | 73.522 |
| Compound | Structure | pIC50 |
|---|---|---|
| 1 |  | −1.569 |
| 2 |  | −1.839 |
| 3 |  | −1.875 |
| 4 |  | −1.934 |
| Compound | Structure | pIC50 |
|---|---|---|
| 5 |  | 0.800 |
| 6 |  | 0.971 |
| 7 |  | 1.066 |
| 8 |  | 1.076 |
| 9 |  | 1.137 |
| 10 |  | −0.360 |
| 11 |  | 1.638 |
| 12 |  | −1.200 |
| 13 |  | 1.276 |
| 14 |  | 0.285 |
| 15 |  | 1.187 |
| 16 |  | −0.375 |
| 17 |  | 0.086 |

| C a | R1 | R2 | R3 | R4 | X | Bond C3-X | Bond C1-C2 | pIC50 |
|---|---|---|---|---|---|---|---|---|
| 18 | Cyclopropyl | H | H | H | C | C3-X | C1-C2 | 0.523 |
| 19 | Cyclohexyl | H | - | H | O | C3-X | C1-C2 | 0.167 |
| 20 | Cyclopropyl | H | - | H | O | C3-X | C1-C2 | 0.276 |
| 21 | Isopropyl | H | –OH | H | C | C3=X | C1=C2 | 1.523 |
| 22 | Cyclopropyl | Br | –OH | H | C | C3=X | C1=C2 | 1.699 |
| 23 | Methylcyclopropyl | Cl | –OH | H | C | C3=X | C1=C2 | 1.699 |
| 24 | Cyclopropyl | Cl | –OCH3 | Cl | C | C3=X | C1=C2 | 1.398 |
| 25 | Ethanol | H | –OH | H | C | C3=X | C1=C2 | 1.301 |
| 26 | 1,3-Benzodioxole | Cl | –OH | H | C | C3=X | C1=C2 | 2.000 |
| 27 | Methylcyclopropyl | H | –OH | H | C | C3=X | C3=X | 1.301 |
Sample Availability: Samples of the compounds are not available from the authors. |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morales-Bayuelo, A. Molecular Quantum Similarity, Chemical Reactivity and Database Screening of 3D Pharmacophores of the Protein Kinases A, B and G from Mycobacterium tuberculosis. Molecules 2017, 22, 1027. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22061027
Morales-Bayuelo A. Molecular Quantum Similarity, Chemical Reactivity and Database Screening of 3D Pharmacophores of the Protein Kinases A, B and G from Mycobacterium tuberculosis. Molecules. 2017; 22(6):1027. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22061027
Chicago/Turabian StyleMorales-Bayuelo, Alejandro. 2017. "Molecular Quantum Similarity, Chemical Reactivity and Database Screening of 3D Pharmacophores of the Protein Kinases A, B and G from Mycobacterium tuberculosis" Molecules 22, no. 6: 1027. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22061027