Regioselectivity in Reactions between Bis(2-benzothiazolyl)ketone and Vinyl Grignard Reagents: C- versus O-alkylation—Part III †



Abstract
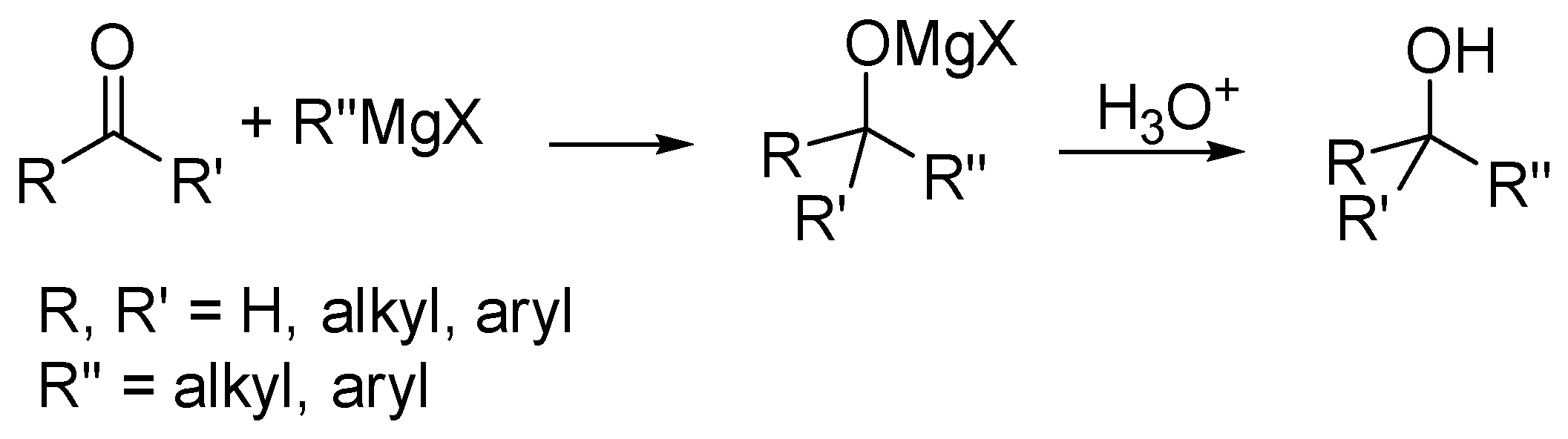
:1. Introduction
2. Results and Discussion
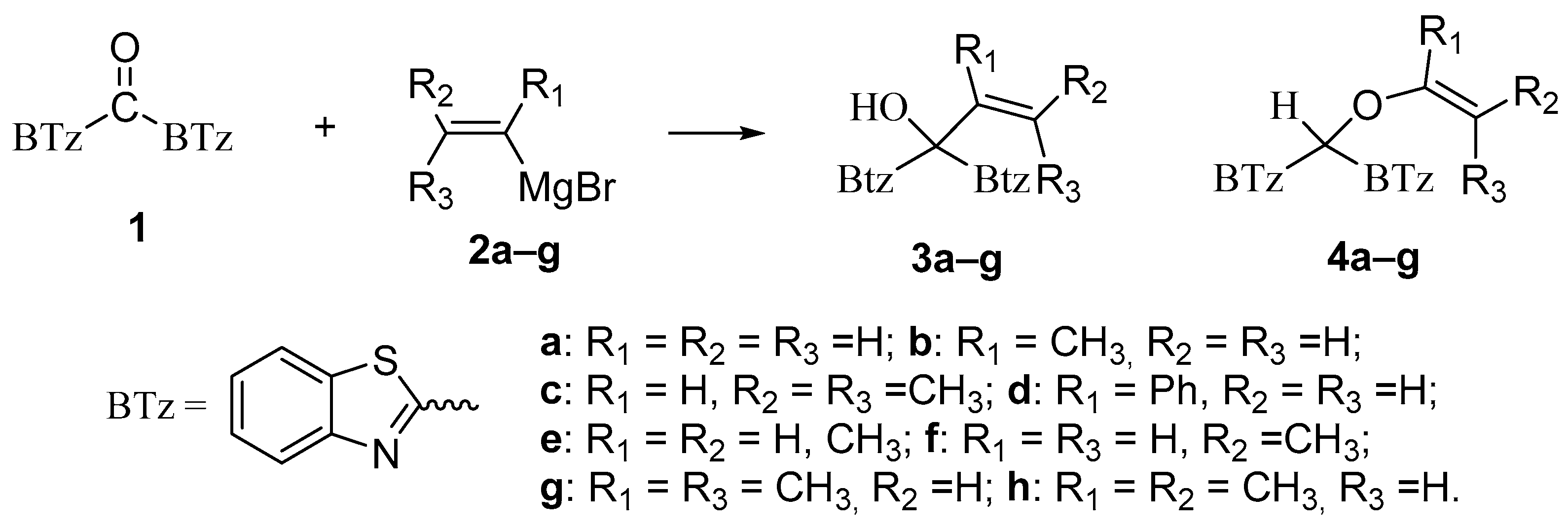
2.1. Influence of the Stereochemistry of Methyl Substituents on Vinyl Moiety
2.1.1. Methyl Substituents in β-Position
2.1.2. Effect of the Substituent in α-Position
2.1.3. Effect of the Substituent in α-Position
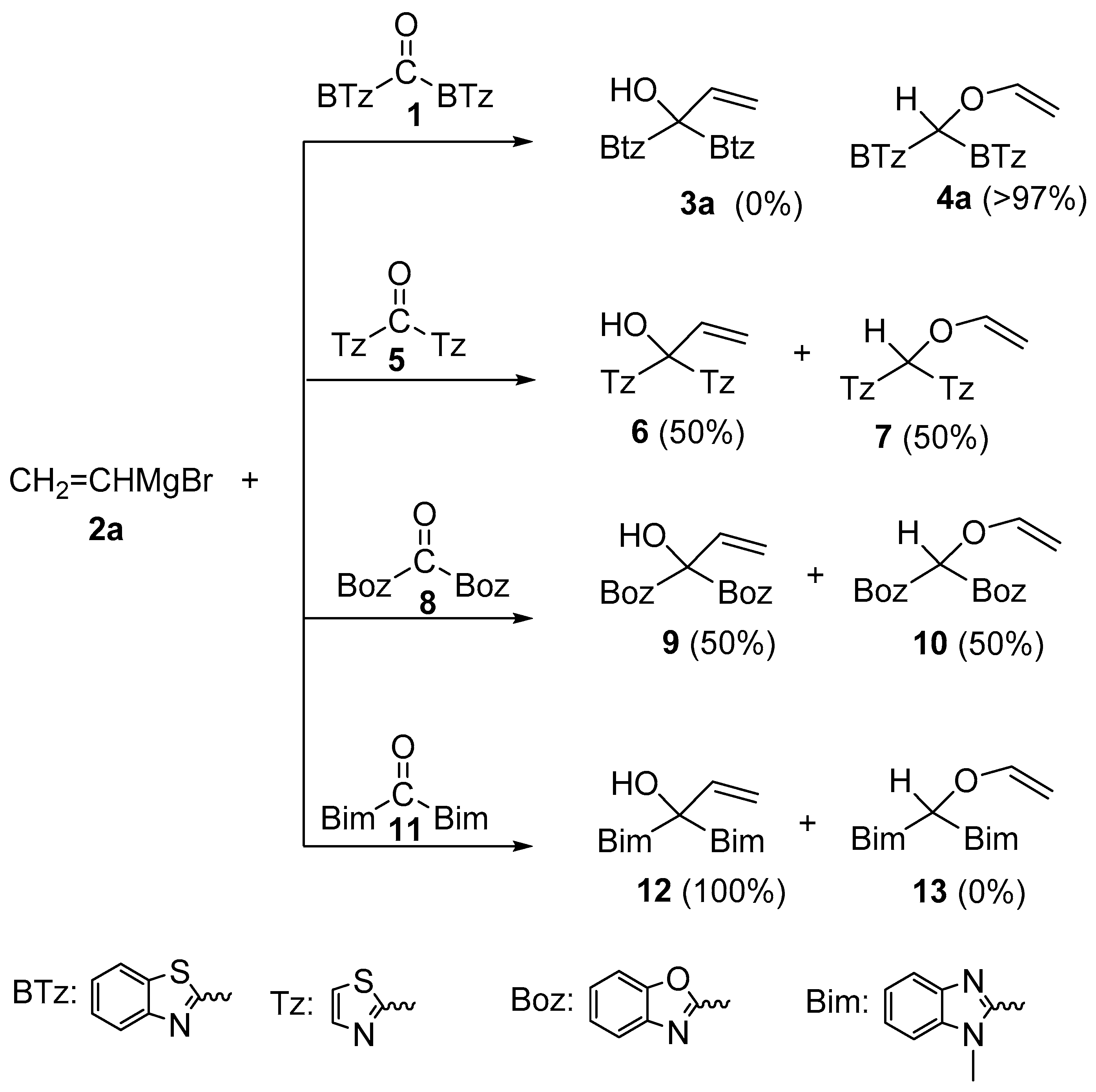
2.2. Influence of the Solvent and of the Metal Bound to Vinyl Group
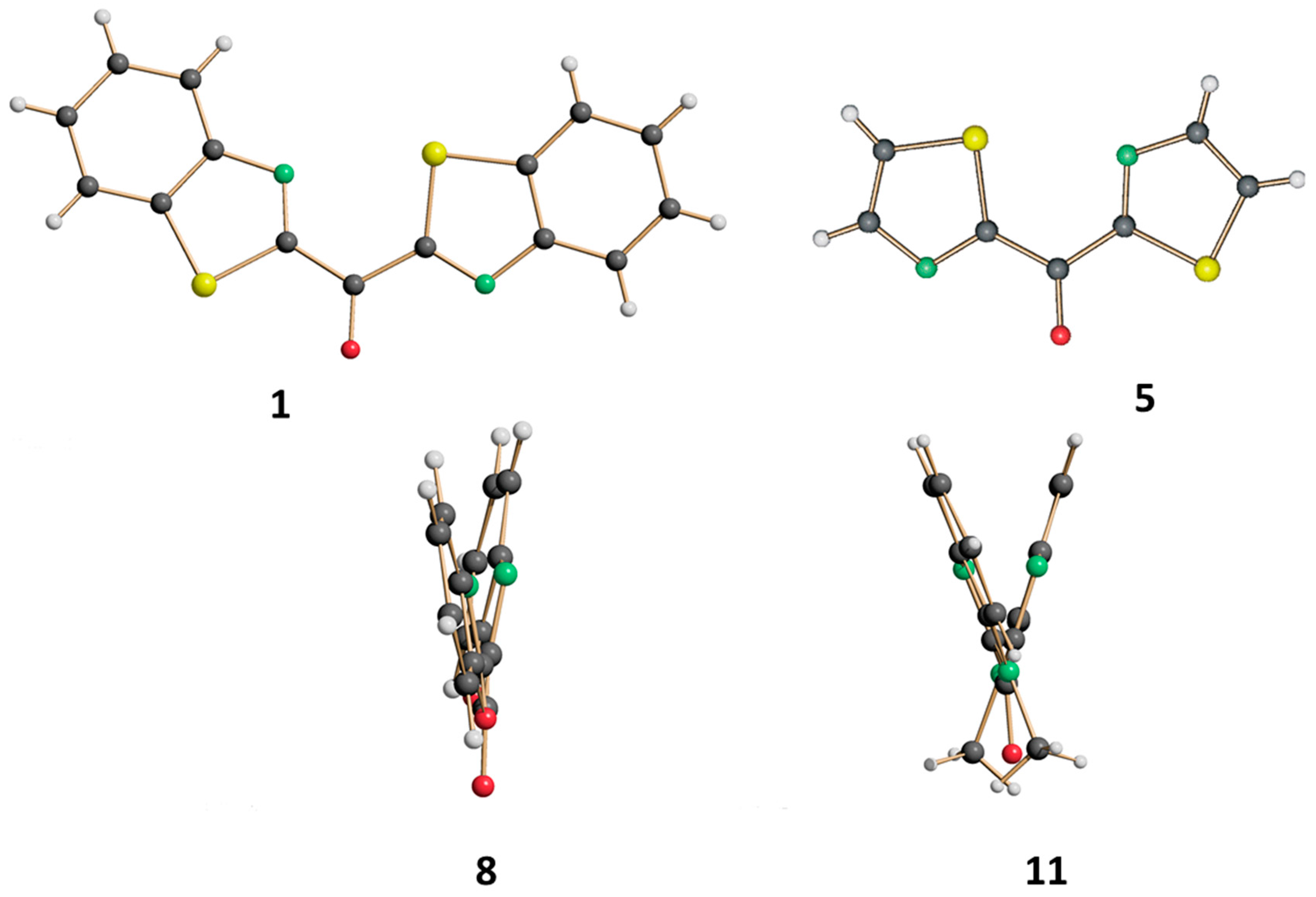
2.3. X-ray Diffraction Analysis of Azaheteroaryl Ketones Showing Different C-/O-Alkylation Regioselectivity towards Vinylmagnesium Bromide
3. Materials and Methods
3.1. General Methods
3.2. Synthesis of 1,1-Di(1,3-benzothiazol-2-yl)-2-propen-1-ol (3a)
3.3. Reactions between 1 and Vinylmagnesium Bromide Derivatives (2a–h)—General Procedure
3.4. Reactions of Propenylmagnesium Bromide with 1 or Benzophenone
3.5. X-ray Diffraction Data
Crystallographic Data Collection and Structure Determination
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Silverman, G.S.; Rakita, P.E. Handbook of Grignard Reagents Dekker; CRC Press: New York, NY, USA, 1996. [Google Scholar]
- Holm, T.; Crossland, I. Mechanistic Features of the Reactions of Organomagnesium Compounds. In Grignard Reagents-New Developments; Richey, H.G., Jr., Ed.; John Wiley and Sons: Chichester, UK, 2000. [Google Scholar]
- Laird, T. Aromatic Ketones in Comprehensive Organic Chemistry; Barton, D., Ollis, W.D., Stoddart, J.F., Eds.; Pergamon Press: Oxford, UK, 1979; Chapter 5.4; Volume 1, pp. 1174–1181. [Google Scholar]
- Blomberg, C.; Grootveld, H.H.; Gerner, T.H.; Bickelhaupt, F. Radical formation during reactions of Grignard reagents with quinones. J. Organomet. Chem. 1970, 24, 549–553. [Google Scholar] [CrossRef]
- Wessely, F.; Kotlan, J. Über die Einwirkung von metallorganischen Verbindungen auf Chinole II. Monatshefte Chem. 1953, 84, 124–133. [Google Scholar] [CrossRef]
- Miller, B. Reactions of cyclohexadienones. 37. Attack of Grignard and lithium reagents at carbonyl oxygens of o-quinol acetates. J. Org. Chem. 1977, 42, 1402–1408. [Google Scholar] [CrossRef]
- Miller, B. Reactions of cyclohexadienones. 38. Substituent effects on reactions of benzylmagnesium chlorides with o-quinol acetates. J. Org. Chem. 1977, 42, 1408–1415. [Google Scholar] [CrossRef]
- Wege, D. Abnormal addition of vinylmagnesium bromide to 9,10-phenanthraquinone. Aust. J. Chem. 1971, 24, 1531–1535. [Google Scholar] [CrossRef]
- Holm, T. The reaction of benzil with Grignard reagents. Acta Chem. Scand. Ser. B 1987, 41, 278–284. [Google Scholar] [CrossRef]
- Boga, C.; Forlani, L.; Todesco, P.E. Unexpected regioselectivity in the attack of vinyl Grignard reagents to bis(2-benzothiazolyl) ketone. Tetrahedron Lett. 1997, 38, 4845–4848. [Google Scholar] [CrossRef]
- Boga, C.; Forlani, L.; Todesco, P.E. A simple synthesis of new carbinols from bis(2-benzothiazolyl)ketone. Gazz. Chim. Ital. 1997, 127, 197–199. [Google Scholar]
- Boga, C.; Stengel, R.; Abdayem, R.; Del Vecchio, E.; Forlani, L.; Todesco, P.E. Regioselectivity in the Addition of Vinylmagnesium Bromide to Heteroarylic Ketones: C-versus O-Alkylation. J. Org. Chem. 2004, 69, 8903–8909. [Google Scholar] [CrossRef] [PubMed]
- Boga, C.; Micheletti, G. Regioselectivity in the Addition of Grignard Reagents to Bis(2-benzothiazolyl)Ketone: C- vs. O-Alkylation Using Aryl Grignard Reagents. Eur. J. Org. Chem. 2010, 2010, 5659–5665. [Google Scholar]
- Forlani, L.; Boga, C.; Del Vecchio, E.; Padovani, M. Spontaneous Oxidation of bis(heteroaryl)methanes and bis(heteroaryl)carbinols to Ketones. Arkivoc 2003, 2003, 75–91. [Google Scholar]
- Galli, C.; Guarnieri, A.; Koch, H.; Mencarelli, P.; Rappoport, Z. Effect of Substituents on the Structure of the Vinyl Radical: Calculations and Experiments. J. Org. Chem. 1997, 62, 4072–4077. [Google Scholar] [CrossRef]
- Goumans, T.P.M.; van Alem, K.; Lodder, G. Photochemical Generation and Structure of Vinyl Radicals. Eur. J. Org. Chem. 2008, 435–443. [Google Scholar] [CrossRef]
- Singer, L.A. Selective Organic Transformations; Thyagarajan, B.S., Ed.; Wiley-Interscience: New York, NY, USA, 1972; Volume 2, pp. 239–268. [Google Scholar]
- Beckwith, A.L.J.; Ingold, K.U. Rearrangements in Ground and Excited States; De Mayo, P., Ed.; Academic Press: New York, NY, USA, 1980; Volume 1, pp. 280–310. [Google Scholar]
- Simumara, O. The Stereochemistry of Cyclohexyl and Vinylic Radicals. Top. Stereochem. 1969, 4, 1–37. [Google Scholar]
- Galli, C.; Rappoport, Z. Unequivocal SRN1 Route of Vinyl Halides with a Multitude of Competing Pathways: Reactivity and Structure of the Vinyl Radical Intermediate. Acc. Chem. Res. 2003, 36, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Bonazzola, L.; Feinstein, S.; Marx, R. Electronic structure of α-substituted vinyl radicals. Mol. Phys. 1971, 22, 689–695. [Google Scholar] [CrossRef]
- Neilson, G.W.; Symons, M.C.R. Radicals formed from acetylenes by high energy radiation and hydrogen atom bombardment: An electron spin resonance study. J. Chem. Soc. Perkin Trans. 2 1973, 1405–1410. [Google Scholar] [CrossRef]
- Ashby, E.C.; Laemmle, J.; Neumann, H.M. The Mechanisms of Grignard Reagent Addition to Ketones. Acc. Chem. Res. 1974, 7, 272–280. [Google Scholar] [CrossRef]
- McKinley, J.; Aponick, A.; Raber, J.C.; Fritz, C.; Montgomery, D.; Wigal, C.T. Reactions of Alkyl lithium and Grignard Reagents with Benzoquinone: Evidence for an Electron-Transfer Mechanism. J. Org. Chem. 1997, 62, 4874–4876. [Google Scholar] [CrossRef]
- Ashby, E.C.; Bowers, J.R., Jr. Organometallic Reaction Mechanisms. 17. Nature of Alkyl Transfer in Reactions of Grignard Reagents with Ketones. Evidence for Radical Intermediates in the Formation of 1,2-Addition Product Involving Tertiary and Primary Grignard Reagents. J. Am. Chem. Soc. 1981, 103, 2242–2250. [Google Scholar] [CrossRef]
- Ashby, E.C. A detailed description of the mechanism of reaction of Grignard reagents with ketones. Pure Appl. Chem. 1980, 52, 545–569. [Google Scholar] [CrossRef]
- Bulak, E.; Sarper, O.; Dogan, A.; Lissner, F.; Schleid, T.; Kaim, W. Dichlorogold(III) complexes of bis(1-methyl-2-imidazolyl)ketone and related ligands: Geometrical and electronic structures. Polyhedron 2006, 25, 2577–2582. [Google Scholar] [CrossRef]
- Bergbreiter, D.E.; Pendergrass, E. Analysis of Organomagnesium and Organolithium Reagents Using N-Phenyl-1-naphthylamin. J. Org. Chem. 1981, 49, 219–220. [Google Scholar] [CrossRef]
- SMART & SAINT Software Reference Manuals; Version 5.051 (Windows NT Version); Bruker Analytical X-ray Instruments Inc.: Madison, WI, USA, 1998.
- Sheldrick, G.M. SADABS, Program for Empirical Absorption Correction; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; De Caro, L.; Giacovazzo, C.; Polidori, G.; Spagna, R. SIR2004: An improved tool for crystal structure determination and refinement. J. Appl. Cryst. 2005, 38, 381–388. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELX-2014; University of Göttingen and Bruker AXS: Karlsruhe, Germany, 2014. [Google Scholar]
Sample Availability: Not available. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
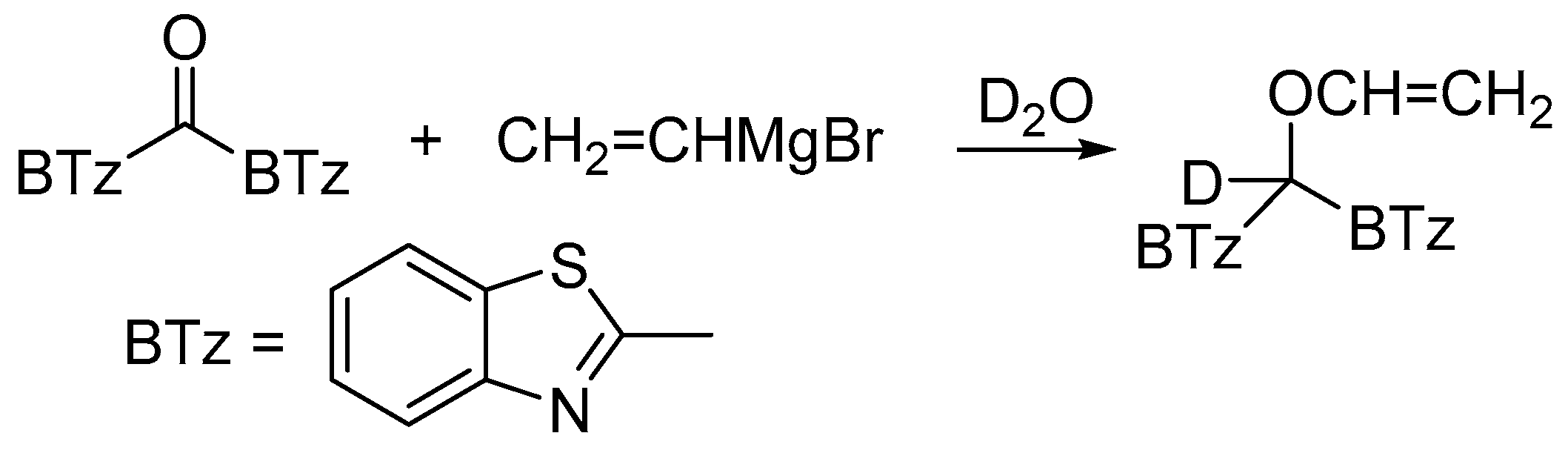{kind=link}
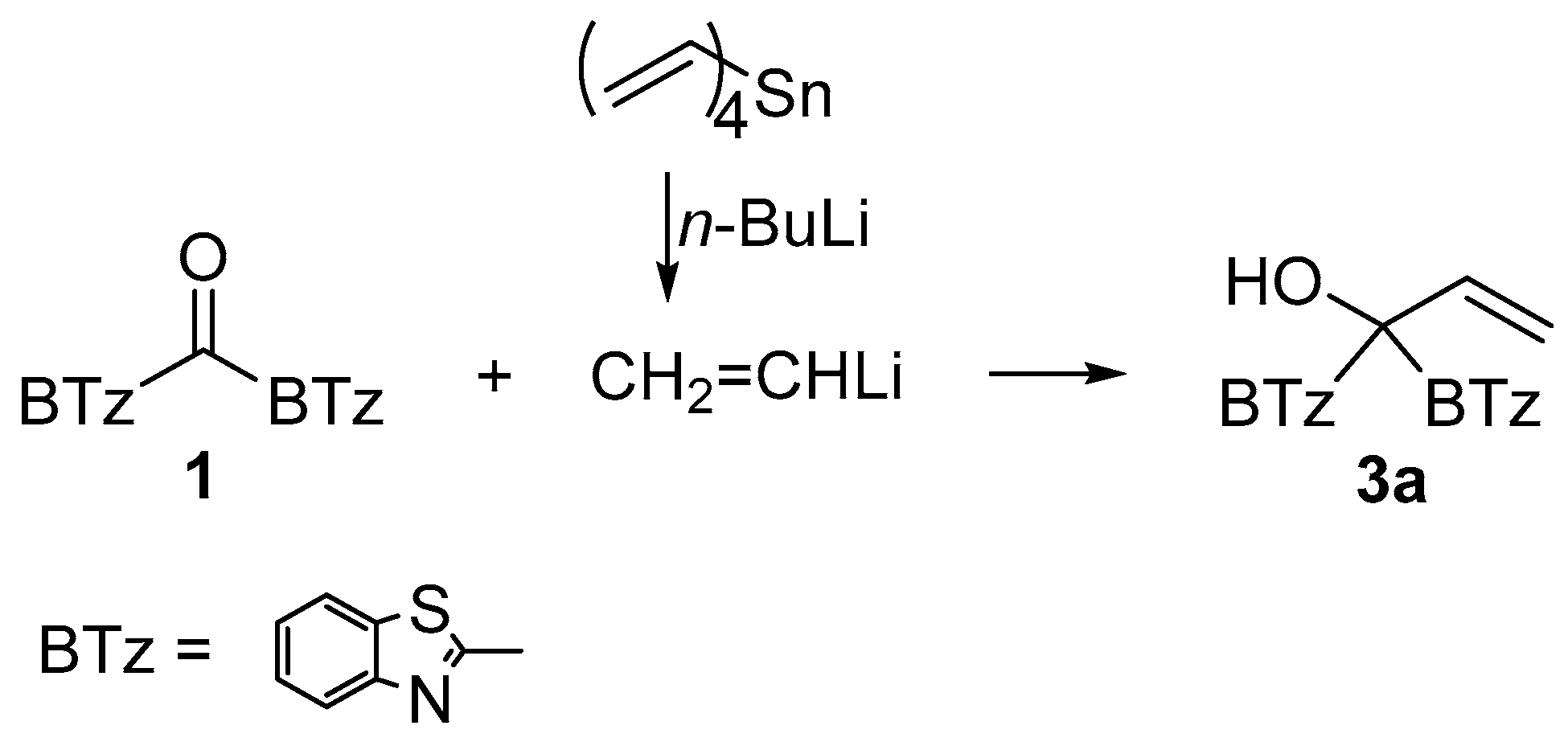{kind=link}
| Entry | Grignard Reagent | C-Alkylation Product (%) | O-Alkylation Product (%) |
|---|---|---|---|
| 1 |  |  3a b,c n.d. d |  4a b > 97 c |
| 2 |  |  3b b,c n.d. d |  4b b > 97 c |
| 3 |  |  3c 57 b,c |  4c b 43 c |
| 4 |  |  3d 70 c |  4d 30 c |
| 5 |  |  3e 70 c,e |  4e 30 c,e |
| 6 |  |  3f 5 |  4f 95 c |
| 7 |  |  3g 90 c |  4g 10 c |
| 8 |  |  3h 5 f |  4h n.d. d |
| Entry | Vinyl Reagent | Solvent | Reaction Time (min) | Radical Scavenger | Conversion% b | C-Alkylation Product (%) c | O-Alkylation Product (%) c |
|---|---|---|---|---|---|---|---|
| 1 | 2a | THF | 10 | - | 80 | 3a (n.d.) d | 4a (pres.) e |
| 2 | 2a | THF | 10 | TEMPO (1.0 eq.) | 60 | 3a (n.d.) d | 4a (pres.) e |
| 3 | 2c | THF | 10 | - | 80 | 3c (67) | 4c (33) |
| 4 | 2c | THF | 10 | TEMPO (0.5 eq.) | 50 | 3c (67) | 4c (33) |
| 5 | 2c | THF | 10 | TEMPO (1.0 eq.) | 30 | 3c (pres.) e | 4c (n.d.) d |
| 6 | 2c | THF | 15–30–45–90 | - | 80 | 3c (67) c,f | 4c (33) c,f |
| 7 | 2a | Et2O | 60 | - | 28 | 3a (50) | 4a (50) |
| Compound | 1 | 5 | 8 | 11.2H2O |
|---|---|---|---|---|
| Empirical formula | C15H8N2OS2 | C7H4N2OS2 | C15H8N2O3 | C17H14N4O.2H2O |
| Formula. weight | 296.35 | 196.24 | 264.23 | 326.35 |
| Temperature | 273(2) K | 273(2) K | 273(2) K | 273(2) K |
| Wavelength | 0.71073 A | 0.71073 A | 0.71073 A | 0.71073 A |
| Crystal system | orthorhombic | Monoclinic | Monoclinic | Monoclinic |
| Space group | Pna21 | Cc | P21/c | P21/n |
| a, Å | 18.3273(12) | 3.7993(3) | 13.1814(14) | 7.3636(7) |
| b, Å | 16.2782(11) | 29.361(2) | 11.9694(13) | 15.5117(14) |
| c, Å | 4.2718(3) | 13.7502(10) | 7.6380(8) | 14.4556(13) |
| α, ° | 90 | 90 | 90 | 90 |
| β, ° | 90 | 91.769(1) | 100.217(2) | 103.544(1) |
| γ , ° | 90 | 90 | 90 | 90 |
| Volume, Å3 | 1274.43(15) | 1533.1(2) | 1186.0(2) | 1605.2(3) |
| Z, Dc, Mg m−3 | 4, 1.545 | 8, 1.700 | 4, 1.480 | 4, 1.350 |
| μ(Mo-Kα), mm−1 | 0.412 | 0.636 | 0.106 | 0.095 |
| F(000) | 608 | 800 | 544 | 688 |
| Crystal size, mm | 0.28 × 0.15 × 0.15 | 0.20 ×0.15 × 0.12 | 0.35 × 0.30 × 0.20 | 0.20 × 0.15 × 0.10 |
| θ limits, ° | 1.67–28.62 | 1.39–28.66 | 1.57–27.989 | 1.96–26.24 |
| Reflections collected | 10765 | 6588 | 9953 | 12377 |
| Unique obs. Reflections [Fo > 4σ(Fo)] | 3045 [R(int) = 0.0188] | 3461 [R(int) = 0.0183] | 2785 [R(int) = 0.0190] | 3230 [R(int) = 0.0242] |
| Goodness-of-fit on F2 | 1.076 | 1.037 | 0.993 | 1.090 |
| R1 (F)a, wR2 (F2) [I > 2σ(I)] | 0.0255, 0.0681 | 0.0239, 0.0592 | 0.0348, 0.0883 | 0.0448, 0.1243 |
| R1 (F)a, wR2 (F2) (all data) | 0.0279, 0.0691 | 0.0254, 0.0597 | 0.0473, 0.0947 | 0.0682, 0.1366 |
| Largest diff. peak and hole, e. Å−3 | 0.291 and −0.155 | 0.339 and −0.337 | 0.165 and −0.134 | 0.355 and −0.195 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boga, C.; Bordoni, S.; Casarin, L.; Micheletti, G.; Monari, M. Regioselectivity in Reactions between Bis(2-benzothiazolyl)ketone and Vinyl Grignard Reagents: C- versus O-alkylation—Part III. Molecules 2018, 23, 171. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23010171
Boga C, Bordoni S, Casarin L, Micheletti G, Monari M. Regioselectivity in Reactions between Bis(2-benzothiazolyl)ketone and Vinyl Grignard Reagents: C- versus O-alkylation—Part III. Molecules. 2018; 23(1):171. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23010171
Chicago/Turabian StyleBoga, Carla, Silvia Bordoni, Lucia Casarin, Gabriele Micheletti, and Magda Monari. 2018. "Regioselectivity in Reactions between Bis(2-benzothiazolyl)ketone and Vinyl Grignard Reagents: C- versus O-alkylation—Part III" Molecules 23, no. 1: 171. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23010171