3.1. Comparison of the Accuracy Obtained Using Particular Types of Descriptors
The results of the prediction gave us an opportunity to compare the usage of short peptides and short nucleotides as descriptors. It is clear that, in general, the RT nucleotide descriptors yield a better performance compared to the peptide descriptors. However, for particular drugs—including Lamivudine (3TC), Abacavir (ABC), and Tenofovir (TDF)—the situation was different. The fact that, in general, nucleotide descriptors yield the better performance has confirmed our hypothesis that they are more descriptive than peptide ones. As to the resistance to PR inhibitors, in general, the performance based on the nucleotide descriptors was not higher in comparison to that based on the short peptides; the performance of prediction was more dependent on the drug than on the type of the descriptors. To explain our findings, we compared the value reflecting the ratio of the number of resistant to the number of susceptible variants in the sets of protease and reverse transcriptase sequences, respectively. The ratio r of resistant to susceptible variants was 0.49 ± 0.56 (average ± standard deviation (SD)) for the set of RT amino acid sequences (the PhenosenseDS-RT set; see the Materials and Methods Section). The same value, r, was 2.73 ± 2.26 for the nucleotide sequences of RT. The value r was 1.61 ± 0.87 for the nucleotide sequence of PR. Therefore, in general, the RT dataset was more imbalanced and biased towards the resistant class. It might be a factor that influences a higher performance of prediction for the RT dataset in comparison to the PR one. Nevertheless, nucleotide descriptors are largely preferable for the prediction of HIV-1 resistance. They do not require any processing procedures (i.e., the translation of a nucleotide sequence to amino acid sequence) or pre-alignment and can be used for prediction using various machine learning approaches. In addition, we did not use the complete set of peptide/nucleotide descriptors generated from the training sets in order to reduce their number and avoid an incorrect ratio between the number of instances and the number of descriptors. Although we selected only a few descriptors, the performance of prediction was still reasonable. It confirmed that we chose correct procedure of descriptors selection.
3.3. Comparison with the Earlier Developed Approaches
The most recent approaches aimed at predicting HIV resistance to RT and/or PR inhibitors are briefly reviewed in References [
19,
20]. There are several studies related to the HIV resistance predictions [
2,
3,
4,
5,
6,
7,
8,
9,
10,
11,
12,
13,
14,
15]. However, References [
13,
14,
15,
16] do not provide a prediction of HIV resistance to protease and reverse transcriptase inhibitors; they are aimed either on the prediction of tropism, the usage of a co-receptor, and HIV maturation inhibition [
13,
14,
15] or prediction of the HIV inhibitors activity against mutated strains. We compared the performances of prediction using our method with the earlier developed machine learning approaches [
3,
4], where decision trees were used as the main computational method. For comparison purposes, we chose two studies that used metrics similar to ours, including sensitivity, specificity, and balanced accuracy. Thus, we were able to compare the performances of the predictions in a comparatively straightforward way. Assuming that we can calculate the balanced accuracy (BA) of prediction if we know the sensitivity and specificity (BA = (Sensitivity + Specificity)/2), we decided to compare the BA values obtained by our method and those achieved by other methods. We compared the accuracy of the prediction obtained by our method using descriptors based on short nucleotides. The results are given in
Table 3. The AUC values were also given and available for comparison.
In the study by Beerenwinkel, N. et al. [
3], the authors used 471 pre-aligned sequences of the
pol gene. The decision tree model was built for each drug. In the study by Rhee, S.-Y. et al. [
5], the authors applied five different statistical methods to classify the isolates as susceptibly/intermediately/highly resistant to a certain drugs. However, it is unclear whether the preliminary alignment was used in this study. For a direct comparison, we used only the parameters of performance obtained by the decision tree algorithm.
We also compared the results of the prediction obtained by our approach with other machine learning methods aimed at predicting the resistance to RT and PR inhibitors [
7,
10].
Table 3 shows the comparison of the prediction performance. For the most common reverse transcriptase inhibitors, such as abacavir (ABC) and nevirapine (NVP), the prediction performance obtained by our approach was the same as that which was reported in the study by Beerenwinkel et al., 2002 [
3]. For zidovudine (AZT), the prediction performance achieved by our method was lower than in Reference [
3], but it was higher than that reported in the study by Rhee S.Y. et al. [
3]. In our approach, the prediction performance for stavudine (D4T) and didanozine (DDI) was much higher whereas, for IDV, NFV, SQV protease inhibitors this value was higher or the same. Hence, this simple method based on the use of short nucleotide descriptors and the Random Forest as a modeling method can be successfully applied for prediction of HIV variants resistance to reverse transcriptase and protease inhibitors.
The AUC values were higher for lamivudine (3TC), zidovudine (AZT), stavudine (D4T), didanosine (DDI), efavirenz (EFV), tenofovir (TDF), atazanavir (ATV), and indinavir (IDV) in comparison with those reported in Reference [
7]. The number of misclassification errors, in general, was higher for the predictions made by our approach for RT inhibitors and they are lower for PR inhibitors compared to Reference [
10]. We would like to emphasize that the usage of misclassification errors without any additional metrics of accuracy is not enough for a proper comparison of accuracy since the number of errors does not include the metrics reflecting the recognition of each class, i.e., “resistant” and “susceptible”. In this case, the number of classification errors might be very low despite the fact that one class—for example, the “susceptible”one—is prevalent in the test set and has a very good prediction, while another class cannot be recognized well.
The results of the prediction reported in the study by Murray et al. [
6] are only given for tenofovir (TDF), so the comparison of these results with those obtained by our method with this study will not provide any significant differences [
6]. Additionally, we could not directly compare our approach with that of Reference [
8] since those models were obtained using another test system (Antivirogram). We showed earlier that the Phenosense data provided a higher accuracy of prediction [
19]. Comparing to our approaches developed earlier [
11,
12], the performance of prediction was higher for several drugs, including AZT, TDF, EFV, and DDI.
For this purpose, we created a training set and a test set of PR sequences. As a training set, we used data on HIV PR sequences and the corresponding data on resistance uploaded to the Stanford HIV Drug Resistance Database (StDB) no later than 2006.
It consisted of 448 sequences with resistance data against 8 HIV protease inhibitors. For the test set, we used all sequences uploaded to StDB after 2006. This set contained 51 sequences. So, we attempted to model a kind of prospective validation. Both sets with sequences, resistance data and the set of descriptors are available in
Supplementary Materials (TrainingPR, ExtTestSetPR). Further, we would have liked to compare the results of prediction with other similar approaches. Unfortunately, we were able to draw a comparison only with the approach by Beerenwinkel, N. et al., 2003 [
3] since the Geno2Pheno web-application is available just for that approach [
4].
We used the training set, created the models, made predictions for the test set using our method and then uploaded the sequences of the test set to the Geno2Pheno web server. The results obtained in Geno2Pheno made it possible to calculate the values of Sensitivity, Specificity, Precision (PPV), Recall (Rec) and Balanced accuracy (BA). The same characteristics were obtained in Weka 3.8 using the set of descriptors and RF model (
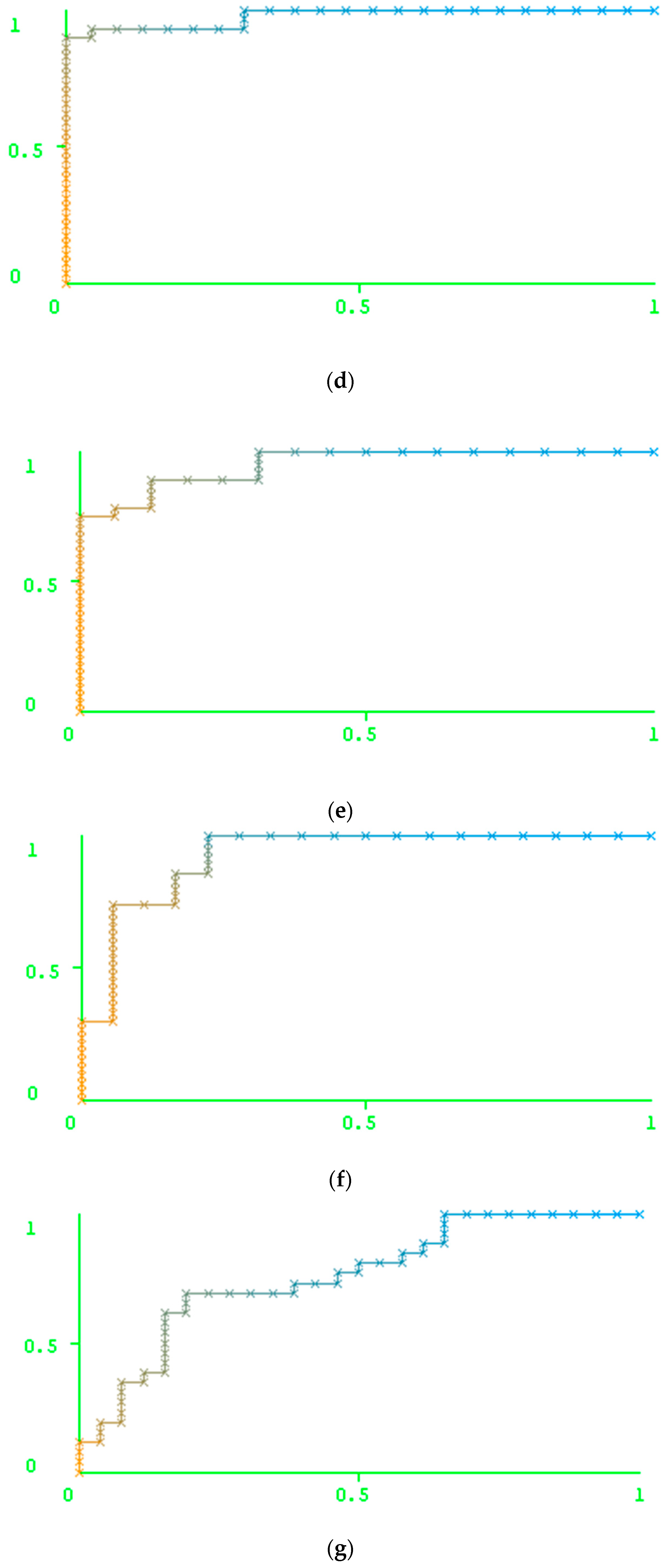
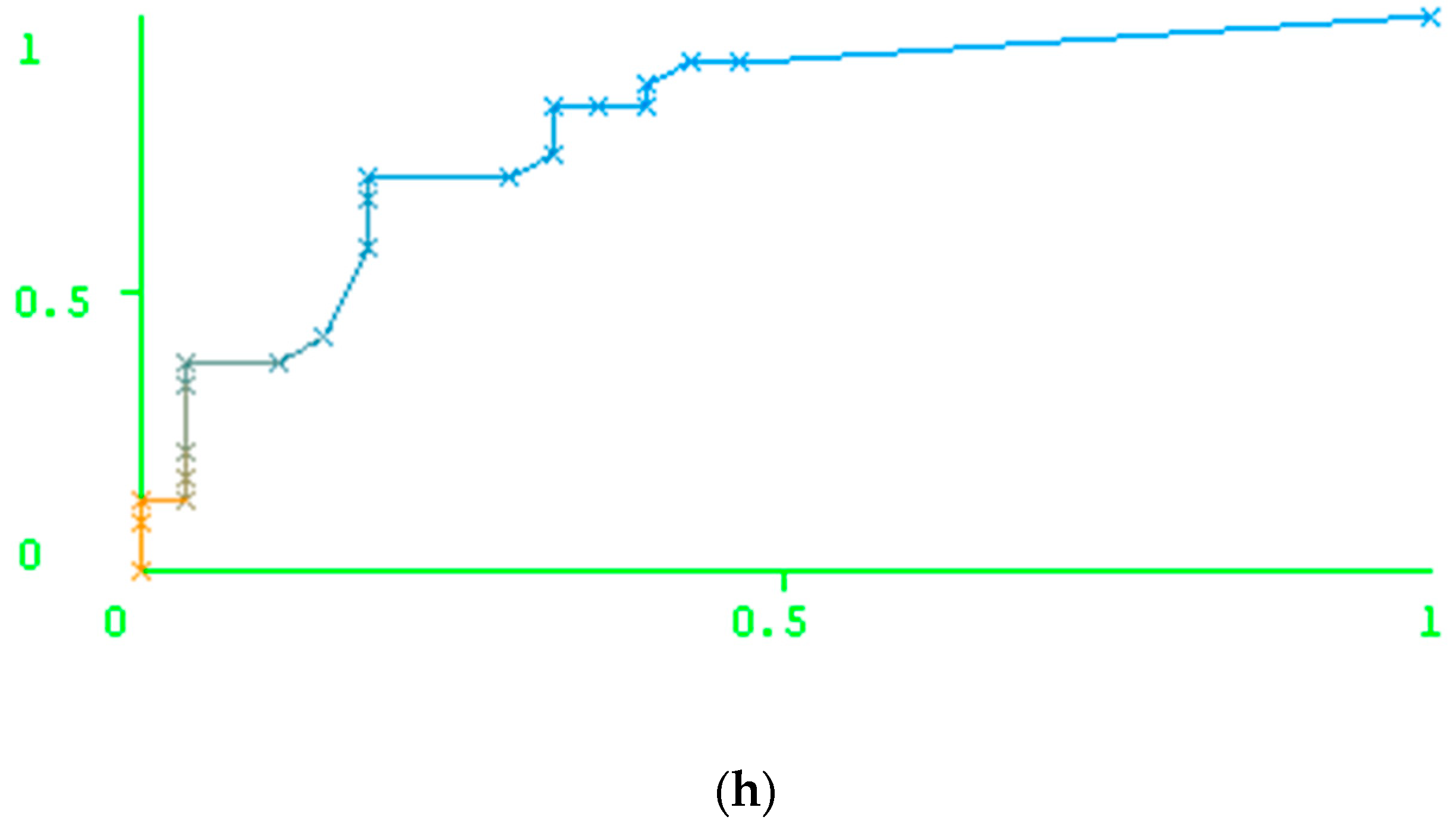
Table 4). We also reported the areas under the ROC curve for each model and the AUC values themselves (
Figure 1). The data used for the ROC curve plotting are also available in the
Supplementary Materials. We suppose that these data can be used for the comparison of our approach with any other method.
As
Table 4 shows, the results of the prediction made by Geno2Pheno are better than the prediction results obtained by our method for the resistance against ATV and DRV, while, for IDV, LPV, NFV, and SQV, the results of the prediction obtained using our method outperform the results of the prediction using Geno2Pheno. The accuracy of prediction was similar for the cases of FPV and TPV.
As
Figure 1 shows, the performance of the prediction is high for indinavir and nelfinavir, while this value is not as high for tipranavir (TPV) and darunavir (DRV). In particular, one can notice a comparatively low number of resistant variants against the latter two drugs in the test set. In this case, the results might be explained by the peculiarities of the test set creation. However, the 5-fold validation also did not reveal the high accuracy of prediction for these two drugs. Therefore, the low accuracy of prediction for these two drugs can be explained by a weak relationship between the features of the amino acid and the nucleotide sequences and the level of resistance of the corresponding variants to the PR inhibitors.
In general, the 5-fold validation and the results of the prediction for the test set allowed for good recognition of resistant HIV variants against the following drugs: zidovudine (AZT), stavudine (D4T), efavirenz (EFV), etravirine (ETR), nevirapine (NVP), indinavir (IDV), lopinavir (LPV), nelfinavir (NFV), and saquinavir (SQV).







{kind=link}
{kind=link}
{kind=link}