
Analytical Profiling of Proanthocyanidins from Acacia mearnsii Bark and In Vitro Assessment of Antioxidant and Antidiabetic Potential


Abstract
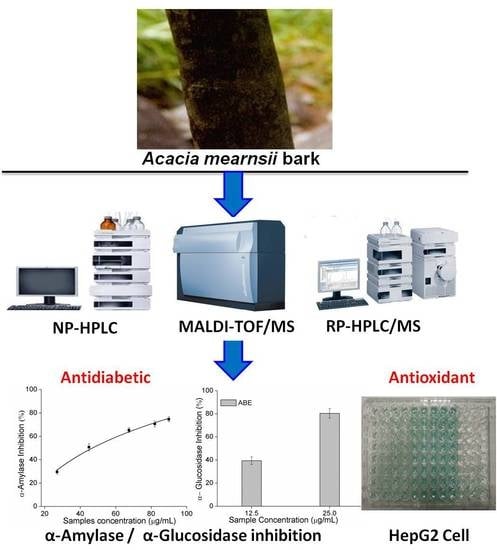
:
1. Introduction
2. Results and Discussion
2.1. Determination of Total Polyphenol Content (TPC), PAs, and Antioxidant Activity
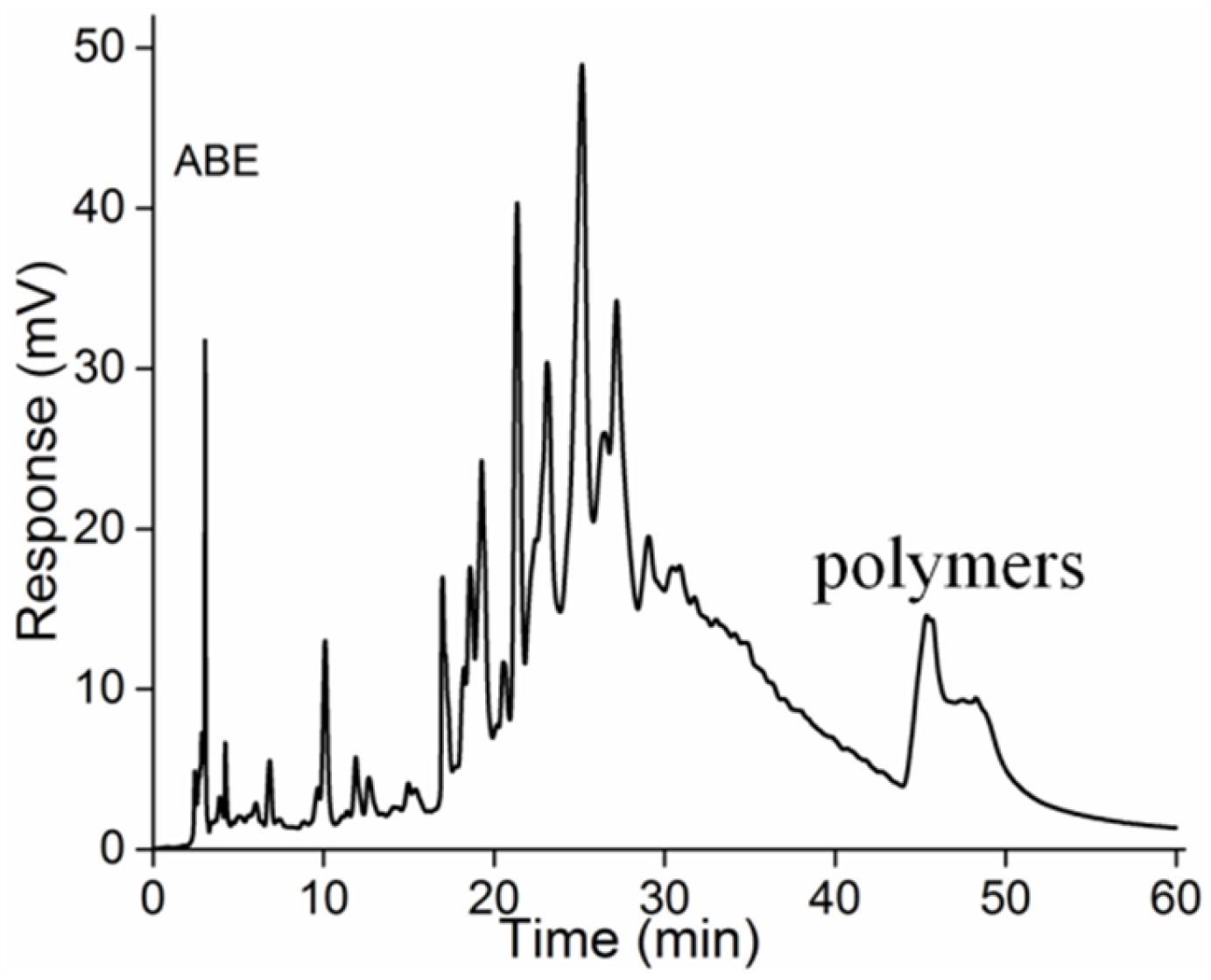
2.2. Normal-Phase HPLC Analysis
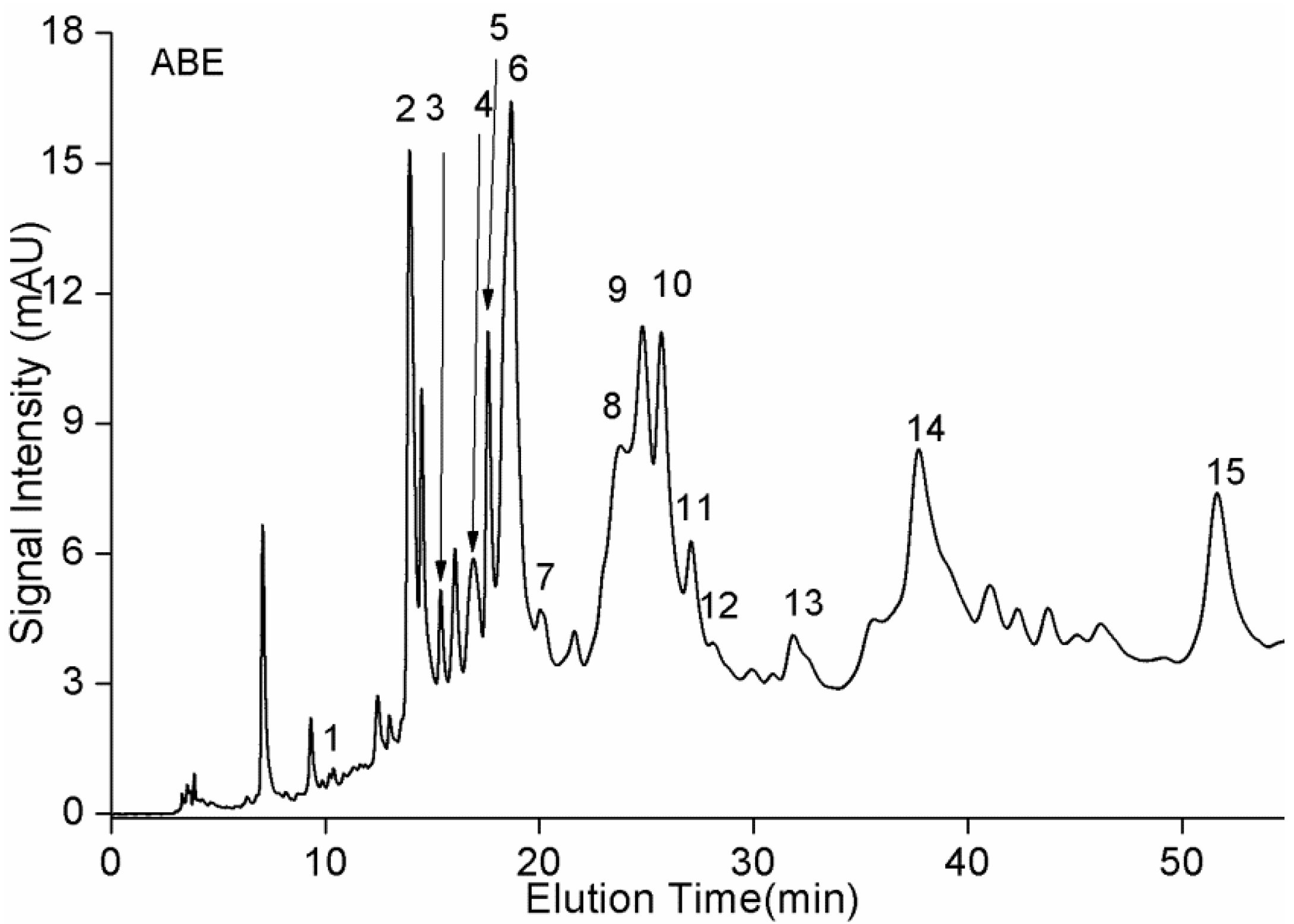
2.3. RP-HPLC/MS Analysis
2.4. MALDI-TOF/TOF-MS Analysis of ABE
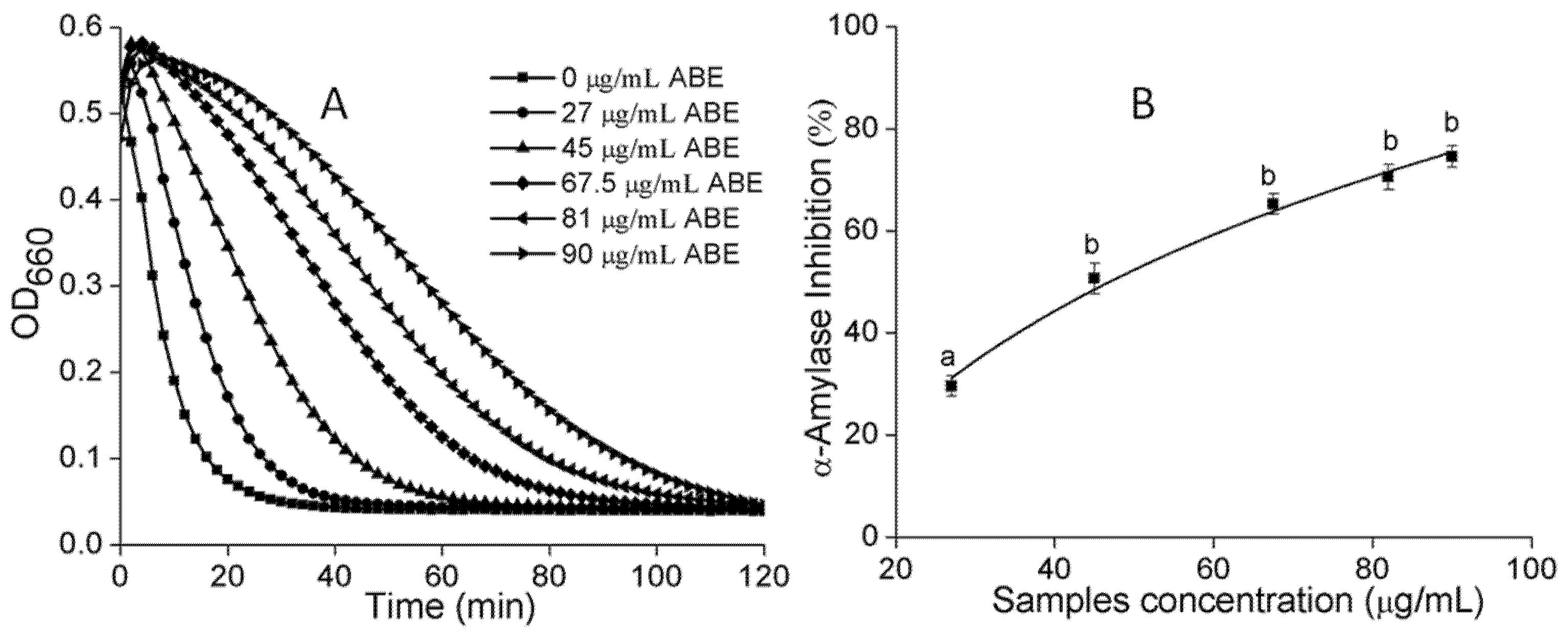
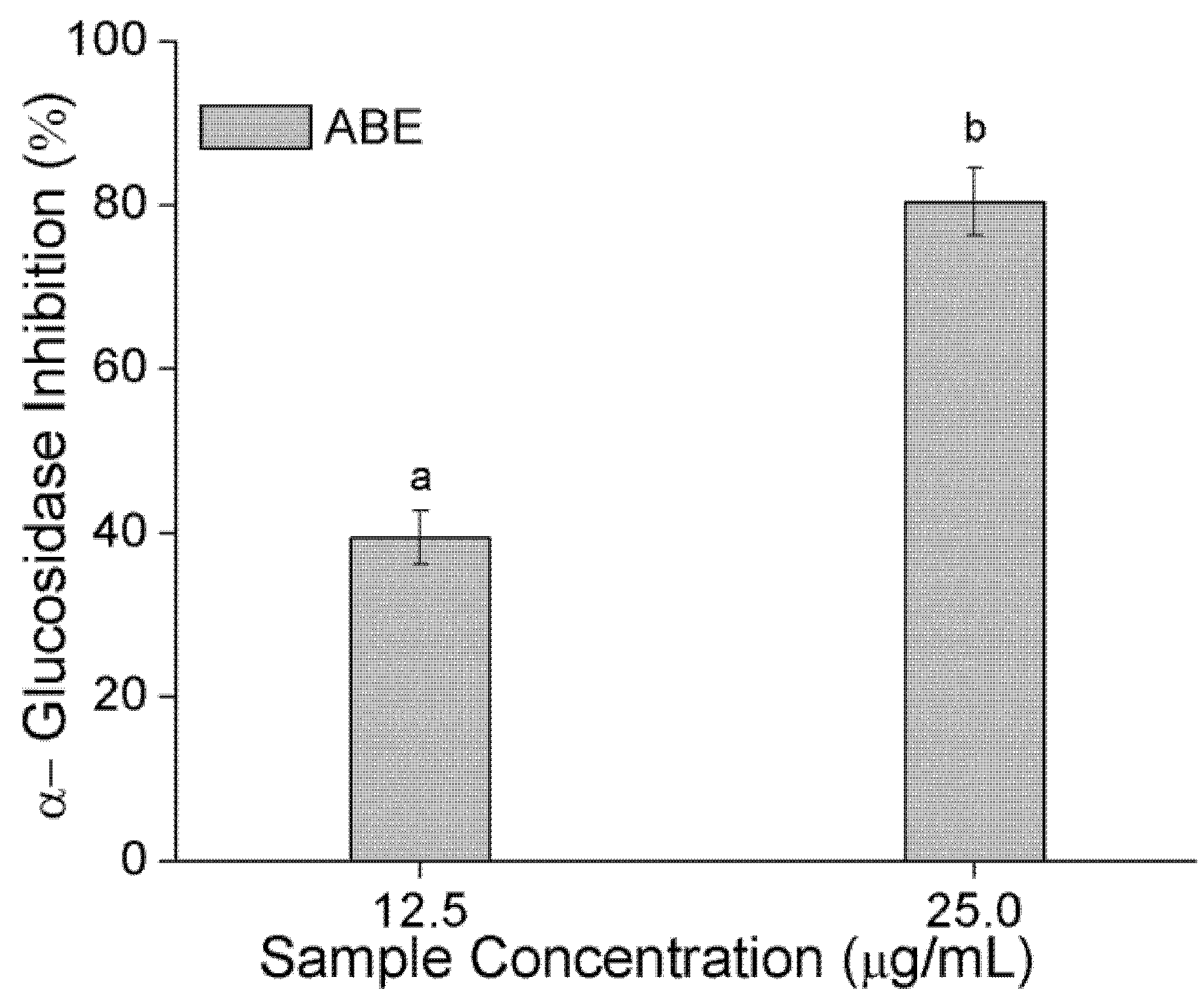
2.5. α-Amylase and α-Glucosidase Inhibition
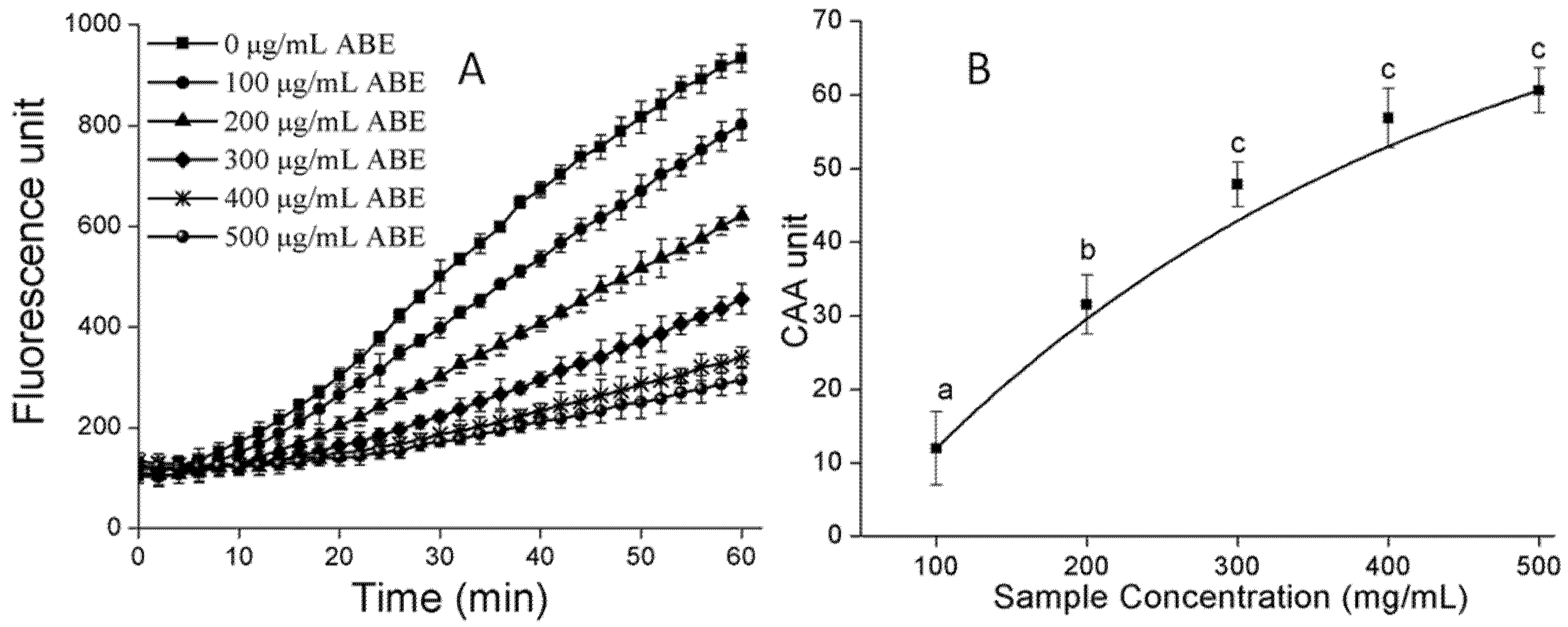
2.6. Cellular Antioxidant Activity (CAA)
3. Materials and Methods
3.1. Materials and Reagents
3.2. Sample Extraction
3.3. Determination of PAs and TPC
3.4. NP-HPLC/ Variable Wavelength Detector (VWD) Analysis
3.5. RP-HPLC/MS Analysis
3.6. MALDI-TOF/TOF-MS Analysis of ABE
3.7. Chemical Antioxidant Activity
3.7.1. DPPH and ABTS Assays
3.7.2. Oxygen Radical Absorbance Capacity (ORAC) Assay
3.7.3. Assay of Rapid Peroxyl Radical Scavenging Capacity (PSC)
3.8. α-Amylase and α-Glucosidase Inhibition
3.9. Cell Culture and Treatment
3.10. Cytotoxicity
3.11. Cellular Antioxidant Activity
3.12. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rodrigues, A.S.; Pérez-Gregorio, M.R.; García-Falcón, M.S.; Simal-Gándara, J.; Almeida, D.P.F. Effect of post-harvest practices on flavonoid content of red and white onion cultivars. Food Control 2010, 21, 878–884. [Google Scholar] [CrossRef]
- Yang, H.; Ye, X.; Liu, D.; Chen, J.; Zhang, J.; Shen, Y.; Yu, D. Characterization of unusual proanthocyanidins in leaves of bayberry (Myrica rubra Sieb. Et zucc.). J. Agric. Food Chem. 2011, 59, 1622–1629. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, X.; Chen, J.; Liu, C.; Li, T.; Mcclements, D.J.; Dai, T.; Liu, J. Antioxidant activity of proanthocyanidins-rich fractions from choerospondias axillaris peels using a combination of chemical-based methods and cellular-based assay. Food Chem. 2016, 208, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Porter, L.J. Flavans and Proanthocyanidins; Springer: New York, NY, USA, 1988; pp. 21–62. [Google Scholar]
- Aron, P.M.; Kennedy, J.A. Flavan-3-ols: Nature, occurrence and biological activity. Mol. Nutr. Food Res. 2008, 52, 79–104. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Xiong, J.; Wang, F.; Grace, M.H.; Lila, M.A.; Xu, R. α-Amylase and α-glucosidase inhibitory activities of phenolic extracts from Eucalyptus grandis x e. Urophylla bark. J. Chem. 2017, 2017, 8516964. [Google Scholar] [CrossRef]
- Xiong, J.; Grace, M.H.; Esposito, D.; Komarnytsky, S.; Wang, F.; Lila, M.A. Polyphenols isolated from Acacia mearnsii bark with anti-inflammatory and carbolytic enzyme inhibitory activities. Chin. J. Nat. Med. 2017, 15, 816–824. [Google Scholar] [CrossRef]
- Xiong, J.; Graceb, M.H.; Esposito, D.; Wang, F.; Lila, M.A. Phytochemical characterization and anti-inflammatory properties of Acacia mearnsii leaves. Nat. Prod. Commun. 2016, 11, 649–653. [Google Scholar] [PubMed]
- Okuda, T. Systematics and health effects of chemically distinct tannins in medicinal plants. Phytochemistry 2005, 66, 2012–2031. [Google Scholar] [CrossRef] [PubMed]
- Rocío, D.L.I.; Milagro, F.I.; Campión, J.; Boqué, N.; Martínez, J.A. Healthy properties of proanthocyanidins. BioFactors 2010, 36, 159–168. [Google Scholar]
- Neilson, A.P.; O’Keefe, S.F.; Bolling, B.W. High-molecular-weight proanthocyanidins in foods: Overcoming analytical challenges in pursuit of novel dietary bioactive components. Annu. Rev. Food Sci. Technol. 2015, 7, 43–64. [Google Scholar] [CrossRef] [PubMed]
- Venter, P.B.; Senekal, N.D.; Kemp, G.; Amra-Jordaan, M.; Khan, P.; Bonnet, S.L.; Westhuizen, J.H.V.D. Analysis of commercial proanthocyanidins. Part 3: The chemical composition of wattle (Acacia mearnsii) bark extract. Phytochemistry 2012, 83, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Pasch, H.; Pizzi, A.; Rode, K. MALDI–TOF mass spectrometry of polyflavonoid tannins. Polymer 2001, 42, 7531–7539. [Google Scholar] [CrossRef]
- Kusano, R.; Ogawa, S.; Matsuo, Y.; Tanaka, T.; Yazaki, Y.; Kouno, I. α-Amylase and lipase inhibitory activity and structural characterization of Acacia bark proanthocyanidins. J. Nat. Prod. 2011, 74, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Yazaki, Y. Tannins from Acacia mearnsii de wild. Bark: Tannin determination and biological activities. Molecules 2018, 23, 837. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Wang, Y.; Wang, F. Characterisation and biological activities of proanthocyanidins from the barks of Pinus massonian and Acacia mearnsii. Nat. Prod. Res. 2010, 24, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xiong, J.; He, L.; Zhang, Y.; Li, X.; Zhang, L.; Wang, F. Effects of in vitro digestion on the content and biological activity of polyphenols from Acacia mearnsii bark. Molecules 2018, 23, 1804. [Google Scholar] [CrossRef] [PubMed]
- Dudonné, S.; Vitrac, X.; Coutière, P.; Woillez, M.; Mérillon, J.M. Comparative study of antioxidant properties and total phenolic content of 30 plant extracts of industrial interest using DPPH, ABTS, FRAP, SOD, and ORAC assays. J. Agric. Food Chem. 2009, 57, 1768–1774. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.A.; Taylor, A.W. Analysis of proanthocyanidins by high-performance gel permeation chromatography. J. Chromatogr. 2003, 995, 99–107. [Google Scholar] [CrossRef]
- Roux, D.G.; Maihs, E.A. Condensed tannins. 3. Isolation and estimation of (-)-7:3′:4′:5′-tetrahydroxyflavan-3-ol, (+)-catechin and (+)-gallocatechin from black-wattle-bark extract. Biochem. J. 1960, 74, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Hoong, Y.B.; Pizzi, A.; Tahir, P.M.; Pasch, H. Characterization of acacia mangium polyflavonoid tannins by maldi-tof mass spectrometry and CP-MAS 13C NMR. Eur. Polym. J. 2010, 46, 1268–1277. [Google Scholar] [CrossRef]
- Pérez-Jiménez, J.; Torres, J.L. Analysis of proanthocyanidins in almond blanch water by HPLC–ESI–QqQ–MS/MS and MALDI–TOF/TOF MS. Food Res. Int. 2012, 49, 798–806. [Google Scholar] [CrossRef]
- Roux, D.G.; Ferreira, D.; Hundt, H.K.L.; Malan, E. Structure, stereochemistry, and reactivity of natural condensed tannins as basis for their extended industrial application. Appl. Polym. Symp. 1975, 28, 335–353. [Google Scholar]
- Zhang, Y.; Wong, A.I.C.; Wu, J.E.; Karim, N.B.A.; Huang, D. Lepisanthes alata (Malay cherry) leaves are potent inhibitors of starch hydrolases due to proanthocyanidins with high degree of polymerization. J. Funct. Foods 2016, 25, 568–578. [Google Scholar] [CrossRef]
- Ma, Y.Y.; Zhao, D.G.; Zhou, A.Y.; Zhang, Y.; Du, Z.; Zhang, K. A-glucosidase inhibition and antihyperglycemic activity of phenolics from the flowers of edgeworthia gardneri. J. Agric. Food Chem. 2015, 63, 8162–8169. [Google Scholar] [CrossRef] [PubMed]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Riceevans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
- Huang, D.; Ou, B.; Hampschwoodill, M.; Flanagan, J.A.; Prior, R.L. High-throughput assay of oxygen radical absorbance capacity (ORAC) using a multichannel liquid handling system coupled with a microplate fluorescence reader in 96-well format. J. Agric. Food Chem. 2002, 50, 4437–4444. [Google Scholar] [CrossRef] [PubMed]
- Lordan, S.; Smyth, T.J.; Soler-Vila, A.; Stanton, C.; Ross, R.P. The α-amylase and α-glucosidase inhibitory effects of irish seaweed extracts. Food Chem. 2013, 141, 2170–2176. [Google Scholar] [CrossRef] [PubMed]
- Rubilar, M.; Jara, C.; Poo, Y.; Acevedo, F.; Gutierrez, C.; Sineiro, J.; Shene, C. Extracts of maqui (Aristotelia chilensis) and murta (Ugni molinae Turcz.): Sources of antioxidant compounds and α-glucosidase/α-amylase inhibitors. J. Agric. Food Chem. 2011, 59, 1630–1637. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.H.; Finley, J. Potential cell culture models for antioxidant research. J. Agric. Food Chem. 2005, 53, 4311–4314. [Google Scholar] [CrossRef] [PubMed]
- Granato, D.; Shahidi, F.; Wrolstad, R.; Kilmartin, P.; Melton, L.D.; Hidalgo, F.J.; Miyashita, K.; Camp, J.V.; Alasalvar, C.; Ismail, A.B. Antioxidant activity, total phenolics and flavonoids contents: Should we ban in vitro screening methods? Food Chem. 2018, 264, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Manach, C.; Scalbert, A.; Morand, C.; Rémésy, C.; Jiménez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef] [PubMed]
- Ou, K.; Gu, L. Absorption and metabolism of proanthocyanidins. J. Funct. Foods 2014, 7, 43–53. [Google Scholar] [CrossRef]
- Li, Q.; Chen, J.; Li, T.; Liu, C.; Wang, X.; Dai, T.; Mcclements, D.J.; Liu, J. Impact of in vitro simulated digestion on the potential health benefits of proanthocyanidins from choerospondias axillaris peels. Food Res. Int. 2015, 78, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Adom, K.K.; Liu, R.H. Rapid peroxyl radical scavenging capacity (PSC) assay for assessing both hydrophilic and lipophilic antioxidants. J. Agric. Food Chem. 2005, 53, 6572. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Sun, Y.; Lou, S.; Li, H.; Ye, X. In vitro digestion combined with cellular assay to determine the antioxidant activity in chinese bayberry (Myrica rubra Sieb. Et zucc.) fruits: A comparison with traditional methods. Food Chem. 2014, 146, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, K.L.; Liu, R.H. Cellular antioxidant activity (CAA) assay for assessing antioxidants, foods, and dietary supplements. J. Agric. Food Chem. 2007, 55, 8896–8907. [Google Scholar] [CrossRef] [PubMed]
- Ikarashi, N.; Takeda, R.; Ito, K.; Ochiai, W.; Sugiyama, K. The inhibition of lipase and glucosidase activities by Acacia polyphenol. Evid.-Based Complement. Altern. Med. 2010, 2011, 272075. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Approximate Retention Time from VWD (min) | Peak | Mass (Da) | Proposed Structure |
|---|---|---|---|
| 10.4 | 1 | 306 | gallocatechin |
| 14.0 | 2 | 594 | B dimer |
| 15.6 | 3 | 290 | catechin |
| 17.1 | 4 | 578 | B dimer |
| 17.8 | 5 | 578 | B dimer |
| 18.7 | 6 | 578 | B dimer |
| 19.9 | 7 | 866 | B trimer |
| 23.3 | 8 | 882 | B trimer |
| 24.4 | 9 | 562 | B dimer |
| 25.3 | 10 | 882 | B trimer |
| 26.9 | 11 | 866 | B trimer |
| 28.0 | 12 | 562 | B dimer |
| 33.6 | 13 | 866 | B trimer |
| 39.2 | 14 | 866 | B trimer |
| 52 | 15 | 850 | B trimer |
| Polymer | Number of C/R | Number of GC | Number of F | Number of GAL | Calculated [M + Cs]+ | Observed [M + Cs]+ |
|---|---|---|---|---|---|---|
| Trimer | 1 | 0 | 2 | 0 | 967.1 | 967.43 |
| 2 | 0 | 1 | 0 | 983.1 | 983.43 | |
| 3 | 0 | 0 | 0 | 999.1 | 999.43 | |
| 2 | 1 | 0 | 0 | 1015.1 | 1015.43 | |
| Tetramer | 1 | 0 | 3 | 0 | 1239.2 | 1239.57 |
| 2 | 0 | 2 | 0 | 1255.2 | 1255.58 | |
| 3 | 0 | 1 | 0 | 1271.2 | 1271.58 | |
| 4 | 0 | 0 | 0 | 1287.2 | 1287.58 | |
| 3 | 1 | 0 | 0 | 1303.2 | 1303.58 | |
| 2 | 2 | 0 | 0 | 1319.2 | 1319.58 | |
| Pentamer | 2 | 0 | 3 | 0 | 1527.2 | 1527.72 |
| 3 | 0 | 2 | 0 | 1543.2 | 1543.72 | |
| 4 | 0 | 1 | 0 | 1559.2 | 1559.72 | |
| 5 | 0 | 0 | 0 | 1575.2 | 1575.73 | |
| 4 | 1 | 0 | 0 | 1591.2 | 1591.73 | |
| 3 | 2 | 6 | 0 | 1607.2 | 1607.73 | |
| Hexamer | 3 | 0 | 3 | 0 | 1815.3 | 1815.86 |
| 4 | 0 | 2 | 0 | 1831.3 | 1831.86 | |
| 5 | 0 | 1 | 0 | 1847.3 | 1847.87 | |
| 6 | 0 | 0 | 0 | 1863.3 | 1863.87 | |
| 5 | 1 | 0 | 0 | 1879.3 | 1879.86 | |
| 4 | 2 | 0 | 0 | 1895.3 | 1895.86 | |
| Heptamer | 4 | 0 | 3 | 0 | 2103.3 | 2103.99 |
| 5 | 0 | 2 | 0 | 2119.3 | 2119.99 | |
| 6 | 0 | 1 | 0 | 2135.3 | 2135.99 | |
| 7 | 0 | 0 | 0 | 2151.3 | 2151.99 | |
| 6 | 1 | 0 | 0 | 2167.3 | 2167.99 | |
| 5 | 2 | 0 | 0 | 2183.3 | 2183.99 | |
| Octamer | 5 | 0 | 3 | 0 | 2391.4 | 2392.12 |
| 6 | 0 | 2 | 0 | 2407.4 | 2408.12 | |
| 7 | 0 | 1 | 0 | 2423.4 | 2424.12 | |
| 8 | 0 | 0 | 0 | 2339.4 | 2440.12 | |
| 7 | 1 | 0 | 0 | 2455.4 | 2456.12 | |
| 6 | 2 | 0 | 0 | 2471.4 | 2472.12 | |
| Nonamer | 7 | 0 | 2 | 0 | 2695.5 | 2697.27 |
| 8 | 0 | 1 | 0 | 2711.5 | 2714.29 | |
| 9 | 0 | 0 | 0 | 2727.5 | 2729.30 | |
| 8 | 1 | 0 | 0 | 2743.5 | 2745.28 | |
| Decamer | 7 | 0 | 3 | 0 | 2969.5 | 2969.38 |
| 8 | 0 | 2 | 0 | 2985.5 | 2985.40 | |
| 9 | 0 | 1 | 0 | 3003.5 | 3002.42 | |
| 10 | 0 | 0 | 0 | 3015.5 | 3016.42 | |
| 9 | 1 | 0 | 0 | 3031.5 | 3033.41 | |
| Undecamer | 10 | 0 | 1 | 0 | 3287.6 | 3288.59 |
| 11 | 0 | 0 | 0 | 3303.6 | 3304.59 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Xiong, J.; Huang, S.; Li, X.; Zhang, Y.; Zhang, L.; Wang, F. Analytical Profiling of Proanthocyanidins from Acacia mearnsii Bark and In Vitro Assessment of Antioxidant and Antidiabetic Potential. Molecules 2018, 23, 2891. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23112891
Chen X, Xiong J, Huang S, Li X, Zhang Y, Zhang L, Wang F. Analytical Profiling of Proanthocyanidins from Acacia mearnsii Bark and In Vitro Assessment of Antioxidant and Antidiabetic Potential. Molecules. 2018; 23(11):2891. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23112891
Chicago/Turabian StyleChen, Xiao, Jia Xiong, Shenlin Huang, Xun Li, Yu Zhang, Liping Zhang, and Fei Wang. 2018. "Analytical Profiling of Proanthocyanidins from Acacia mearnsii Bark and In Vitro Assessment of Antioxidant and Antidiabetic Potential" Molecules 23, no. 11: 2891. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23112891