
Synthesis of Jacaranone-Derived Nitrogenous Cyclohexadienones and Their Antiproliferative and Antiprotozoal Activities

 ,
,
Abstract
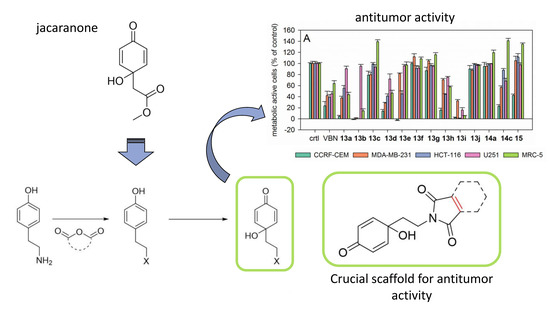
:
1. Introduction
2. Results and Discussion
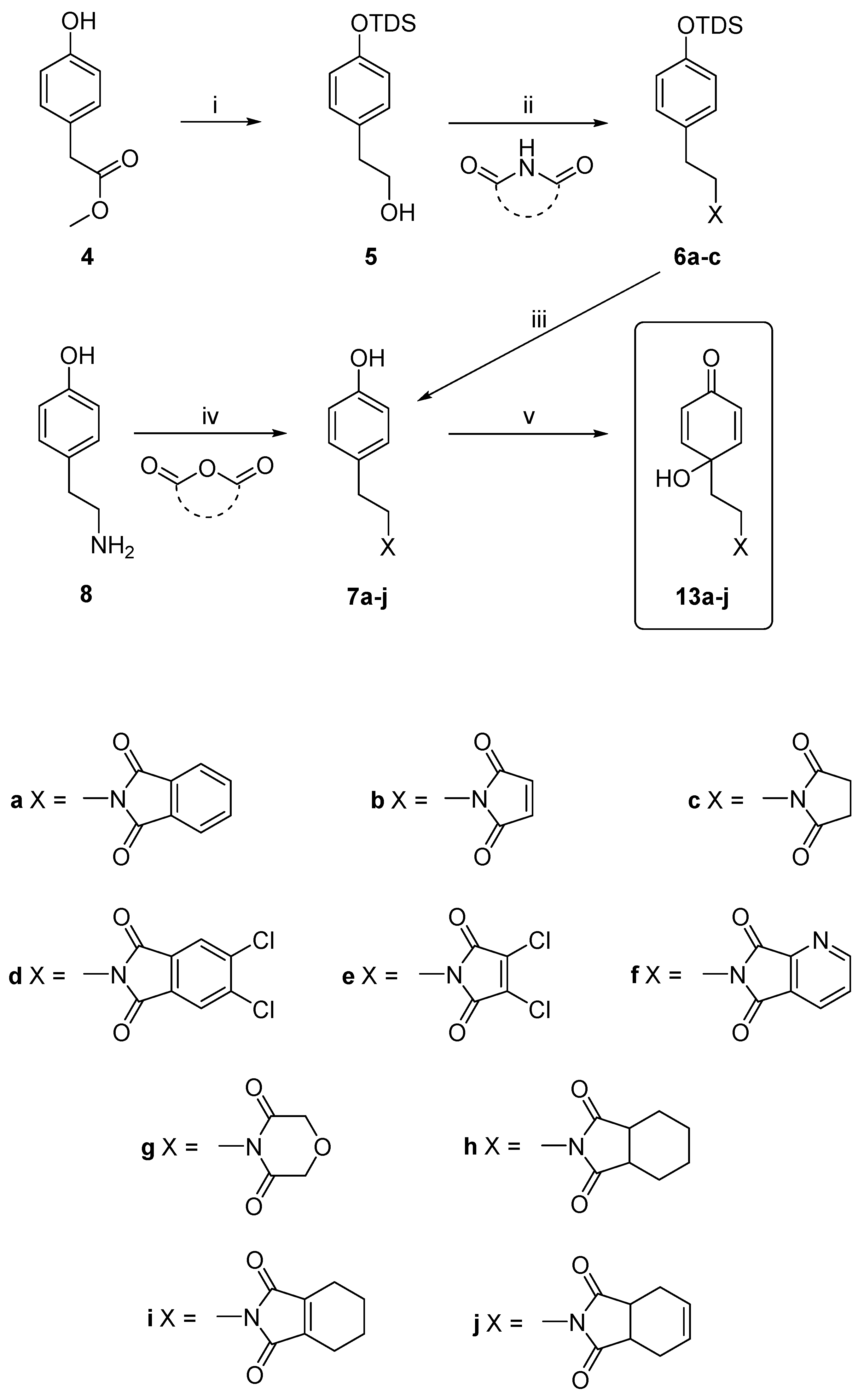
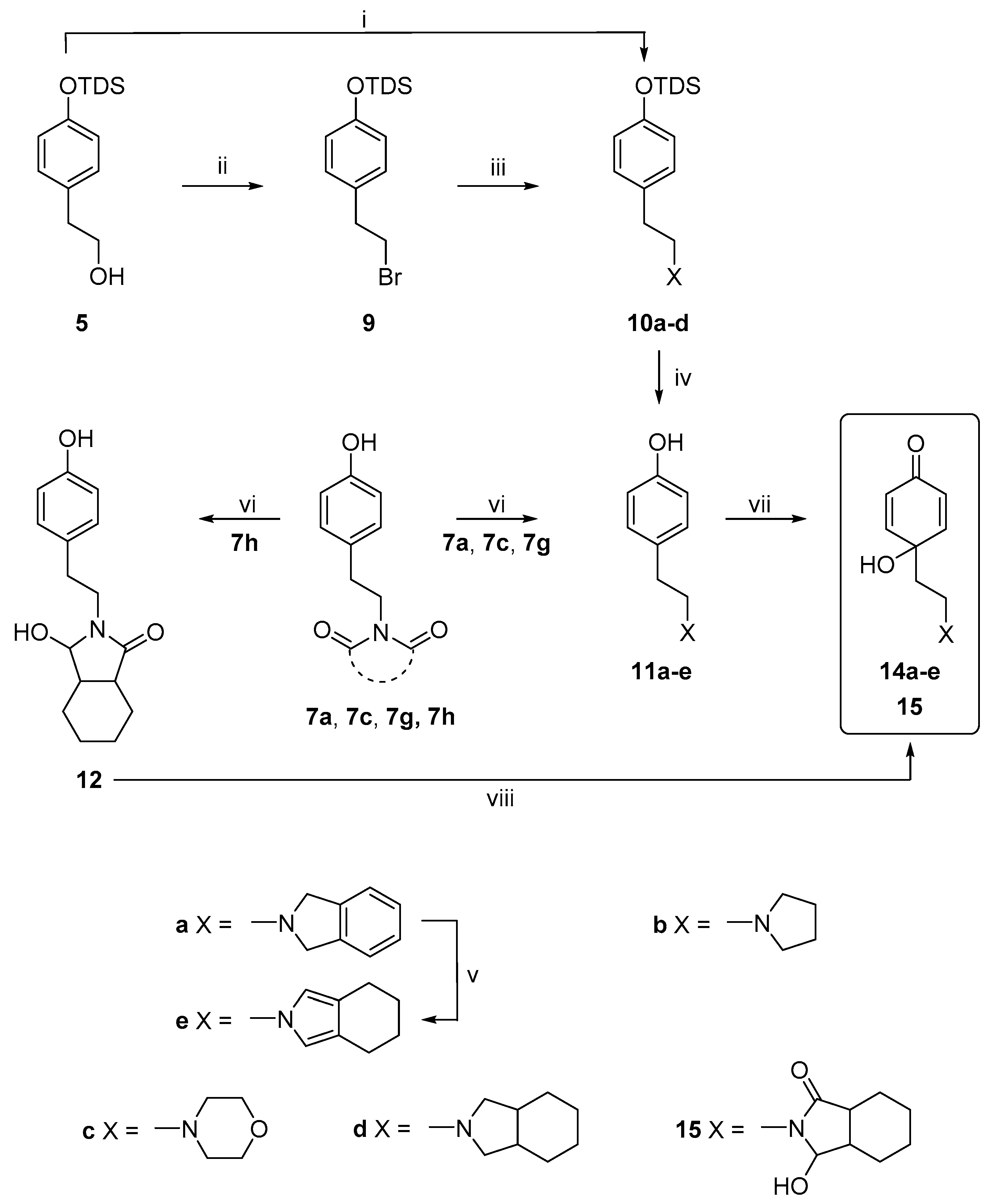
2.1. Chemistry
2.2. Physicochemical Properties
2.3. Biological Evaluation
2.3.1. Antiproliferative Activity
2.3.2. Antiprotozoal Activity
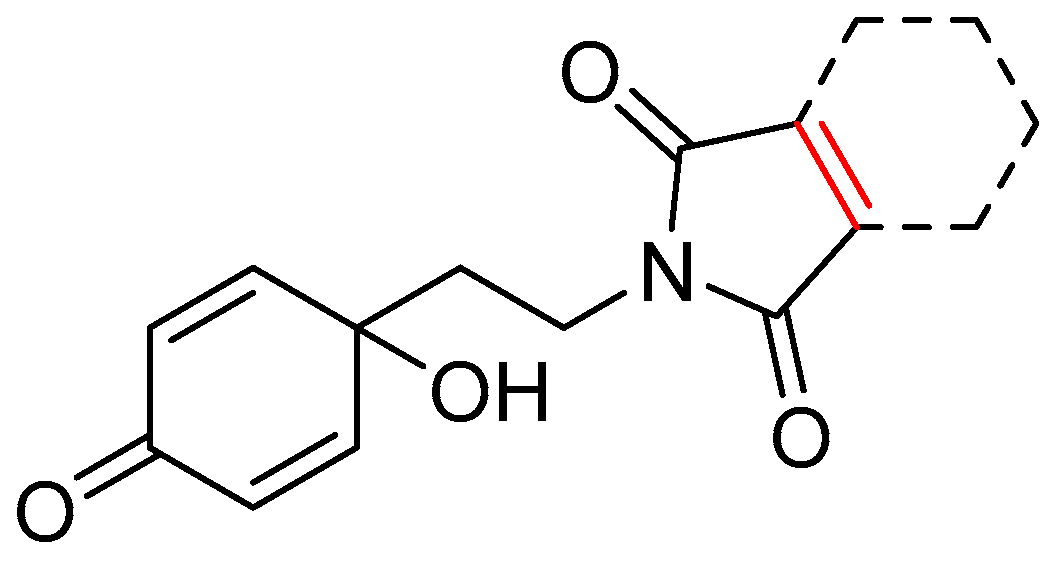
2.3.3. Structure-Activity Relationships (SAR) of the Antiproliferative and Antiprotozoal Activity
3. Experimental Section
3.1. Chemicals and Instruments
3.2. Synthesis
3.2.1. General Procedure for the Synthesis of the Compounds, 7a–7j
3.2.2. General Procedure for the Synthesis of the Compounds, 11a–11d
3.2.3. General Procedure for the Reduction of Heterocyclic N-Imides
3.2.4. Procedures for the Synthesis of Dienones, 13a–j, 14a, 14c, 15
3.3. Cytotoxicity against Human (Cancer) Cells
3.3.1. Cell Culture
3.3.2. XTT Viability Assay
3.4. In Vitro Growth Inhibition Assay of Plasmodium Falciparum NF54
3.5. In vitro Growth Inhibition Assay of Trypanosoma Brucei Rhodesiense
3.6. Cytotoxicity against L6 Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Khazir, J.; Mir, B.A.; Mir, S.A.; Cowan, D. Natural products as lead compounds in drug discovery. J. Asian Nat. Prod. Res. 2013, 15, 764–788. [Google Scholar] [CrossRef] [Green Version]
- Brahmachari, G. Natural products in drug discovery: Impacts and opportunities—An assessment. In Bioactive Natural Products; World Scientific Publishing Co Pte Ltd.: Singapore, 2011; pp. 1–199. [Google Scholar]
- Harvey, A.L.; Clark, R.L.; Mackay, S.P.; Johnston, B.F. Current strategies for drug discovery through natural products. Expert Opin. Drug Discov. 2010, 5, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Kingston, D.G.I. Modern natural products drug discovery and its relevance to biodiversity conservation. J. Nat. Prod. 2011, 74, 496–511. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.B.; Tiwari, V.K. Natural products: An evolving role in future drug discovery. Eur. J. Med. Chem. 2011, 46, 4769–4807. [Google Scholar] [CrossRef] [PubMed]
- Montaser, R.; Luesch, H. Marine natural products: A new wave of drugs? Future Med. Chem. 2011, 3, 1475–1489. [Google Scholar] [CrossRef] [PubMed]
- Koehn, F.E. Biosynthetic medicinal chemistry of natural product drugs. MedChemComm 2012, 3, 854–865. [Google Scholar] [CrossRef]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3670–3695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.; Oh, D.-C. Considerations of the chemical biology of microbial natural products provide an effective drug discovery strategy. Arch. Pharm. Res. 2015, 38, 1591–1605. [Google Scholar] [CrossRef] [PubMed]
- Gachet, M.S.; Kunert, O.; Kaiser, M.; Brun, R.; Munoz, R.A.; Bauer, R.; Schuhly, W. Jacaranone-derived glucosidic esters from Jacaranda glabra and their activity against Plasmodium falciparum. J. Nat. Prod. 2010, 73, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Lozada-Lechuga, J.; Villarreal, M.L.; Fliniaux, M.-A.; Bensaddek, L.; Mesnard, F.; del Carmen Gutiérrez, M.; Cardoso-Taketa, A.T. Isolation of jacaranone, a sedative constituent extracted from the flowers of the Mexican tree Ternstroemia pringlei. J. Ethnopharmacol. 2010, 127, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Massaoka, M.H.; Matsuo, A.L.; Figueiredo, C.R.; Farias, C.F.; Girola, N.; Arruda, D.C.; Scutti, J.A.B.; Romoff, P.; Favero, O.A.; Ferreira, M.J.P.; et al. Jacaranone induces apoptosis in melanoma cells via ROS-mediated downregulation of Akt and p38 MAPK activation and displays antitumor activity in vivo. PLoS ONE 2012, 7, e38698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morais, T.R.; Romoff, P.; Favero, O.A.; Reimao, J.Q.; Lourenco, W.C.; Tempone, A.G.; Hristov, A.D.; Di Santi, S.M.; Lago, J.H.G.; Sartorelli, P.; et al. Anti-malarial, anti-trypanosomal, and anti-leishmanial activities of jacaranone isolated from Pentacalia desiderabilis (Vell.) Cuatrec. (Asteraceae). Parasitol. Res. 2012, 110, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Akendengue, B.; Ngou-Milama, E.; Roblot, F.; Laurens, A.; Hocquemiller, R.; Grellier, P.; Frappier, F. Antiplasmodial activity of Uvaria klaineana. Planta Med. 2002, 68, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.; Wube, A.A.; Bucar, F.; Bauer, R.; Kunert, O.; Haslinger, E. Quinolone alkaloids from Evodia rutaecarpa: A potent new group of antimycobacterial compounds. Int. J. Antimicrob. Agents 2005, 26, 262–264. [Google Scholar] [CrossRef] [PubMed]
- Dijoux, M.-G.; Schnabel, P.C.; Hallock, Y.F.; Boswell, J.L.; Johnson, T.R.; Wilson, J.A.; Ireland, C.M.; van Soest, R.; Boyd, M.R.; Barrows, L.R.; et al. Antitumor activity and distribution of pyrroloiminoquinones in the sponge genus Zyzzya. Bioorg. Med. Chem. 2005, 13, 6035–6044. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.-E.; Na, Z.; Jung, M.; Lee, H.-S.; Sim, C.J.; Nahm, K.; Oh, K.-B.; Shin, J. Discorhabdins from the Korean marine sponge Sceptrella sp. J. Nat. Prod. 2010, 73, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Sallam, A.A.; Ramasahayam, S.; Meyer, S.A.; El Sayed, K.A. Design, synthesis, and biological evaluation of dibromotyrosine analogues inspired by marine natural products as inhibitors of human prostate cancer proliferation, invasion, and migration. Bioorg. Med. Chem. 2010, 18, 7446–7457. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.-H.; Hwang, T.-L.; Thang, T.D.; Leu, Y.-L.; Kuo, P.-C.; Nguyet, B.T.M.; Dai, D.N.; Wu, T.-S. Isolation and synthesis of melodamide A, a new anti-inflammatory phenolic amide from the leaves of Melodorum fruticosum. Planta Med. 2013, 79, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aziz, A.A.-M. Novel and versatile methodology for synthesis of cyclic imides and evaluation of their cytotoxic, DNA binding, apoptotic inducing activities and molecular modeling study. Eur. J. Med. Chem. 2007, 42, 614–626. [Google Scholar] [CrossRef] [PubMed]
- Mitsunobu, O. The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural products. Synthesis 1981, 1–28. [Google Scholar] [CrossRef]
- Wetter, H.; Oertle, K. Thexyldimethylsilyl chloride, an easily accessible reagent for the protection of alcohols. Tetrahedron Lett. 1985, 26, 5515–5518. [Google Scholar] [CrossRef]
- Corey, E.J.; Venkateswarlu, A. Protection of hydroxyl groups as tert-butyldimethylsilyl derivatives. J. Am. Chem. Soc. 1972, 94, 6190–6191. [Google Scholar] [CrossRef]
- Walker, M.A. A High Yielding Synthesis of N-Alkyl Maleimides using a novel modification of the mitsunobu reaction. J. Org. Chem. 1995, 60, 5352–5355. [Google Scholar] [CrossRef]
- Lima, L.M.; Barreiro, E.J.; Fraga, C.A.M. O-alkylation of bioactive phthalimide derivatives under microwave irradiation in dry media. Synth. Commun. 2000, 30, 3291–3306. [Google Scholar] [CrossRef]
- Chen, J.; Spear, S.K.; Huddleston, J.G.; Rogers, R.D. Polyethylene glycol and solutions of polyethylene glycol as green reaction media. Green Chem. 2005, 7, 64–82. [Google Scholar] [CrossRef]
- Liang, J.; Lv, J.; Fan, J.-C.; Shang, Z.-C. Polyethylene Glycol as a Nonionic Liquid Solvent for the Synthesis of N-Alkyl and N-Arylimides. Synth. Commun. 2009, 39, 2822–2828. [Google Scholar] [CrossRef]
- Jain, S.L.; Singhal, S.; Sain, B. PEG-assisted solvent and catalyst free synthesis of 3,4-dihydropyrimidinones under mild reaction conditions. Green Chem. 2007, 9, 740–741. [Google Scholar] [CrossRef]
- Bähn, S.; Imm, S.; Neubert, L.; Zhang, M.; Neumann, H.; Beller, M. The Catalytic Amination of Alcohols. ChemCatChem 2011, 3, 1853–1864. [Google Scholar] [CrossRef]
- Smith, M.B.; March, J. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7 rev. ed.; Wiley: Hoboken, NJ, USA, 2012. [Google Scholar]
- Watson, T.J.; Ayers, T.A.; Shah, N.; Wenstrup, D.; Webster, M.; Freund, D.; Horgan, S.; Carey, J.P. Process improvements for the preparation of kilo quantities of a series of isoindoline compounds. Org. Process Res. Dev. 2003, 7, 521–532. [Google Scholar] [CrossRef]
- Artamonov, O.S.; Slobodyanyuk, E.Y.; Shishkin, O.V.; Komarov, I.V.; Mykhailiuk, P.K. Synthesis of isomeric 6-trifluoromethyl-3-azabicyclo[3.1.0]hexanes: Conformationally restricted analogues of 4-trifluoromethylpiperidine. Synthesis 2013, 45, 225–230. [Google Scholar] [CrossRef]
- Pan, S.; Shibata, T. Recent advances in iridium-catalyzed alkylation of C-H and N-H bonds. ACS Catal. 2013, 3, 704–712. [Google Scholar] [CrossRef]
- Fujita, K.-I.; Enoki, Y.; Yamaguchi, R. Cp*Ir-catalyzed N-alkylation of amines with alcohols. A versatile and atom economical method for the synthesis of amines. Tetrahedron 2008, 64, 1943–1954. [Google Scholar] [CrossRef]
- Iranpoor, N.; Firouzabadi, H.; Aghapour, G.; Vaez zadeh, A.R. Triphenylphosphine/2,3-dichloro-5,6-dicyanobenzoquinone as a new, selective and neutral system for the facile conversion of alcohols, thiols and selenols to alkyl halides in the presence of halide ions. Tetrahedron 2002, 58, 8689–8693. [Google Scholar] [CrossRef]
- Li, D.; Chen, S.; Bellomo, E.A.; Tarasov, A.I.; Kaut, C.; Rutter, G.A.; Li, W.-H. Imaging dynamic insulin release using a fluorescent zinc indicator for monitoring induced exocytotic release (ZIMIR). Proc. Natl. Acad. Sci. USA 2011, 108, 21063–21068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ankala, S.V.; Fenteany, G. Selective deprotection of either alkyl or aryl silyl ethers from aryl, alkyl bis-silyl ethers. Tetrahedron Lett. 2002, 43, 4729–4732. [Google Scholar] [CrossRef]
- Hou, D.-R.; Hsieh, Y.-D.; Hsieh, Y.-W. New formation of 4,5,6,7-tetrahydroisoindoles. Tetrahedron Lett. 2005, 46, 5927–5929. [Google Scholar] [CrossRef]
- Hou, D.-R.; Wang, M.-S.; Chung, M.-W.; Hsieh, Y.-D.; Tsai, H.-H.G. Formation of 4,5,6,7-Tetrahydroisoindoles by Palladium-Catalyzed Hydride Reduction. J. Org. Chem. 2007, 72, 9231–9239. [Google Scholar] [CrossRef] [PubMed]
- Felpin, F.-X. Oxidation of 4-arylphenol trimethylsilyl ethers to p-arylquinols using hypervalent iodine(III) reagents. Tetrahedron Lett. 2007, 48, 409–412. [Google Scholar] [CrossRef]
- Magdziak, D.; Meek, S.J.; Pettus, T.R.R. Cyclohexadienone ketals and quinols: Four building blocks potentially useful for enantioselective synthesis. Chem. Rev. 2004, 104, 1383–1430. [Google Scholar] [CrossRef] [PubMed]
- Quideau, S.; Pouysegu, L.; Deffieux, D. Oxidative dearomatization of phenols. Why, how and what for? Synlett 2008, 467–495. [Google Scholar] [CrossRef]
- Roche, S.P.; Porco, J.A., Jr. Dearomatization strategies in the synthesis of complex natural products. Angew. Chem. Int. Ed. 2011, 50, 4068–4093. [Google Scholar] [CrossRef] [PubMed]
- Harned, A.M. Asymmetric oxidative dearomatizations promoted by hypervalent iodine(III) reagents: An opportunity for rational catalyst design? Tetrahedron Lett. 2014, 55, 4681–4689. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Hoveyda, A.H.; Snapper, M.L. Catalytic enantioselective silylation of acyclic and cyclic triols: Application to total syntheses of cleroindicins D, F, and C. Angew. Chem. Int. Ed. 2009, 48, 547–550. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, M.P.; Hersey, A.; Montanari, D.; Overington, J. Probing the links between in vitro potency, ADMET and physicochemical parameters. Nat. Rev. Drug Discov. 2011, 10, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Leeson, P.D.; Oprea, T.I. Drug-like physicochemical properties. In Drug Design Strategies: Quantitative Approaches; Livingstone, D.J., Davis, A.M., Eds.; Royal Society of Chemistry: Cambridge, UK, 2012; pp. 35–59. [Google Scholar]
- Tian, S.; Wang, J.; Li, Y.; Li, D.; Xu, L.; Hou, T. The application of in silico drug-likeness predictions in pharmaceutical research. Adv. Drug Delivery Rev. 2015, 86, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Pajouhesh, H.; Lenz, G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2005, 2, 541–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bytheway, I.; Darley, M.G.; Popelier, P.L.A. The calculation of polar surface area from first principles: An application of quantum chemical topology to drug design. ChemMedChem 2008, 3, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Ferrins, L.; Gazdik, M.; Rahmani, R.; Varghese, S.; Sykes, M.L.; Jones, A.J.; Avery, V.M.; White, K.L.; Ryan, E.; Charman, S.A.; et al. Pyridyl benzamides as a novel class of potent inhibitors for the kinetoplastid Trypanosoma brucei. J. Med. Chem. 2014, 57, 6393–6402. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Keserü, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 2014, 13, 105–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavalluzzi, M.M.; Mangiatordi, G.F.; Nicolotti, O.; Lentini, G. Ligand efficiency metrics in drug discovery: The pros and cons from a practical perspective. Expert Opin. Drug Discov. 2017, 12, 1087–1104. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Zoete, V. A BOILED-Egg to Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Egan, W.J.; Merz, K.M., Jr.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef] [PubMed]
- Nwaka, S.; Ramirez, B.; Brun, R.; Maes, L.; Douglas, F.; Ridley, R. Advancing drug innovation for neglected diseases-criteria for lead progression. PLoS Negl. Trop. Dis. 2009, 3, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.-O.; Hou, X.; Jacob, C. 1,4-naphthoquinones: From oxidative damage to cellular and inter-cellular signaling. Molecules 2014, 19, 14902–14918. [Google Scholar] [CrossRef] [PubMed]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carneiro, P.F.; Pinto, M.C.R.F.; Marra, R.K.F.; da Silva, F.d.C.; Resende, J.A.L.C.; e Silva, L.F.R.; Alves, H.G.; Barbosa, G.S.; de Vasconcellos, M.C.; Lima, E.S.; et al. Synthesis and antimalarial activity of quinones and structurally-related oxirane derivatives. Eur. J. Med. Chem. 2016, 108, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Rinner, B.; Kretschmer, N.; Knausz, H.; Mayer, A.; Boechzelt, H.; Hao, X.-J.; Heubl, G.; Efferth, T.; Schaider, H.; Bauer, R. A petrol ether extract of the roots of Onosma paniculatum induces cell death in a caspase dependent manner. J. Ethnopharmacol. 2010, 129, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Matile, H.; Richard, J.; Pink, L. Plasmodium falciparum malaria parasite cultures and their use in immunology. In Immunological Methods, Volume IV; Lefkovits, I., Pernis, B., Eds.; Academic Press: Cambridge, MA, USA, 1990; pp. 221–234. [Google Scholar]
- Baltz, T.; Baltz, D.; Giroud, C.; Crockett, J. Cultivation in a semi-defined medium of animal infective forms of Trypanosoma brucei, T. equiperdum, T. evansi, T. rhodesiense and T. gambiense. EMBO J. 1985, 4, 1273–1277. [Google Scholar] [CrossRef] [PubMed]
- Räz, B.; Iten, M.; Grether-Bühler, Y.; Kaminsky, R.; Brun, R. The Alamar Blue assay to determine drug sensitivity of African trypanosomes (T.b. rhodesiense and T.b. gambiense) in vitro. Acta Trop. 1997, 68, 139–147. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 13a–j, 14a, 14c and 15 are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | P. falc.a | SI b | T.b.rhod.c | SI b | Cyt. L6 d | Chemical Structure | logP | tPSA (7.4) |
|---|---|---|---|---|---|---|---|---|
| IC50 (μM) | IC50 (μM) | IC50 (μM) | ||||||
| Chl. | 0.002 | |||||||
| Mel. | 0.004 | |||||||
| Pod. | 0.007 | |||||||
| 13a | 3.78 | 1.3 | 1.58 | 3.2 | 5.08 |  | 0.85 | 74.68 |
| 13b | 4.42 | 6.1 | 2.15 | 12.5 | 26.80 |  | −0.18 | 74.68 |
| 13c | 8.63 | 1.1 | 6.93 | 1.4 | 9.90 |  | −0.60 | 74.68 |
| 13d | 3.24 | 1.9 | 5.71 | 1.1 | 6.28 |  | 1.89 | 74.68 |
| 13e | 74.38 | 0.1 | 2.26 | 4.5 | 10.26 |  | −0.38 | 74.68 |
| 13f | 28.04 | 5.5 | 44.75 | 3.4 | 153.48 |  | −0.06 | 87.57 |
| 13g | 11.82 | 6.6 | 15.32 | 5.1 | 77.54 |  | −1.13 | 83.91 |
| 13h | 3.04 | 1.0 | 1.70 | 1.7 | 2.96 |  | 0.81 | 74.68 |
| 13i | 1.28 | 2.7 | 0.80 | 4.4 | 3.52 |  | 0.66 | 74.68 |
| 13j | 3.93 | 2.0 | 0.97 | 8.3 | 8.04 |  | 0.55 | 74.68 |
| 14a | 2.91 | 1.0 | 0.57 | 4.9 | 2.81 |  | 1.89 | 41.74 |
| 14c | 10.93 | 0.3 | 0.27 | 12.9 | 3.46 |  | 0.04 | 49.77 |
| 15 | 6.18 | 2.0 | 1.52 | 8.3 | 12.63 |  | 0.58 | 77.84 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Presser, A.; Lainer, G.; Kretschmer, N.; Schuehly, W.; Saf, R.; Kaiser, M.; Kalt, M.-M. Synthesis of Jacaranone-Derived Nitrogenous Cyclohexadienones and Their Antiproliferative and Antiprotozoal Activities. Molecules 2018, 23, 2902. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23112902
Presser A, Lainer G, Kretschmer N, Schuehly W, Saf R, Kaiser M, Kalt M-M. Synthesis of Jacaranone-Derived Nitrogenous Cyclohexadienones and Their Antiproliferative and Antiprotozoal Activities. Molecules. 2018; 23(11):2902. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23112902
Chicago/Turabian StylePresser, Armin, Gunda Lainer, Nadine Kretschmer, Wolfgang Schuehly, Robert Saf, Marcel Kaiser, and Marc-Manuel Kalt. 2018. "Synthesis of Jacaranone-Derived Nitrogenous Cyclohexadienones and Their Antiproliferative and Antiprotozoal Activities" Molecules 23, no. 11: 2902. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23112902