3.3. Deprotometalation Followed by Trapping with Electrophiles
3.3.1. General Procedure 1
To a solution of 2,2,6,6-tetramethylpiperidine (0.51 mL, 3.0 mmol) in THF (3 mL) at 0 °C were successively added BuLi (about 1.6 M hexanes solution, 3.0 mmol) and, 15 min later, ZnCl
2·TMEDA [
53] (0.25 g, 1.0 mmol). After 15 min at 0 °C, the pyrazine (2.0 mmol) was introduced, and the mixture was stirred for 2 h at rt before addition of I
2 (0.76 g, 3.0 mmol) in THF (3 mL) at 0 °C. The mixture was stirred at this temperature for 1 h before addition of an aqueous saturated solution of Na
2S
2O
3 (10 mL) and extraction with EtOAc (3 × 20 mL). The combined organic layers were dried over MgSO
4, filtered and concentrated under reduced pressure. The crude product was purified by chromatography over silica gel (the eluent is given in the product description).
3.3.2. 5-Iodo-2,3-diphenylquinoxaline (1b)
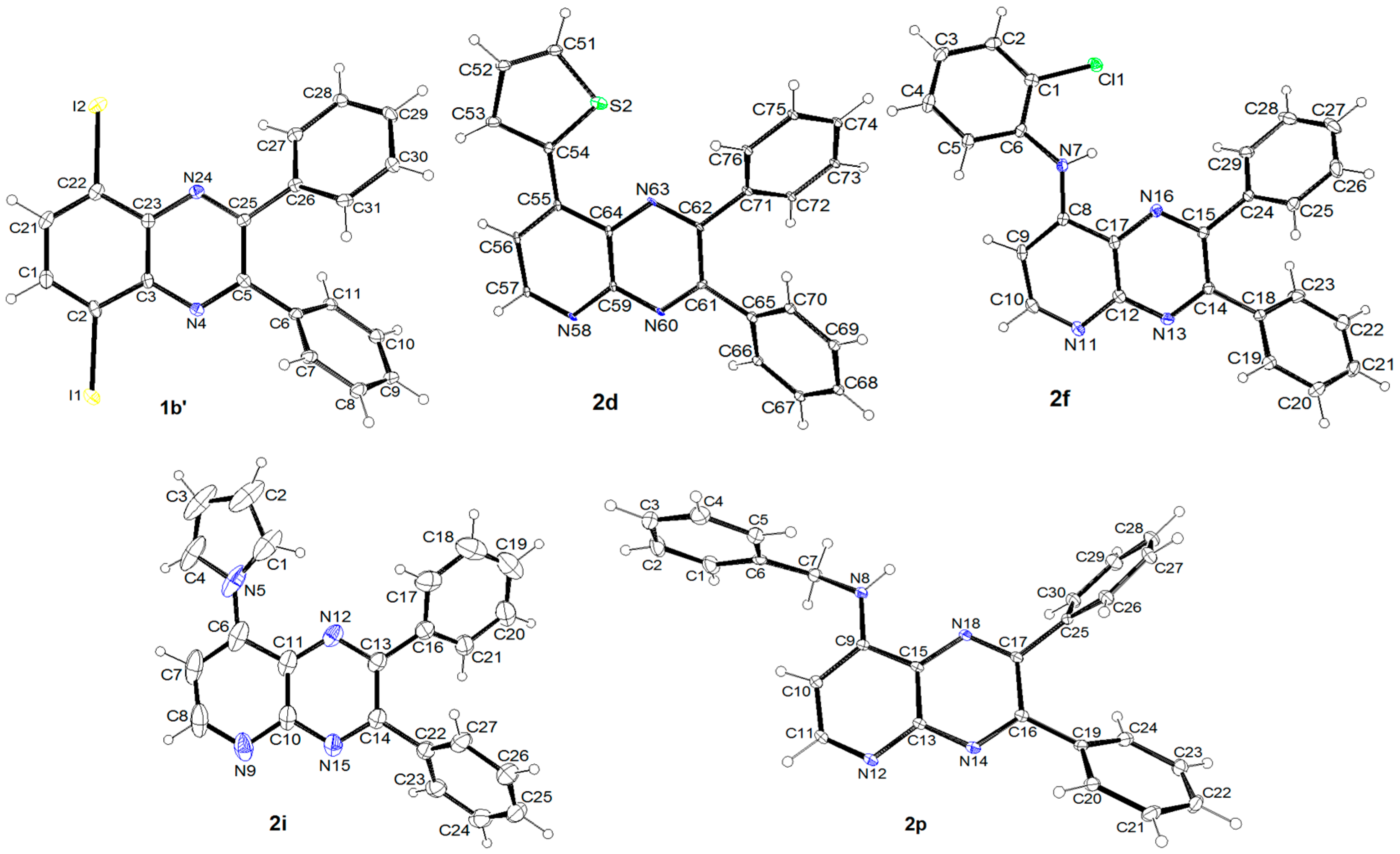
The general procedure 1 using 2,3-diphenylquinoxaline (1a, 0.56 g) gave 1b (eluent: heptane- CH2Cl2 60:40; Rf = 0.55) in 74% yield as a pale yellow powder. Mp: 148 °C. IR: 486, 529, 551, 602, 689, 695, 701, 763, 776, 796, 892, 978, 1023, 1068, 1079, 1184, 1281, 1336, 1384, 1497, 1534, 3051 cm−1. 1H-NMR (CDCl3): 7.31–7.41 (m, 6H), 7.48 (dd, 1H, J = 8.3 and 7.4 Hz), 7.54–7.57 (m, 2H), 7.64–7.67 (m, 2H), 8.15 (dd, 1H, J = 8.4 and 1.3 Hz), 8.36 (dd, 1H, J = 7.4 and 1.3 Hz). 13C-NMR (CDCl3): 102.8 (C), 128.3 (2CH), 128.5 (2CH), 129.2 (CH), 129.3 (CH), 129.9 (2CH), 130.0 (CH), 130.5 (2CH), 131.0 (CH), 138.2 (C), 138.7 (C), 140.1 (CH), 140.9 (C), 141.3 (C), 153.9 (C), 154.1 (C). Anal. Calc. for C20H13IN2 (408.24): C 58.84, H 3.21, N, 6.86. Found: C 59.05, H 3.27, N, 6.70. 5,8-Diiodo-2,3-diphenylquinoxaline (1b′) was similarly isolated (eluent: heptane-CH2Cl2 60:40; Rf = 0.69) in 7% yield as a yellow powder. Mp: 222 °C. IR: 533, 572, 613, 649, 692, 771, 824, 893, 978, 1025, 1055, 1077, 1169, 1209, 1325, 1383, 1447, 2930, 3059 cm−1. 1H-NMR (CDCl3): 7.34–7.45 (m, 6H), 7.64–7.76 (m, 4H), 8.02 (s, 2H). 13C-NMR (CDCl3): 103.5 (2C), 128.4 (4CH), 129.7 (2CH), 130.4 (4CH), 137.7 (2C), 140.6 (2CH), 140.8 (2C), 154.5 (2C). Crystal data for 1b′. C20H12I2N2, M = 534.12, T = 150(2) K, monoclinic, P 21, a = 10.1153(9), b = 5.8725(5), c = 14.9603(14) Å, β = 98.489(4) °, V = 878.94(14) Å3, Z = 2, d = 2.018 g cm−3, μ = 3.581 mm−1. A final refinement on F2 with 3888 unique intensities and 217 parameters converged at ωR(F2) = 0.0701 (R(F) = 0.0343) for 3602 observed reflections with I > 2σ(I). CCDC 1858478.
3.3.3. 8-Iodo-2,3-diphenylpyrido[2,3-b]pyrazine (2b-I)
The general procedure 1 using 2,3-diphenylpyrido[2,3-b]pyrazine (2a, 0.57 g) gave 2b-I (eluent: CH2Cl2; Rf = 0.34) in 70% yield as a whitish powder. Mp: 220 °C. IR: 534, 562, 613, 624, 637, 699, 980, 1023, 1336, 1416, 1519, 1570, 3068 cm−1. 1H-NMR (CDCl3): 7.32–7.44 (m, 6H), 7.64–7.69 (m, 4H), 8.28 (d, 1H, J = 4.5 Hz), 8.70 (d, 1H, J = 4.6 Hz). 13C-NMR (CDCl3): 116.1 (C), 128.3 (2CH), 128.4 (2CH), 129.7 (CH), 129.8 (CH), 130.3 (2CH), 130.3 (2CH), 135.6 (CH), 136.6 (C), 137.6 (C), 137.6 (C), 149.1 (C), 153.6 (CH), 155.0 (C), 157.1 (C). Anal. Calc. for C19H12IN3 (409.23): C 55.77, H 2.96, N, 10.27. Found: C 55.91, H 3.06, N, 10.03.
3.3.4. 8-Bromo-2,3-diphenylpyrido[2,3-b]pyrazine (2b-Br)
To a stirred mixture of 2,3-diphenyl pyrido[2,3-
b]pyrazine (
2a, 0.28 g, 1.0 mmol) and ZnCl
2· TMEDA [
53] (0.26 g, 1.0 mmol) in THF (1 mL) at −20 °C was added dropwise a solution of LiTMP (prepared by adding BuLi (about 1.6 M hexanes solution, 1.2 mmol) to a stirred, cooled (−20 °C) solution of 2,2,6,6-tetramethylpiperidine (0.24 mL, 1.2 mmol) in THF (2 mL) and stirring for 15 min) cooled at −20 °C. After 30 min at −20 °C, Br
2 (97 μL, 2.0 mmol) was introduced, and the mixture was stirred for 1 h before addition of an aqueous saturated solution of Na
2S
2O
3 (5 mL) and extraction with EtOAc (3 × 20 mL). The combined organic layers were dried over MgSO
4, filtered and concentrated under reduced pressure. The crude product was purified by chromatography over silica gel (eluent: CH
2Cl
2-EtOAc 90:10; R
f = 0.50) to give
2b-Br in 60% yield as a whitish powder. Mp: 183 °C. IR: 491, 538, 563, 615, 625, 649, 698, 767, 839, 985, 1021, 1049, 1090, 1179, 1241, 1336, 1387, 1421, 1460, 1524, 1584, 3067 cm
−1.
1H-NMR (CDCl
3): 7.32–7.42 (m, 6H), 7.63–7.66 (m, 4H), 8.00 (d, 1H,
J = 4.7 Hz), 8.91 (d, 1H,
J = 4.7 Hz).
13C-NMR (CDCl
3): 128.3 (CH), 128.4 (CH), 128.7 (CH), 129.7 (CH), 129.8 (CH), 130.3 (CH), 130.3 (CH), 134.7 (C), 136.3 (C), 137.7 (C), 137.9 (C), 150.1 (C), 153.4 (CH), 154.9 (C), 157.0 (C). Anal. Calc. for C
19H
12BrN
3 (362.23): C 63.00, H 3.34, N, 11.60. Found: C 63.24, H 3.58, N, 11.43.
3.3.5. 8-Chloro-2,3-diphenylpyrido[2,3-b]pyrazine (2b-Cl)
To a stirred mixture of 2,3-diphenyl pyrido[2,3-
b]pyrazine (
2a, 0.28 g, 1.0 mmol) and ZnCl
2· TMEDA [
53] (0.26 g, 1.0 mmol) in THF (1 mL) at −20 °C was added dropwise a solution of LiTMP (prepared by adding BuLi (about 1.6 M hexanes solution, 1.2 mmol) to a stirred, cooled (−20 °C) solution of 2,2,6,6-tetramethylpiperidine (0.24 mL, 1.2 mmol) in THF (2 mL) and stirring for 15 min) cooled at −20 °C. After 30 min at −20 °C, trichloroisocyanuric acid (0.30 g, 1.3 mmol) was introduced (CAUTION: dissolution of trichloroisocyanuric acid in THF at a temperature above 0 °C produces intense heat), and the mixture was stirred at this temperature for 1 h before addition of water (5 mL) and extraction with EtOAc (3 × 20 mL). The combined organic layers were dried over MgSO
4, filtered and concentrated under reduced pressure. The crude product was purified by chromatography over silica gel (eluent: CH
2Cl
2-EtOAc 90:10; R
f = 0.60) to give
2b-Cl in 62% yield as a whitish powder. Mp: 180 °C. IR: 534, 544, 617, 658, 699, 770, 851, 991, 1025, 1055, 1193, 1242, 1341, 1388, 1422, 1442, 1452, 1532, 1583, 3034, 3051 cm
−1.
1H-NMR (CDCl
3): 7.31–7.44 (m, 6H), 7.62–7.66 (m, 4H), 7.79 (d, 1H,
J = 4.7 Hz), 9.02 (d, 1H,
J = 4.7 Hz).
13C-NMR (CDCl
3): 125.1 (CH), 128.3 (CH), 128.5 (CH), 129.7 (CH), 129.8 (CH), 130.2 (CH), 130.3 (CH), 133.7 (C), 137.8 (C), 138.1 (C), 144.5 (C), 150.5 (C), 153.3 (CH), 154.8 (C), 157.1 (C). Anal. Calc. for C
19H
12ClN
3 (317.78): C 71.81, H 3.81, N, 13.22. Found: C 71.77, H 3.85, N, 13.14.
3.3.6. General Procedure 2
To a stirred mixture of the pyrazine (1.0 mmol) and ZnCl
2·TMEDA [
53] (0.26 g, 1.0 mmol) in THF (1 mL) at −20 °C was added dropwise a solution of LiTMP (prepared by adding BuLi (about 1.6 M hexanes solution, 1.2 mmol) to a stirred, cooled (−20 °C) solution of 2,2,6,6-tetramethylpiperidine (0.24 mL, 1.2 mmol) in THF (2 mL) and stirring for 15 min) cooled at −20 °C. After 30 min at −20 °C, I
2 (0.37 g, 1.5 mmol) in THF (2 mL) was introduced, and the mixture was stirred at this temperature for 1 h before addition of an aqueous saturated solution of Na
2S
2O
3 (5 mL) and extraction with EtOAc (3 × 20 mL). The combined organic layers were dried over MgSO
4, filtered and concentrated under reduced pressure. The crude product was purified by chromatography over silica gel (the eluent is given in the product description).
3.3.7. 8-Bromo-7-iodo-2,3-diphenylpyrido[3,4-b]pyrazine (3b)
The general procedure 2 using 8-bromo-2,3-diphenylpyrido[3,4-
b]pyrazine (
3a [
25,
26], 0.36 g) gave
3b (eluent: CH
2Cl
2-petroleum ether 80:20; R
f = 0.44) in 67% yield as a red powder. Mp: 186–188 °C. IR: 493, 529, 559, 600, 658, 695, 765, 984, 1025, 1055, 1117, 1238, 1315, 1373, 1399, 1446, 1493, 1551, 3034, 3060 cm
−1.
1H-NMR (CDCl
3): 7.34–7.47 (m, 6H), 7.54–7.57 (m, 2H), 7.62–7.65 (m, 2H), 9.27 (s, 1H).
13C-NMR (CDCl
3): 121.7 (C), 128.6 (2CH), 128.7 (2CH), 129.7 (C), 129.8 (2CH), 130.1 (CH), 130.5 (2CH), 130.5 (CH), 136.0 (C), 137.4 (C), 137.7 (C), 142.3 (C), 152.5 (CH), 156.2 (C), 158.6 (C). Anal. Calc. for C
19H
11BrIN
3 (488.13): C 46.75, H 2.27, N, 8.61. Found: C 46.89, H 2.49, N, 8.55. 8-Bromo-5,7-diiodo-2,3-diphenyl pyrido[3,4-
b]pyrazine, also formed in <5% yield, was identified by its
1H-NMR (CDCl
3): 7.36–7.47 (m, 6H), 7.54–7.57 (m, 2H), 7.65–7.68 (m, 4H).
3.3.8. 7-Bromo-6-iodo-2,3-diphenylpyrido[2,3-b]pyrazine (4b)
The general procedure 2 using 7-bromo-2,3-diphenylpyrido[2,3-
b]pyrazine (
4a [
54], prepared in 90% yield [
6], 0.36 g) gave
4b (eluent: CH
2Cl
2-heptane 70:30; R
f (heptane-CH
2Cl
2 80:20) = 0.80) in 5% yield as a yellow powder. Mp: 150–152 °C. IR: 495, 547, 596, 615, 697, 731, 770, 778, 903, 937, 1025, 1060, 1107, 1178, 1274, 1332, 1390, 1448, 1502, 1562, 1603, 1699, 1768, 2734, 2940, 3064 cm
−1.
1H-NMR (CDCl
3): 7.30–7.43 (m, 6H), 7.52–7.55 (m, 2H), 7.59–7.62 (m, 2H), 8.62 (s, 1H).
13C-NMR (CDCl
3): 128.3 (2CH), 128.6 (2CH), 128.7 (C), 129.9 (CH), 129.9 (2CH), 130.0 (CH), 130.0 (C), 130.3 (2CH), 135.5 (C), 137.6 (C), 138.0 (C), 139.5 (CH), 148.0 (C), 155.9 (C), 157.1 (C). Anal. Calc. for C
19H
11BrIN
3 (488.13): C 46.75, H 2.27, N, 8.61. Found: C 46.93, H 2.38, N, 8.49.
3.4. Suzuki Coupling Reactions
3.4.1. General Procedure 3
To a stirred mixture of the iodide (0.50 mmol) and Pd(PPh3)4 (29 mg, 25 μmol) in degassed 1,2-dimethoxyethane (5 mL) was added the boronic acid (0.60 mmol) and NaHCO3 (2.0 mmol) in degassed water (1.6 mL). The resulting mixture was heated at 80 °C for 3 h and cooled to rt before addition of water (5 mL) and extraction with EtOAc (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by chromatography over silica gel (the eluent is given in the product description).
3.4.2. 2,3,5-Triphenylquinoxaline (1c)
The general procedure 3 using 5-iodo-2,3-diphenyl quinoxaline (1b, 0.20 g) and phenylboronic acid (73 mg) gave 1c (eluent: CH2Cl2-heptane 60:40; Rf = 0.35) in 42% yield as a white powder. Mp: 150 °C. IR: 763, 804, 841, 927, 984, 1023, 1081, 1128, 1233, 1336, 1388, 1433, 1444, 1491, 1566, 2858, 2927, 2965, 3064 cm−1. 1H-NMR (CDCl3): 7.26–7.34 (m, 3H), 7.36–7.48 (m, 4H), 7.52–7.64 (m, 6H), 7.81–7.89 (m, 4H), 8.21 (dd, 1H, J = 7.3 and 2.5 Hz). 13C-NMR (CDCl3): 127.7 (CH), 128.0 (2CH), 128.1 (2CH), 128.5 (2CH), 128.7 (CH), 128.8 (CH), 129.0 (CH), 129.8 (CH), 129.9 (2CH), 130.3 (2CH), 130.4 (CH), 131.1 (2CH), 138.4 (C), 139.0 (C), 139.1 (C), 139.4 (C), 140.6 (C), 141.3 (C), 152.4 (C), 152.9 (C). Anal. Calc. for C26H18N2 (358.44): C 87.12, H 5.06, N, 7.82. Found: C 87.25, H 5.22, N, 7.70.
3.4.3. 2,3-Diphenyl-5-(2-thienyl)quinoxaline (1d)
The general procedure 3 using 5-iodo-2,3-diphenylquinoxaline (1b, 0.20 g) and 2-thienylboronic acid (77 mg) gave 1d (eluent: CH2Cl2-heptane 60:40; Rf = 0.20) in 97% yield as a yellow powder. Mp: 210 °C. IR: 738, 766, 796, 828, 854, 916, 933, 969, 1025, 1053, 1083, 1163, 1238, 1336, 1390, 1442, 1495, 1562, 1592, 3064 cm−1. 1H-NMR (CDCl3): 7.18 (dd, 1H, J = 5.1 and 3.7 Hz), 7.32–7.40 (m, 6H), 7.51 (dd, 1H, J = 5.1 and 1.2 Hz), 7.58–7.61 (m, 2H), 7.67–7.70 (m, 2H), 7.76 (dd, 1H, J = 8.3 and 7.4 Hz), 7.88 (dd, 1H, J = 3.7 and 1.2 Hz), 8.08 (dd, 1H, J = 8.3 and 1.3 Hz), 8.13 (dd, 1H, J = 7.4 and 1.3 Hz). 13C-NMR (CDCl3): 126.7 (CH), 126.9 (CH), 127.5 (CH), 128.1 (CH), 128.2 (2CH), 128.5 (2CH), 128.8 (CH), 129.0 (CH), 129.0 (CH), 129.9 (2CH), 129.9 (CH), 130.6 (2CH), 133.0 (C), 137.6 (C), 138.8 (C), 138.9 (C), 139.2 (C), 141.4 (C), 152.3 (C), 153.2 (C). Anal. Calc. for C24H16N2S (364.47): C 79.09, H 4.43, N, 7.69. Found: C 79.11, H 4.48, N, 7.72.
3.4.4. 2,3-Diphenyl-8-(2-thienyl)pyrido[2,3-b]pyrazine (2d)
The general procedure 3 using 8-iodo-2,3-diphenylpyrido[2,3-b]pyrazine (2b-I, 0.20 g) and 2-thienylboronic acid (77 mg) gave 2d (eluent: CH2Cl2-EtOAc 95:5; Rf = 0.50) in 75% yield as a pale yellow powder. Mp: 215 °C. IR: 540, 695, 744, 1025, 1096, 1120, 1188, 1238, 1336, 1384, 1435, 1480, 551, 1568, 2927, 2965, 3060 cm−1. 1H-NMR (CDCl3): 7.23 (dd, 1H, J = 5.1 and 3.8 Hz), 7.32–7.45 (m, 6H), 7.66–7.72 (m, 5H), 7.98 (d, 1H, J = 4.8 Hz), 8.10 (dd, 1H, J = 3.8 and 1.2 Hz), 9.10 (d, 1H, J = 4.8 Hz). 13C-NMR (CDCl3): 120.3 (CH), 127.2 (CH), 128.3 (2CH), 128.4 (2CH), 129.2 (CH), 129.4 (CH), 129.5 (CH), 130.3 (2CH), 130.5 (2CH), 132.4 (CH), 132.4 (C), 136.0 (C), 138.2 (C), 138.3 (C), 140.9 (C), 150.2 (C), 153.1 (C), 153.9 (CH), 155.8 (C). Crystal data for 2d. C23H15N3S, M = 365.44, T = 150(2) K, triclinic, P 1, a = 6.6311(18), b = 9.939(3), c = 13.655(4) Å, α = 81.914(12), β = 80.405(11), γ = 89.955(10) °, V = 878.3(4) Å3, Z = 2, d = 1.382 g cm−3, μ = 0.197 mm−1. A final refinement on F2 with 7113 unique intensities and 236 parameters converged at ωR(F2) = 0.3351 (R(F) = 0.1327) for 6147 observed reflections with I > 2σ(I). CCDC 1858479.
3.4.5. 5-(2-Aminophenyl)-2,3-diphenylquinoxaline (1e)
The general procedure 3 using 5-iodo-2,3-diphenylquinoxaline (1b, 0.20 g) and 2-amino- phenylboronic acid (82 mg) gave 1e (eluent: heptane-CH2Cl2 70:30; Rf = 0.31) in 92% yield as a yellow powder. Mp: 178 °C. IR: 689, 702, 740, 771, 977, 1307, 1342, 1492, 1626, 3025, 3060, 3212, 3328, 3468 cm−1. 1H-NMR (CDCl3): 3.87 (br s, 2H, NH2), 6.85 (dd, 1H, J = 7.9 and 1.1 Hz), 6.92 (td, 1H, J = 7.4 and 1.2 Hz), 7.21–7.30 (m, 5H), 7.35–7.40 (m, 3H), 7.47–7.50 (m, 2H), 7.55–7.58 (m, 2H), 7.78–7.86 (m, 2H), 8.20 (dd, 1H, J = 7.8 and 2.1 Hz). 13C-NMR (CDCl3): 116.5 (CH), 118.8 (CH), 125.7 (C), 128.1 (2CH), 128.5 (2CH), 128.9 (CH), 129.0 (CH), 129.0 (CH), 129.0 (CH), 129.9 (2CH), 130.2 (CH), 130.3 (2CH), 132.0 (CH), 132.3 (CH), 138.8 (C), 139.2 (C), 139.3 (C), 139.6 (C), 141.3 (C), 145.0 (C), 152.5 (C), 153.2 (C). Anal. Calc. for C26H19N3 (373.46): C 83.62, H 5.13, N, 11.25. Found: C 83.81, H 5.26, N, 11.17.
3.4.6. 8-(2-Aminophenyl)-2,3-diphenylpyrido[2,3-b]pyrazine (2e)
The general procedure 3 using 8-iodo-2,3-diphenylpyrido[2,3-b]pyrazine (2b-I, 0.20 g) and 2-aminophenylboronic acid (82 mg) gave 2e (eluent: CH2Cl2-EtOAc 70:30; Rf = 0.50) in 73% yield as a yellow powder. Mp: 205 °C. IR: 687, 742, 766, 854, 981, 1015, 1047, 1237, 1307, 1382, 1489, 1623, 3024, 3055, 3345 cm−1. 1H-NMR (CDCl3): 3.99 (br s, 2H, NH2), 6.87 (dd, 1H, J = 8.4 and 1.2 Hz), 6.94 (td, 1H, J = 7.4 and 1.2 Hz), 7.25–7.40 (m, 8H), 7.51–7.54 (m, 2H), 7.65–7.68 (m, 2H), 7.73 (d, 1H, J = 4.5 Hz), 9.19 (d, 1H, J = 4.4 Hz). 13C-NMR (CDCl3): 116.9 (CH), 118.7 (CH), 122.6 (C), 126.3 (CH), 128.2 (2CH), 128.2 (2CH), 129.3 (CH), 129.5 (CH), 130.1 (CH), 130.1 (2CH), 130.1 (2CH), 132.0 (CH), 134.3 (C), 138.1 (C), 138.2 (C), 144.9 (C), 149.0 (C), 149.8 (C), 153.4 (C), 154.1 (CH), 155.7 (C). Anal. Calc. for C25H18N4 (374.45): C 80.19, H 4.85, N, 14.96. Found: C 80.07, H 4.87, N, 14.85.
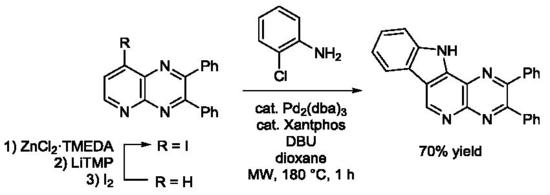
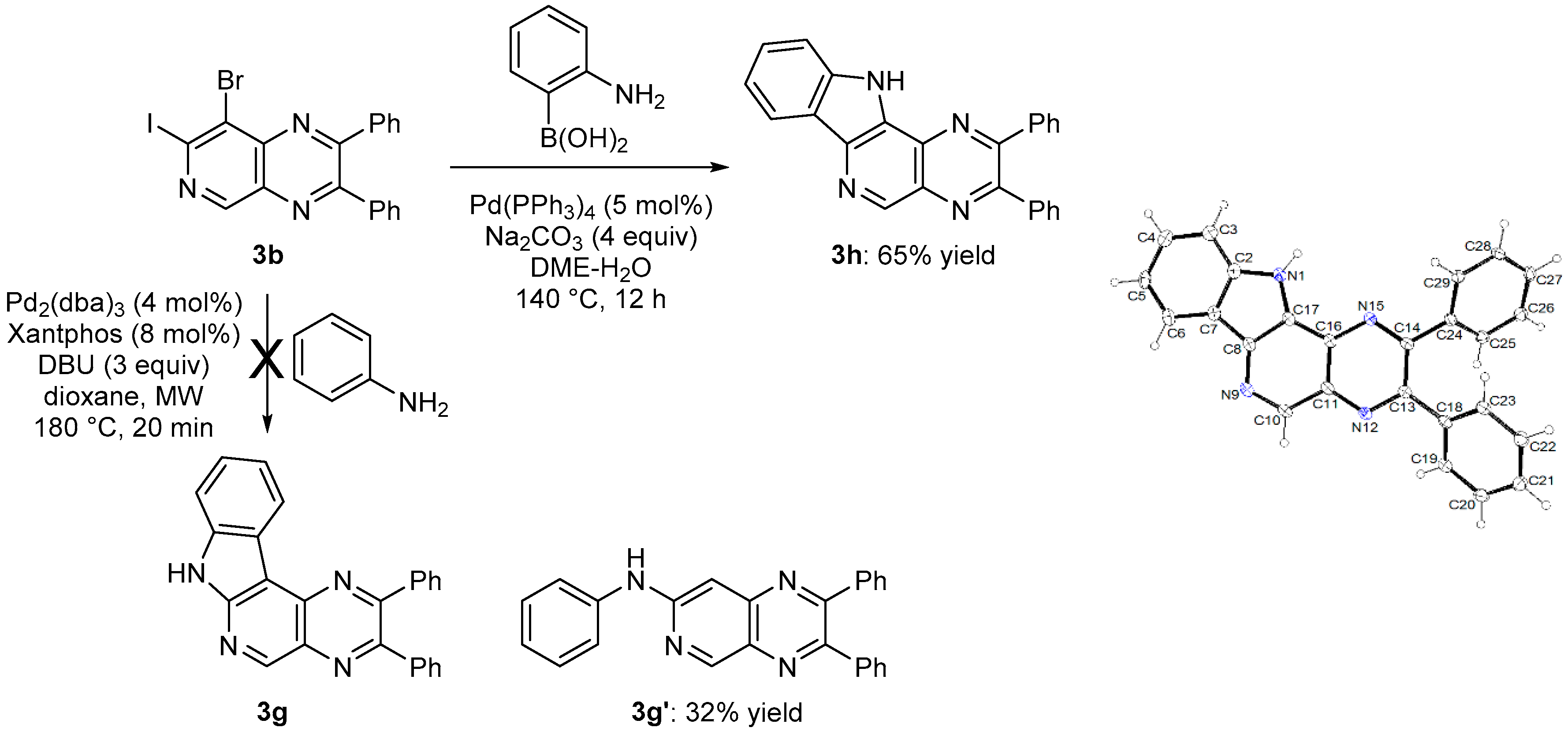
3.4.7. 2,3-Diphenyl-11H-pyrazino[2′,3′:4,5]pyrido[2,3-d]indole (3h)
In a tube containing a stirred mixture of 8-bromo-7-iodo-2,3-diphenylpyrido[3,4-b]pyrazine (3b, 0.24 g, 0.50 mmol) and Pd(PPh3)4 (29 mg, 25 μmol) in degassed 1,2-dimethoxyethane (5 mL) was introduced 2-aminophenylboronic acid (82 mg, 0.60 mmol) and Na2CO3 (2.0 mmol) in degassed water (1.6 mL). The sealed tube was heated overnight at 140 °C and cooled to rt before addition of saturated aqueous NaHCO3 (5 mL) and extraction with EtOAc (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by chromatography over silica gel (eluent: CH2Cl2-EtOAc 90:10; Rf = 0.28) to give 3h in 65% yield as a yellow powder. Mp: 284–286 °C. IR: 695, 748, 763, 1025, 1092, 1190, 1236, 1315, 1328, 1336, 1376, 1446, 1495, 1540, 1624, 3034, 3064, 3420 cm−1. 1H-NMR (CDCl3): 7.30–7.42 (m, 7H), 7.50–7.60 (m, 6H), 8.45 (d, 1H, J = 7.9 Hz), 9.47 (s, 1H), 9.78 (br s, 1H). 13C-NMR (CDCl3): 111.9 (CH), 120.6 (CH), 121.3 (CH), 123.1 (C), 126.7 (C), 127.2 (CH), 128.4 (2CH), 128.5 (2CH), 129.2 (CH), 129.6 (CH), 130.0 (2CH), 130.1 (2CH), 132.4 (C), 134.9 (C), 138.4 (C), 138.6 (C), 138.8 (C), 139.5 (C), 146.5 (CH), 153.6 (C), 155.9 (C). Crystal data for 3h. C25H16N4, M = 372.42, T = 150(2) K, orthorhombic, Pbca, a = 7.1524(9), b = 16.3313(17), c = 33.798(4) Å, V = 3947.9(8) Å3, Z = 8, d = 1.253 g cm−3, μ = 0.076 mm−1. A final refinement on F2 with 4429 unique intensities and 265 parameters converged at ωR(F2) = 0.1564 (R(F) = 0.0739) for 3511 observed reflections with I > 2σ(I). CCDC 1858477. This compound was also obtained in 64% yield under microwave irradiation (300 W; Monowave 300, Anton Paar, Graz, Austria) for 30 min at 150 °C.
3.5. 8-(2-Azidophenyl)-2,3-diphenylpyrido[2,3-b]pyrazine
To a stirred solution of 8-(2-aminophenyl)-2,3-diphenylpyrido[2,3-b]pyrazine (2e, 94 mg, 0.25 mmol) in acetic acid (1.5 mL) at 0 °C was added 1M aqueous NaNO2 (0.35 mL, 0.35 mmol). After stirring for 1 h at rt, the solution was cooled to 0 °C before addition of 1M aqueous NaN3 (0.35 mL, 0.35 mmol). After stirring overnight at rt, 3 mL of saturated aqueous NaHCO3 were added. Extraction with EtOAc (3 × 10 mL), washing of the combined organic layers with brine (10 mL), drying over MgSO4, filtration and concentration under reduced pressure afforded a brown powder which was purified by chromatography over silica gel (eluent: CH2Cl2-EtOAc 95:5; Rf = 0.50) to afford the azide in 64% yield. IR: 685, 745, 1288, 1440, 1577, 2088, 2124, 3064 cm−1. 1H-NMR (CDCl3): 7.23–7.40 (m, 8H), 7.45–7.57 (m, 4H), 7.66–7.69 (m, 3H), 9.18 (d, 1H, J = 4.4 Hz). 13C-NMR (CDCl3): 118.8 (CH), 124.7 (CH), 126.0 (CH), 127.9 (C), 128.2 (2CH), 128.2 (2CH), 129.2 (CH), 129.5 (CH), 130.1 (2CH), 130.3 (2CH), 130.4 (CH), 132.5 (CH), 134.5 (C), 138.3 (C), 138.5 (C), 138.7 (C), 146.7 (C), 149.8 (C), 153.5 (CH), 153.7 (C), 155.8 (C).
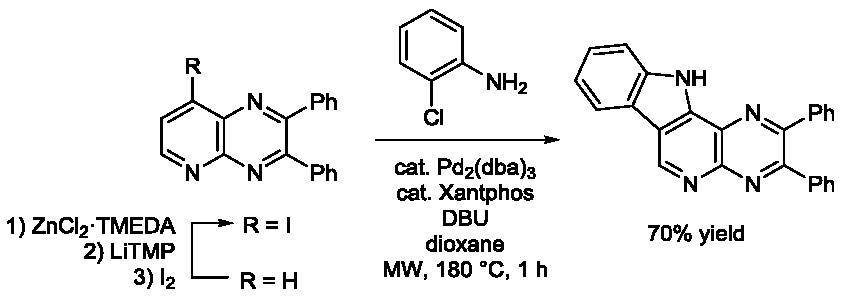
3.6. Palladium-Catalyzed N-arylation
3.6.1. General Procedure 4
To a stirred mixture of the halide (0.50 mmol) and Cs2CO3 (0.48 g, 1.5 mmol) in 2-chloroaniline (63 μL, 0.60 mmol) was added a solution of the catalyst prepared by stirring Pd2(dba)3 (11 mg, 12.5 μmol) and Xantphos (16 mg, 27.5 μmol) in degassed dioxane (2 mL) for 10 min at rt. The resulting mixture was heated at 110 °C for 24 h and cooled to rt before addition of water (5 mL) and extraction with EtOAc (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by chromatography over silica gel (the eluent is given in the product description).
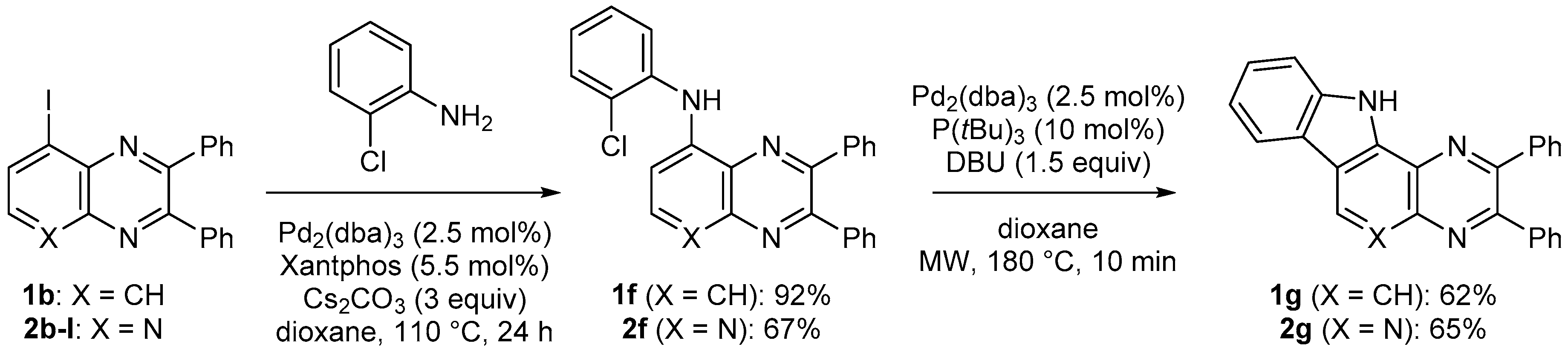
3.6.2. 5-(2-Chlorophenylamino)-2,3-diphenylquinoxaline (1f)
The general procedure 4 using 5-iodo-2,3-diphenylquinoxaline (1b, 0.20 g) gave 1f (eluent: heptane-CH2Cl2 60:40; Rf = 0.42) in 92% yield as a yellow powder. Mp: 182 °C. IR: 695, 729, 748, 959, 1021, 1055, 1072, 1098, 1182, 1218, 1317, 1343, 1356, 1394, 1442, 1454, 1497, 1534, 1562, 1579, 1594, 1613, 3060, 3347 cm−1. 1H-NMR (CDCl3): 6.95 (td, 1H, J = 7.7 and 1.4 Hz), 7.27–7.39 (m, 7H), 7.46–7.52 (m, 2H), 7.55–7.62 (m, 4H), 7.64–7.66 (m, 2H), 7.71 (dd, 1H, J = 8.2 and 1.4 Hz), 8.56 (br s, 1H). 13C-NMR (CDCl3): 109.1 (CH), 118.9 (CH), 118.9 (CH), 122.6 (CH), 125.1 (C), 127.6 (CH), 128.2 (2CH), 128.4 (2CH), 128.9 (CH), 128.9 (CH), 129.9 (2CH), 130.1 (2CH), 130.2 (CH), 130.9 (CH), 132.1 (C), 138.4 (C), 138.9 (C), 139.2 (C), 139.3 (C), 141.9 (C), 150.4 (C), 153.9 (C). Anal. Calc. for C26H18ClN3 (407.90): C 76.56, H 4.45, N, 10.30. Found: C 76.89, H 4.58, N, 10.13.
3.6.3. 8-(2-Chlorophenylamino)-2,3-diphenylpyrido[2,3-b]pyrazine (2f)
The general procedure 4 using 8-iodo-2,3-diphenylpyrido[2,3-b]pyrazine (2b-I, 0.20 g) gave 2f (eluent: CH2Cl2-EtOAc 90:10; Rf = 0.32) in 67% yield as a yellow powder. Mp: 202 °C. IR: 542, 699, 755, 1021, 1102, 1242, 1313, 1336, 1356, 1437, 1452, 1534, 1558, 1583, 1646, 3060, 3322, 3631 cm−1. 1H-NMR (CDCl3): 7.13–7.19 (m, 2H), 7.30–7.41 (m, 7H), 7.53 (dd, 1H, J = 8.0 and 1.5 Hz), 7.57–7.65 (m, 4H), 7.68 (dd, 1H, J = 8.1 and 1.5 Hz), 8.77 (br s, 1H, NH), 8.81 (d, 1H, J = 5.4 Hz, H6). 13C-NMR (CDCl3): 102.8 (CH), 122.2 (CH), 125.5 (CH), 127.2 (C), 127.5 (C), 127.8 (CH), 128.2 (2CH), 128.4 (2CH), 129.2 (CH), 129.4 (CH), 130.0 (2CH), 130.3 (2CH), 130.5 (CH), 136.2 (C), 138.4 (C), 138.5 (C), 147.0 (C), 150.3 (C), 151.1 (C), 155.0 (CH), 156.5 (C). Crystal data for 2f. C25H17ClN4, M = 408.88, T = 150(2) K, orthorhombic, P c a 21, a = 15.3485(15), b = 18.8937(16), c = 6.9936(7) Å, V = 2028.1(3) Å3, Z = 4, d = 1.339 g cm−3, μ = 0.208 mm−1. A final refinement on F2 with 4578 unique intensities and 274 parameters converged at ωR(F2) = 0.1478 (R(F) = 0.0583) for 4133 observed reflections with I > 2σ(I). CCDC 1858474.
3.7. Palladium-Catalyzed N-arylation
2,3-Diphenyl-11
H-pyrazino[2,3-
a]carbazole (
1g) was prepared by adapting a reported procedure [
40]. To a stirred mixture of 5-(2-chlorophenylamino)-2,3-diphenylquinoxaline (
1f, 0.24 g, 0.60 mmol) and 1,8-diazabicyclo[5.4.0]undec-7-ene (0.13 mL, 0.90 mmol), was added a solution of the catalyst prepared by stirring Pd
2(dba)
3 (14 mg, 15 μmol) and P(
tBu)
3 (12 mg, 60 μmol) in degassed dioxane (1 mL) for 10 min at rt. The resulting mixture was heated by microwave irradiation (300 W; Monowave 300, Anton Paar, Graz, Austria) for 10 min at 180 °C before addition of water (5 mL) and extraction with EtOAc (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over MgSO
4, filtered and concentrated under reduced pressure. The crude product was purified by chromatography over silica gel (eluent: heptane-CH
2Cl
2 60:40; R
f = 0.48) to give
1g in 62% yield as a yellow powder. Mp: 260 °C. IR: 1025, 1087, 1102, 1175, 1242, 1326, 1347, 1362, 1384, 1444, 1459, 1624, 1731, 2854, 2922, 3420 cm
−1.
1H-NMR ((CD
3)
2SO): 6.86 (ddd, 1H,
J = 8.0, 7.1 and 1.0 Hz), 6.91–6.97 (m, 6H), 7.01–7.10 (m, 3H), 7.14–7.17 (m, 2H), 7.28 (d, 1H,
J = 8.3 Hz), 7.39 (d, 1H,
J = 8.7 Hz), 7.84 (d, 1H,
J = 7.8 Hz), 8.14 (d, 1H,
J = 8.7 Hz), 12.13 (br s, 1H).
13C-NMR ((CD
3)
2SO): 112.2 (CH), 118.8 (CH), 119.7 (CH), 120.3 (CH), 120.7 (C), 122.7 (C), 124.2 (CH), 125.6 (CH), 128.0 (2CH), 128.0 (2CH), 128.5 (CH), 128.6 (CH), 129.7 (2CH), 129.9 (2CH), 130.0 (C), 134.3 (C), 139.0 (C), 139.2 (C), 139.8 (C), 139.9 (C), 150.7 (C), 151.4 (C). Anal. Calc. for C
26H
17N
3 (371.44): C 84.07, H 4.61, N, 11.31. Found: C 84.19, H 4.52, N, 11.12.
3.8. One-Pot Palladium-Catalyzed N-arylation/C-H Arylation
3.8.1. General Procedure 5
To a mixture of the halide (0.25 mmol), 1,8-diazabicyclo[5.4.0]undec-7-ene (118 μL, 0.75 mmol), 2-chloroaniline (38 mg, 0.30 mmol), Pd2(dba)3 (9.2 mg, 10 μmol) and Xantphos (13 mg, 22 μmol), was added degassed 1,4-dioxane (1 mL). The mixture was heated by microwave irradiation (150 W; Monowave 300, Anton Paar, Graz, Austria) under the conditions given in the product description. The cooled residue was taken up with EtOAc (20 mL). The organic layer was washed with brine (10 mL), dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by chromatography over silica gel (the eluent is given in the product description).
3.8.2. 2,3-Diphenyl-11H-pyrazino[2′,3′:5,6]pyrido[4,3-b]indole (2g)
The general procedure 5 (1 h at 180 °C) using 8-iodo-2,3-diphenylpyrido[2,3-b]pyrazine (2b-I, 0.10 g) gave 2g (eluent: CH2Cl2-EtOAc 90:10; Rf = 0.43) in 70% yield as a white powder. Mp > 260 °C. IR: 525, 542, 551, 626, 699, 750, 768, 1025, 1045, 1075, 1100, 1236, 1339, 1373, 1444, 1555, 1736, 2665, 3056 cm−1. 1H-NMR ((CD3)2SO): 7.40–7.45 (m, 7H), 7.55–7.63 (m, 5H), 7.77 (dd, 1H, J = 8.2 and 0.9 Hz), 8.42 (dt, 1H, J = 7.8 and 1.0 Hz), 9.87 (s, 1H), 13.19 (s, 1H). 13C-NMR ((CD3)2SO): 112.6 (CH), 118.2 (C), 120.7 (CH), 121.3 (CH), 121.4 (C), 126.4 (C), 126.5 (CH), 128.0 (CH), 128.0 (2CH), 128.1 (2CH), 128.8 (CH), 129.8 (2CH), 129.9 (2CH), 138.6 (C), 138.7 (C), 139.4 (C), 140.0 (C), 147.4 (C), 148.5 (CH), 151.4 (C), 153.4 (C). Anal. Calc. for C25H16N4 (372.43): C 80.63, H 4.33, N, 15.04. Found: C 80.54, H 4.28, N, 14.89.
3.8.3. 7-(Phenylamino)-2,3-diphenylpyrido[3,4-b]pyrazine (3g′)
The general procedure 5 (40 min at 180 °C) using 8-bromo-7-iodo-2,3-diphenylpyrido[3,4-b] pyrazine (3b, 0.12 g) gave 3g′ (eluent: CH2Cl2-MeOH 99:1; Rf = 0.27) in 32% yield as a yellow powder. Mp: 224–226 °C. IR: 699, 750, 770, 978, 1025, 1057, 1077, 1169, 1197, 1261, 1336, 1349, 1435, 1450, 1527, 1555, 1588, 1613, 2854, 2927, 2961, 3025, 3232 cm−1. 1H-NMR (CDCl3): 7.14 (p, 1H, J = 4.4 Hz), 7.23 (br s, 1H), 7.29–7.49 (m, 15H), 9.26 (s, 1H). 13C-NMR (CDCl3): 158.3 (C), 155.8 (C), 154.0 (CH), 151.4 (C), 146.3 (C), 139.8 (C), 138.8 (C), 138.7 (C), 132.4 (C), 129.8 (2CH), 129.7 (2CH), 129.7 (2CH), 129.5 (CH), 128.9 (CH), 128.4 (2CH), 128.4 (2CH), 124.1 (CH), 121.4 (2CH), 98.3 (CH). Anal. Calc. for C25H18N4 (374.45): C 80.19, H 4.85, N, 14.96. Found: C 80.17, H 4.99, N, 14.84.
3.9. Copper-Catalyzed N-arylation
3.9.1. General Procedure 6
A mixture containing the iodide (0.50 mmol) and azole (1.0 mmol), Cu2O (6.0 mg, 0.10 mmol), Cs2CO3 (0.33 g, 1.0 mmol) and DMSO (0.5 mL) was stirred at 110 °C for 24 h. The cooled residue was taken up with EtOAc (20 mL) and filtered through a Celite pad. The organic layer was washed with water (10 mL) and brine (10 mL), dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by chromatography over silica gel (the eluent is given in the product description).
3.9.2. 2,3-Diphenyl-8-(N-pyrrolyl)pyrido[2,3-b]pyrazine (2i)
The general procedure 6 using 8-iodo-2,3-diphenylpyrido[2,3-b]pyrazine (2b-I, 0.20 g) and pyrrole (67 mg) gave 2i (eluent: CH2Cl2-EtOAc 90:10; Rf = 0.47) in 67% yield as a yellow powder. Mp: 210 °C. IR: 946, 1025, 1072, 1096, 1107, 1173, 1238, 1289, 1328, 1362, 1388, 1433, 1454, 1482, 1549, 1588, 3025, 3060, 3111, 3141, 3180 cm−1. 1H-NMR (CDCl3): 6.39–6.40 (m, 2H), 7.24–7.35 (m, 6H), 7.49–7.53 (m, 3H), 7.59–7.62 (m, 2H), 7.65–7.66 (m, 2H), 9.01 (d, 1H, J = 5.0 Hz). 13C-NMR (CDCl3): 112.0 (2CH), 115.1 (CH), 122.7 (2CH), 128.3 (2CH), 128.4 (2CH), 129.4 (C), 129.5 (CH), 129.8 (CH), 130.0 (2CH), 130.2 (2CH), 137.8 (C), 138.1 (C), 144.6 (C), 150.7 (C), 153.2 (C), 153.9 (CH), 156.0 (C). Crystal data for 2i. C23H16N4, M = 348.40, T = 150(2) K, orthorhombic, P 21 21 21, a = 6.3672(5), b = 13.0997(10), c = 21.5377(18) Å, V = 1796.4(2) Å3, Z = 4, d = 1.288 g cm−3, μ = 0.079 mm−1. A final refinement on F2 with 2367 unique intensities and 245 parameters converged at ωR(F2) = 0.1207 (R(F) = 0.0498) for 1679 observed reflections with I > 2σ(I). CCDC 1858475.
3.9.3. 8-(N-indolyl)-2,3-diphenylpyrido[2,3-b]pyrazine (2j)
The general procedure 6 using 8-iodo-2,3-diphenylpyrido[2,3-b]pyrazine (2b-I, 0.20 g) and indole (0.12 g) gave 2j (eluent: CH2Cl2; Rf = 0.36) in 51% yield as a red powder. Mp: 136 °C. IR: 1023, 1154, 1208, 1236, 1324, 1356, 1379, 1442, 1454, 1478, 1519, 1555, 1577, 1592, 3240, 3339, 3639 cm−1. 1H-NMR (CDCl3): 6.82 (d, 1H, J = 3.4 Hz), 7.22–7.44 (m, 8H), 7.53–7.56 (m, 2H), 7.67 (d, 1H, J = 8.3 Hz), 7.70–7.72 (m, 3H), 7.86 (dd, 1H, J = 4.9 and 1.2 Hz), 7.94 (d, 1H, J = 3.4 Hz), 9.17 (d, 1H, J = 4.9 Hz). 13C-NMR (CDCl3): 106.1 (CH), 111.4 (CH), 118.0 (CH), 121.5 (CH), 122.1 (CH), 123.2 (CH), 128.4 (2CH), 128.4 (2CH), 128.5 (C), 129.7 (CH), 129.9 (CH), 130.1 (2CH), 130.3 (C), 130.3 (2CH), 130.5 (C), 130.8 (CH), 136.2 (C), 137.8 (C), 138.0 (C), 144.8 (C), 150.8 (C), 153.6 (CH), 156.4 (C). Anal. Calc. for C27H18N4 (398.47): C 81.39, H 4.55, N, 14.06. Found: C 81.26, H 4.67, N, 13.84.
3.9.4. 2,3-Diphenyl-8-(N-pyrazolyl)pyrido[2,3-b]pyrazine (2k)
The general procedure 6 using 8-iodo-2,3-diphenylpyrido[2,3-b]pyrazine (2b-I, 0.20 g) and pyrazole (68 mg) gave 2k (eluent: CH2Cl2-EtOAc 80:20; Rf = 0.47) in 71% yield as a pale yellow powder. Mp: 200 °C. IR: 1027, 1032, 1092, 1164, 1229, 1324, 1356, 1388, 1532, 1549, 1592, 3034, 3060, 3159 cm−1. 1H-NMR (CDCl3): 6.55 (d, 1H, J = 2.2 Hz), 7.30–7.41 (m, 6H), 7.55–7.58 (m, 2H), 7.64–7.66 (m, 2H), 7.82 (s, 1H), 8.34 (dd, 1H, J = 5.3 and 2.4 Hz), 9.12 (dd, 1H, J = 5.2 and 2.2 Hz), 9.46 (t, 1H, J = 2.5 Hz). 13C-NMR (CDCl3): 109.2 (CH), 115.0 (CH), 127.9 (C), 128.3 (2CH), 128.6 (2CH), 129.6 (CH), 129.8 (CH), 129.9 (2CH), 130.3 (2CH), 134.4 (CH), 137.6 (C), 138.3 (C), 142.5 (CH), 143.2 (C), 150.5 (C), 153.4 (C), 154.3 (CH), 156.1 (C). Anal. Calc. for C22H15N5 (349.40): C 75.63, H 4.33, N, 20.04. Found: C 75.71, H 4.42, N, 19.86.
3.9.5. 8-(N-imidazolyl)-2,3-diphenylpyrido[2,3-b]pyrazine (2l)
The general procedure 6 using 8-iodo-2,3-diphenylpyrido[2,3-b]pyrazine (2b-I, 0.20 g) and imidazole (68 mg) gave 2l (eluent: EtOAc-MeOH 95:5; Rf = 0.48) in 69% yield as a yellow powder. Mp: 209 °C. IR: 1019, 1053, 1075, 1105, 1115, 1169, 1236, 1319, 1334, 1379, 1429, 1446, 1459, 1482, 1549, 1594, 3064, 3124, 3639 cm−1. 1H-NMR (CDCl3): 7.32–7.45 (m, 7H), 7.56 (d, 2H, J = 6.6 Hz), 7.65–7.71 (m, 3H), 7.80 (br s, 1H), 8.82 (br s, 1H), 9.20 (d, 1H, J = 4.8 Hz). 13C-NMR (CDCl3): 115.6 (CH), 119.5 (CH), 128.3 (2CH), 128.4 (2CH), 128.9 (C), 129.7 (CH), 129.9 (CH), 129.9 (2CH), 130.1 (2CH), 130.3 (CH), 137.5 (C), 137.6 (C), 138.8 (CH), 141.4 (C), 150.7 (C), 154.1 (C), 154.2 (CH), 156.6 (C). Anal. Calc. for C22H15N5 (349.40): C 75.63, H 4.33, N, 20.04. Found: C 75.74, H 4.37, N, 19.92.
3.9.6. 2,3-Diphenyl-8-[1-(1,2,4-triazolyl)]pyrido[2,3-b]pyrazine (2m)
The general procedure 6 using 8-iodo-2,3-diphenylpyrido[2,3-b]pyrazine (2b-I, 0.20 g) and 1,2,4-triazole (69 mg) gave 2m (eluent: CH2Cl2-EtOAc 80:20; Rf = 0.35) in 79% yield as an orange powder. Mp: 205 °C. IR: 708, 995, 1025, 1049, 1079, 1124, 1158, 1223, 1242, 1276, 1332, 1386, 1403, 1459, 1508, 1551, 1590, 3064, 3146 cm−1. 1H-NMR (CDCl3): 7.35–7.49 (m, 6H), 7.59 (d, 2H, J = 7.0 Hz), 7.68 (d, 2H, J = 7.2 Hz), 8.22 (s, 1H), 8.37 (br s, 1H), 9.27 (br s, 1H), 10.16 (br s, 1H). 13C-NMR (CDCl3): 115.1 (CH), 127.3 (C), 128.2 (2CH), 128.5 (2CH), 129.8 (CH), 129.8 (2CH), 129.9 (CH), 130.1 (2CH), 137.3 (C), 137.7 (C), 140.4 (C), 147.2 (CH), 150.4 (C), 152.0 (CH), 154.1 (C), 154.5 (CH), 156.7 (C). Anal. Calc. for C21H14N6 (350.39): C 71.99, H 4.03, N, 23.99. Found: C 72.19, H 4.15, N, 23.81.
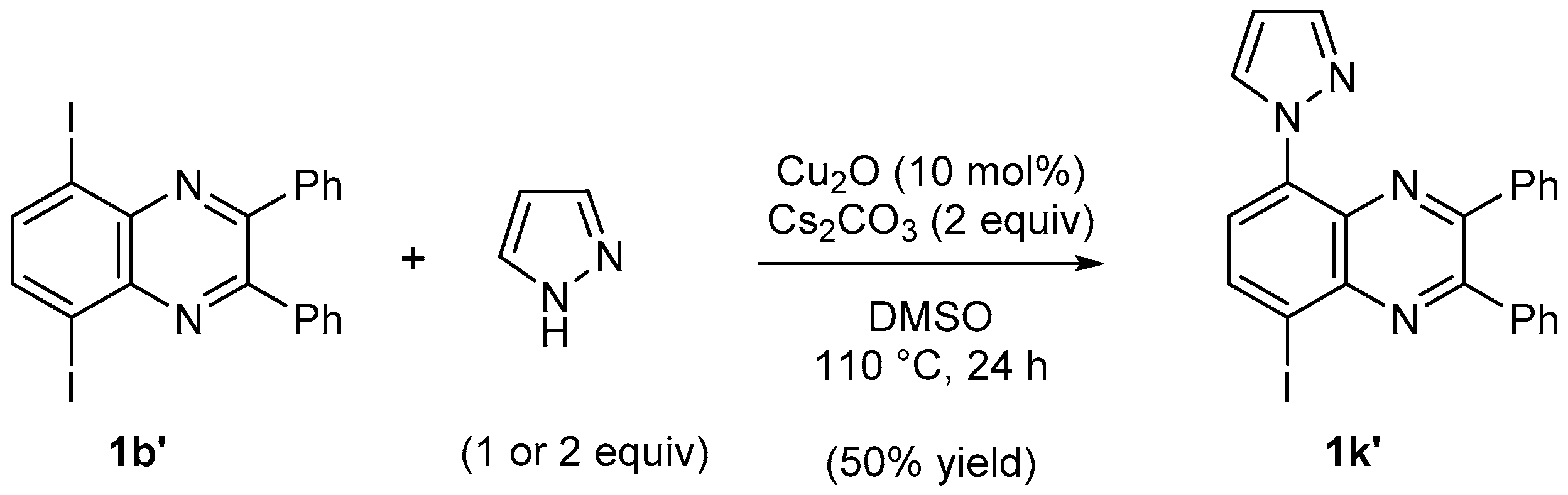
3.9.7. 5-Iodo-2,3-diphenyl-8-(N-pyrazolyl)quinoxaline (1k′)
The general procedure 6 using 5-iodo-2,3-diphenylquinoxaline (1b′, 0.27 g) and pyrazole (68 mg) gave 1k′ (eluent: CH2Cl2-heptane 80:20; Rf = 0.45) in 50% yield as an pale yellow powder. Mp: 200–202 °C. IR: 536, 585, 602, 692, 696, 755, 843, 894, 946, 1040, 1092, 1182, 1193, 1221, 1336, 1397, 1465, 1519, 1543, 1592, 3060, 3159 cm−1. 1H-NMR (CDCl3): 6.56 (dd, 1H, J = 2.6, 1.8 Hz), 7.35–7.45 (m, 6H), 7.57–7.60 (m, 2H), 7.70–7.73 (m, 2H), 7.82 (d, 1H, J = 1.8 Hz), 8.12 (d, 1H, J = 8.2 Hz), 8.43 (d, 1H, J = 8.3 Hz), 8.97–8.98 (m, 1H). 13C-NMR (CDCl3): 99.3 (C), 107.6 (CH), 123.7 (CH), 128.4 (2CH), 128.5 (2CH), 129.5 (CH), 129.6 (CH), 130.0 (2CH), 130.4 (2CH), 133.2 (C), 133.5 (CH), 137.1 (C), 137.8 (C), 138.2 (C), 139.7 (CH), 140.7 (C), 141.1 (CH), 153.0 (C), 153.6 (C). Anal. Calc. for C23H15IN4 (474.31): C 58.24, H 3.19, N, 11.81. Found: C 58.33, H 3.26, N, 11.68.
3.10. Nucleophilic Substitution Using Amines
3.10.1. General Procedure 7
A sealed tube containing the iodide (0.50 mmol) and amine (amount given in the product description) in ethanol (2 mL) was heated (conditions given in the product description). The cooled residue was concentrated before chromatography over silica gel (eluent given in the product description).
3.10.2. 8-(Isopropylamino)-2,3-diphenylpyrido[2,3-b]pyrazine (2n)
The general procedure 7 (150 °C, 18 h) using 8-iodo-2,3-diphenylpyrido[2,3-b]pyrazine (2b-I, 0.20 g) and isopropylamine (51 μL, 0.60 mmol) gave 2n (eluent: CH2Cl2-EtOAc 50:50; Rf = 0.20) in 69% yield as a beige powder. Mp: 179 °C. IR: 699, 703, 772, 804, 1156, 1178, 1236, 1313, 1336, 1538, 1564, 1592, 2965, 3038, 3064, 3390 cm−1. 1H-NMR (CDCl3): 1.27 (d, 6H, J = 6.4 Hz, Me), 3.77 (dp, 1H, J = 7.9 and 6.4 Hz, CHMe2), 6.40 (br d, 1H, J = 8.0 Hz, NH), 6.45 (d, 1H, J = 5.5 Hz), 7.15–7.28 (m, 6H), 7.39–7.42 (m, 2H), 7.46–7.49 (m, 2H), 8.59 (dd, 1H, J = 5.4, 0.6 Hz). 13C-NMR (CDCl3): 22.3 (2CH3), 44.1 (CH), 100.7 (CH), 127.1 (C), 128.0 (2CH), 128.2 (2CH), 128.7 (CH), 129.0 (CH), 129.9 (2CH), 130.2 (2CH), 138.4 (C), 138.9 (C), 149.8 (C), 150.1 (C), 150.1 (C), 154.8 (CH), 155.8 (C). Anal. Calc. for C22H20N4 (340.43): C 77.62, H 5.92, N, 16.46. Found: C 77.72, H 6.14, N, 16.19.
3.10.3. 8-(4-Methoxybenzylamino)-2,3-diphenylpyrido[2,3-b]pyrazine (2o)
The general procedure 7 (150 °C, 24 h) using 8-iodo-2,3-diphenylpyrido[2,3-b]pyrazine (2b-I, 0.20 g) and 4-methoxybenzylamine (78 μL, 0.60 mmol) gave 2o (eluent: CH2Cl2-EtOAc 50:50; Rf = 0.48) in 71% yield as a yellow powder. Mp: 190 °C. IR: 697, 832, 1175, 1236, 1302, 1341, 1437, 1459, 1510, 1585, 2828, 2910, 3064, 3232 cm−1. 1H-NMR (CDCl3): 3.81 (s, 3H, OMe), 4.55 (d, 2H, J = 5.9 Hz), 6.58 (d, 1H, J = 5.3 Hz), 6.90 (d, 2H, J = 8.7 Hz), 7.02 (t, 1H, J = 5.7 Hz), 7.27–7.35 (m, 8H), 7.49–7.51 (m, 2H), 7.58–7.61 (m, 2H), 8.69 (d, J = 5.3 Hz, 1H). 13C-NMR (CDCl3): 46.5 (CH2), 55.3 (CH3), 101.1 (CH), 114.3 (2CH), 127.2 (C), 128.0 (2CH), 128.2 (2CH), 128.6 (2CH), 128.8 (CH), 129.1 (CH), 129.1 (C), 129.9 (2CH), 130.3 (2CH), 138.5 (C), 138.8 (C), 150.0 (C), 150.2 (C), 150.9 (C), 154.9 (CH), 155.9 (C), 159.2 (C). Anal. Calc. for C27H22N4O (418.50): C 77.49, H 5.30, N, 13.39. Found: C 77.58, H 5.44, N, 13.20.
3.10.4. 8-(Benzylamino)-2,3-diphenylpyrido[2,3-b]pyrazine (2p)
The general procedure 7 (150 °C, 24 h) using 8-iodo-2,3-diphenylpyrido[2,3-b]pyrazine (2b-I, 0.20 g) and benzylamine (66 μL, 0.60 mmol) gave 2p (eluent: CH2Cl2-EtOAc 50:50; Rf = 0.50) in 79% yield as a yellow powder. Mp: 238 °C. IR: 697, 768, 873, 1150, 1238, 1300, 1324, 1339, 1439, 1538, 1590, 2910, 3064, 3201 cm−1. 1H-NMR (CDCl3): 4.63 (d, 2H, J = 6.0 Hz), 6.56 (d, 1H, J = 5.4 Hz), 7.11 (t, 1H, J = 6.0 Hz), 7.27–7.39 (m, 11H), 7.49–7.52 (m, 2H), 7.58–7.61 (m, 2H), 8.68 (d, 1H, J = 5.4 Hz). 13C-NMR (CDCl3): 47.0 (CH2), 101.2 (CH), 127.2 (2CH), 127.2 (C), 127.8 (CH), 128.1 (2CH), 128.3 (2CH), 128.9 (CH), 128.9 (2CH), 129.1 (CH), 129.9 (2CH), 130.3 (2CH), 137.2 (C), 138.5 (C), 138.8 (C), 150.0 (C), 150.3 (C), 151.0 (C), 154.9 (CH), 156.0 (C). Crystal data for 2p. C26H20N4, M = 388.46, T = 150(2) K, monoclinic, P 21/n, a = 6.0721(6), b = 12.8640(10), c = 25.460(2) Å, β = 91.436(4) °, V = 1988.1(3) Å3, Z = 4, d = 1.298 g cm−3, μ = 0.078 mm−1. A final refinement on F2 with 4438 unique intensities and 274 parameters converged at ωR(F2) = 0.1432 (R(F) = 0.0626) for 3710 observed reflections with I > 2σ(I). CCDC 1858476.
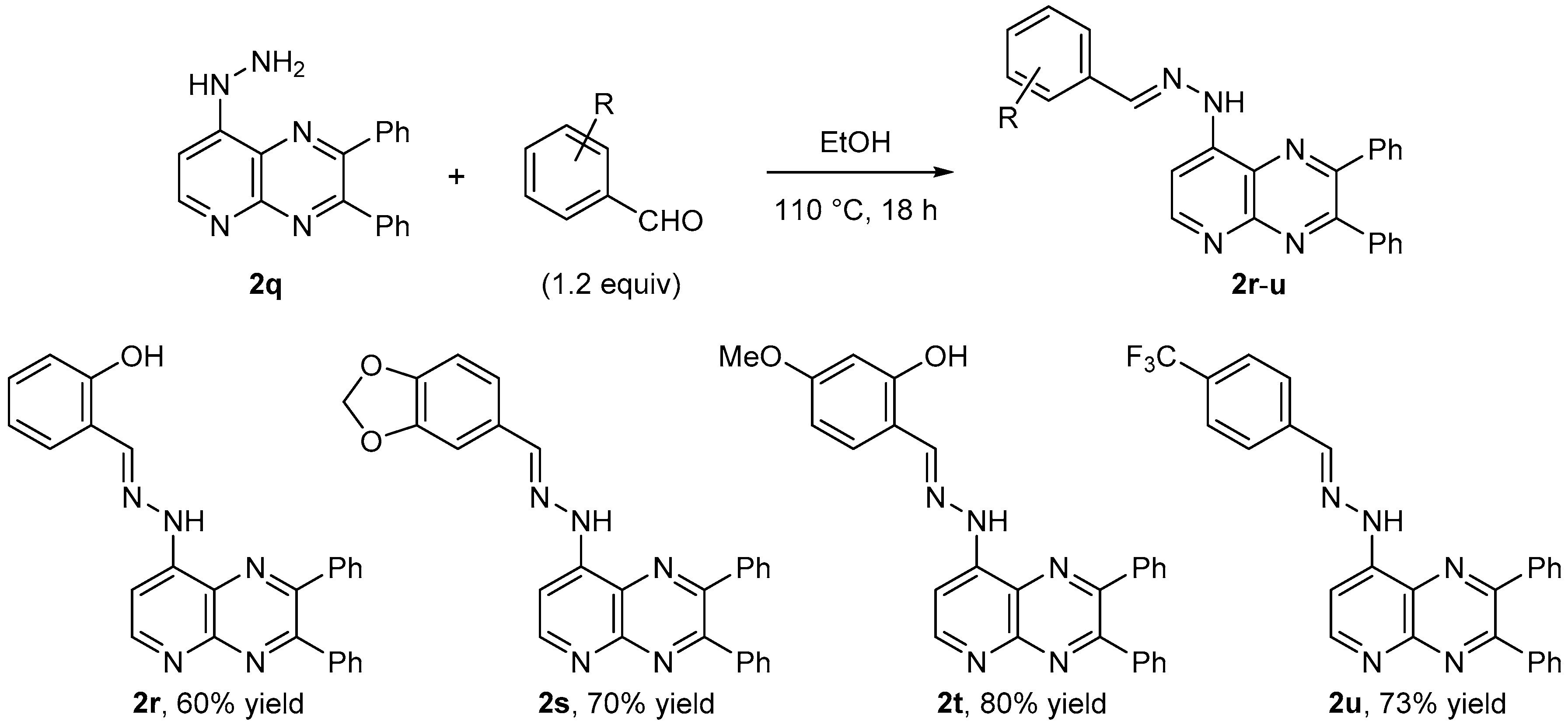
3.11. Nucleophilic Substitution using Hydrazine Hydrate: 8-Hydrazino-2,3-diphenylpyrido[2,3-b]pyrazine (2q)
A solution of 8-iodo-2,3-diphenyl pyrido [2,3-b]pyrazine (2b-I, 0.20 g, 0.50 mmol) and hydrazine hydrate (0.25 mL, 5.0 mmol) in isopropanol (2 mL) was heated under reflux for 4 h. The cooled residue was concentrated and taken up with EtOAc (20 mL). The organic layer was washed with water (10 mL), dried over MgSO4, filtered and concentrated under reduced pressure to give the title compound 2q in 92% yield as a red powder. Mp > 250 °C. 1H-NMR (CDCl3): 4.22 (br s, 2H, NH), 6.91 (d, 1H, J = 5.6 Hz), 7.22–7.36 (m, 7H), 7.40–7.43 (m, 2H), 7.48–7.51 (m, 2H), 8.60 (d, 1H, J = 5.6 Hz). 13C-NMR (CDCl3): 101.2 (CH), 126.2 (C), 128.2 (2CH), 128.3 (2CH), 129.0 (CH), 129.2 (CH), 129.9 (2CH), 130.3 (2CH), 138.4 (C), 138.7 (C), 149.7 (C), 150.3 (C), 153.0 (C), 155.0 (CH), 156.1 (C). Anal. Calc. for C19H15N5 (313.36): C 72.83, H 4.83, N, 22.35. Found: C 72.96, H 4.89, N, 22.31.
3.12. Condensation Reactions from the Hydrazine 2q
3.12.1. General Procedure 8
A sealed tube containing 8-hydrazino-2,3-diphenylpyrido[2,3-b] pyrazine (2q, 0.16 g, 0.50 mmol) and the aldehyde (0.55 mmol) in ethanol (2 mL) was heated at 110 °C overnight. The cooled residue was concentrated under vacuum, washed with methanol and isolated by filtration.
3.12.2. 2-Hydroxybenzaldehyde 2-[8-(2,3-diphenylpyrido[2,3-b]pyrazinyl)]hydrazone (2r)
General Procedure 8 using 2-hydroxybenzaldehyde (67 mg) gave 2r (Rf (CH2Cl2-EtOAc 80:20) = 0.44) in 60% yield as a yellow powder. Mp > 260 °C. IR: 952, 1019, 1096, 1163, 1233, 1270, 1309, 1328, 1422, 1540, 1562, 1594, 1618, 3064, 3317 cm−1. 1H-NMR (CDCl3): 6.96 (td, 1H, J = 7.5 and 1.1 Hz), 7.07 (d, 1H, J = 8.2 Hz), 7.23–7.44 (m, 9H), 7.51–7.54 (m, 2H), 7.58–7.62 (m, 2H), 8.26 (s, 1H), 8.91 (d, 1H, J = 5.3 Hz), 9.71 (br s, 1H), 10.60 (br s, 1H). The 13C spectra could not be recorded due to low solubility in CDCl3 and DMSO. Anal. Calc. for C26H19N5O (417.47): C 74.80, H 4.59, N, 16.78. Found: C 74.72, H 4.39, N, 16.67.
3.12.3. Piperonal 2-[8-(2,3-diphenylpyrido[2,3-b]pyrazinyl)]hydrazone (2s)
General Procedure 8 using piperonal (83 mg) gave 2s (Rf (CH2Cl2-EtOAc 80:20) = 0.37) in 70% yield as a yellow powder. Mp: 254 °C. IR: 933, 1038, 1150, 1255, 1339, 1450, 1489, 1501, 1545, 1568, 1590, 2901, 3060, 3322, 3648 cm−1. 1H-NMR (CDCl3): 6.03 (s, 2H), 6.84 (d, 1H, J = 8.0 Hz), 7.07 (dd, 1H, J = 8.1 and 1.6 Hz), 7.28–7.42 (m, 7H), 7.50 (t, 3H, J = 6.6 Hz), 7.59 (d, 2H, J = 6.8 Hz), 7.97 (s, 1H), 8.85 (d, 1H, J = 5.3 Hz), 9.66 (s, 1H). 13C-NMR ((CD3)2SO): 101.5 (CH2), 103.5 (CH), 104.9 (CH), 108.5 (CH), 123.0 (CH), 125.6 (C), 128.1 (2CH), 128.2 (2CH), 128.8 (CH), 129.1 (CH), 129.2 (C), 129.7 (2CH), 130.1 (2CH), 138.3 (C), 138.6 (C), 145.1 (CH), 147.7 (C), 148.1 (C), 148.8 (C), 149.6 (C), 150.0 (C), 154.5 (CH), 155.5 (C). Anal. Calc. for C27H19N5O2 (445.48): C 72.80, H 4.30, N, 15.72. Found: C 72.95, H 4.44, N, 15.83.
3.12.4. 2-Hydroxy-4-methoxybenzaldehyde 2-[8-(2,3-diphenylpyrido[2,3-b]pyrazinyl)]hydrazone (2t)
General Procedure 8 using 2-hydroxy-4-methoxybenzaldehyde (84 mg) gave 2t (Rf (CH2Cl2- EtOAc 80:20) = 0.58) in 80% yield as a yellow powder. Mp > 260 °C. IR: 1032, 1135, 1163, 1238, 1291, 1339, 1431, 1439, 1461, 1510, 1543, 1566, 1631, 2845, 2931, 3004, 3056, 3176, 3317 cm−1. 1H-NMR (CDCl3): 3.85 (s, 3H), 6.52 (dd, 1H, J = 8.5 and 2.5 Hz), 6.58 (d, 1H, J = 2.5 Hz), 7.15 (d, 1H, J = 8.6 Hz), 7.19 (d, 1H, J = 5.2 Hz), 7.28–7.43 (m, 6H), 7.50–7.54 (m, 2H), 7.58–7.61 (m, 2H), 8.19 (s, 1H), 8.88 (br s, 1H), 9.59 (br s, 1H), 10.81 (s, 1H). The 13C spectra could not be recorded due to low solubility in CDCl3 and DMSO. Anal. Calc. for C27H21N5O2 (447.50): C 72.47, H 4.73, N, 15.65. Found: C 72.53, H 4.89, N, 15.60.
3.12.5. 4-(Trifluoromethyl)benzaldehyde 2-[8-(2,3-diphenylpyrido[2,3-b]pyrazinyl)]hydrazone (2u)
General Procedure 8 using 4-(trifluoromethyl)benzaldehyde (87 mg) gave 2u (Rf (CH2Cl2- EtOAc 80:20) = 0.51) in 73% yield as a yellow powder. Mp: 258–260 °C. IR: 1017, 1066, 1109, 1124, 1145, 1236, 1300, 1321, 1512, 1545, 1562, 1588, 3060, 3184, 3317, 3652 cm−1. 1H-NMR (CDCl3): 7.28–7.43 (m, 6H), 7.50–7.53 (m, 2H), 7.56–7.61 (m, 3H), 7.68 (d, 2H, J = 8.2 Hz), 7.87 (d, 2H, J = 7.8 Hz), 8.11 (s, 1H), 8.91 (d, 1H, J = 5.2 Hz), 9.90 (br s, 1H). 13C-NMR ((CD3)2SO, 333 K): 103.8 (CH), 124.0 (q, CF3, J = 272 Hz), 125.4 (C), 125.5 (q, 2CH, J = 3.7 Hz), 127.1 (2CH), 127.8 (2CH), 127.9 (2CH), 128.7 (CH), 128.8 (CH), 129.2 (q, C-CF3, J = 31.7 Hz), 129.5 (2CH), 129.8 (2CH), 138.1 (C), 138.4 (C), 138.5 (C), 143.2 (CH), 147.4 (C), 149.4 (C), 150.3 (C), 154.3 (CH), 155.5 (C). Anal. Calc. for C27H18F3N5 (469.47): C 69.08, H 3.86, N, 14.92. Found: C 69.25, H 3.97, N, 14.78.
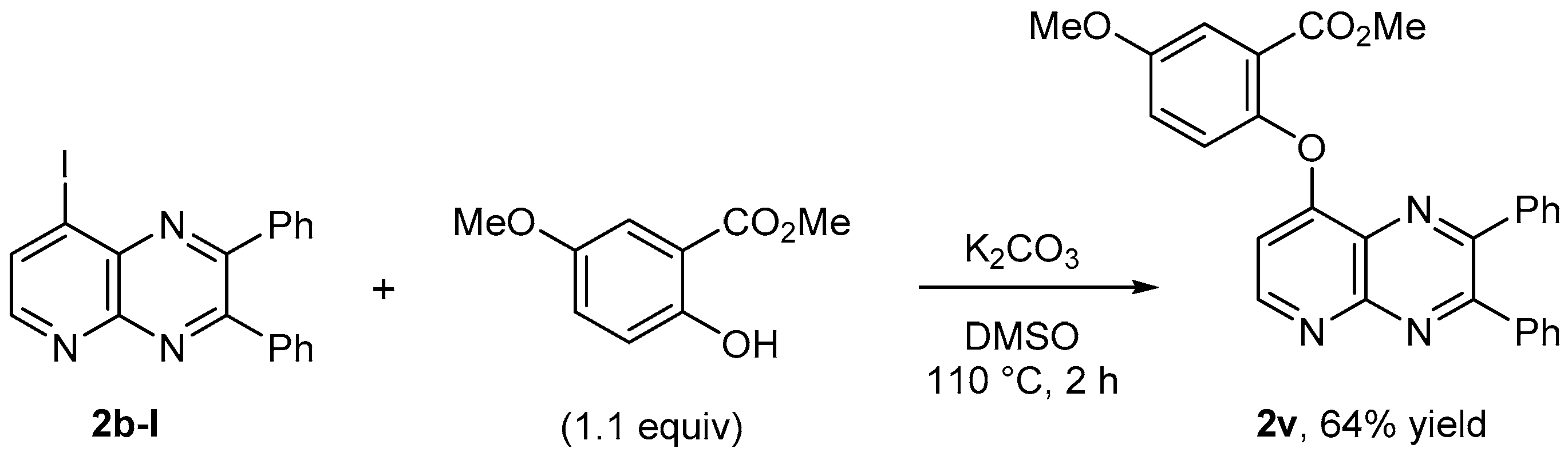
3.13. Nucleophilic Substitution Using a Phenolate: Methyl 2-[8-(2,3-diphenylpyrido[2,3-b]pyrazinyl)]oxy- 5-methoxybenzoate (2v)
A mixture of 8-iodo-2,3-diphenylpyrido[2,3-b]pyrazine (2b-I, 0.20 g, 0.50 mmol), methyl 2-hydroxy-5-methoxy-benzoate (0.10 g, 0.55 mmol), K2CO3 (77 mg, 0.55 mmol) and DMSO (1 mL) was heated at 110 °C for 2 h. The cooled residue was treated by an aqueous solution of Na2CO3 (10 mL) before extraction with Et2O (3 × 10 mL). The organic layer was dried over MgSO4, filtered and concentrated under reduced pressure, and the residue was chromatographed over silica gel (eluent: CH2Cl2-MeOH 95:5; Rf (CH2Cl2-EtOAc 95:5) = 0.50) to give the title compound 2v in 64% yield as a beige powder. Mp: 206 °C. IR: 542, 698, 773, 856, 1021, 1072, 1109, 1205, 1235, 1263, 1333, 1350, 1434, 1468, 1496, 1554, 1594, 1719, 2845, 2956, 3041 cm−1. 1H-NMR (CDCl3): 3.65 (s, 3H), 3.90 (s, 3H), 6.64 (d, J = 5.2 Hz, 1H), 7.19 (dd, 1H, J = 8.9, 3.0 Hz), 7.24 (d, 1H, J = 9.5 Hz), 7.30–7.40 (m, 6H), 7.55–7.58 (m, 3H), 7.61–7.64 (m, 2H), 8.86 (d, 1H, J = 5.2 Hz). 13C-NMR (CDCl3): 52.4 (CH3), 56.0 (CH3), 107.6 (CH), 116.3 (CH), 120.6 (CH), 124.6 (C), 125.0 (CH), 128.2 (2CH), 128.4 (2CH), 129.0 (C), 129.1 (CH), 129.4 (CH), 130.2 (2CH), 130.3 (2CH), 138.2 (C), 138.7 (C), 147.0 (C), 151.1 (C), 153.6 (C), 154.4 (CH), 156.6 (C), 157.4 (C), 163.0 (C), 164.7 (C). Anal. Calc. for C28H21N3O4 (463.49): C 72.56, H 4.57, N, 9.07. Found: C 72.49, H 4.65, N, 9.01.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




















