
Virtual Screening, Biological Evaluation, and 3D-QSAR Studies of New HIV-1 Entry Inhibitors That Function via the CD4 Primary Receptor

 ,
,
Abstract
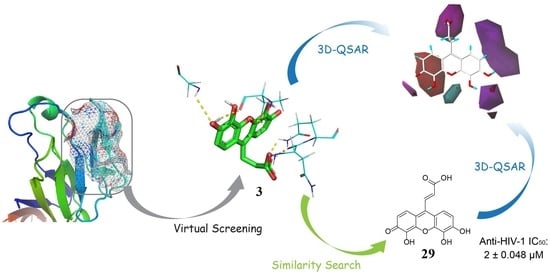
:
1. Introduction
2. Results
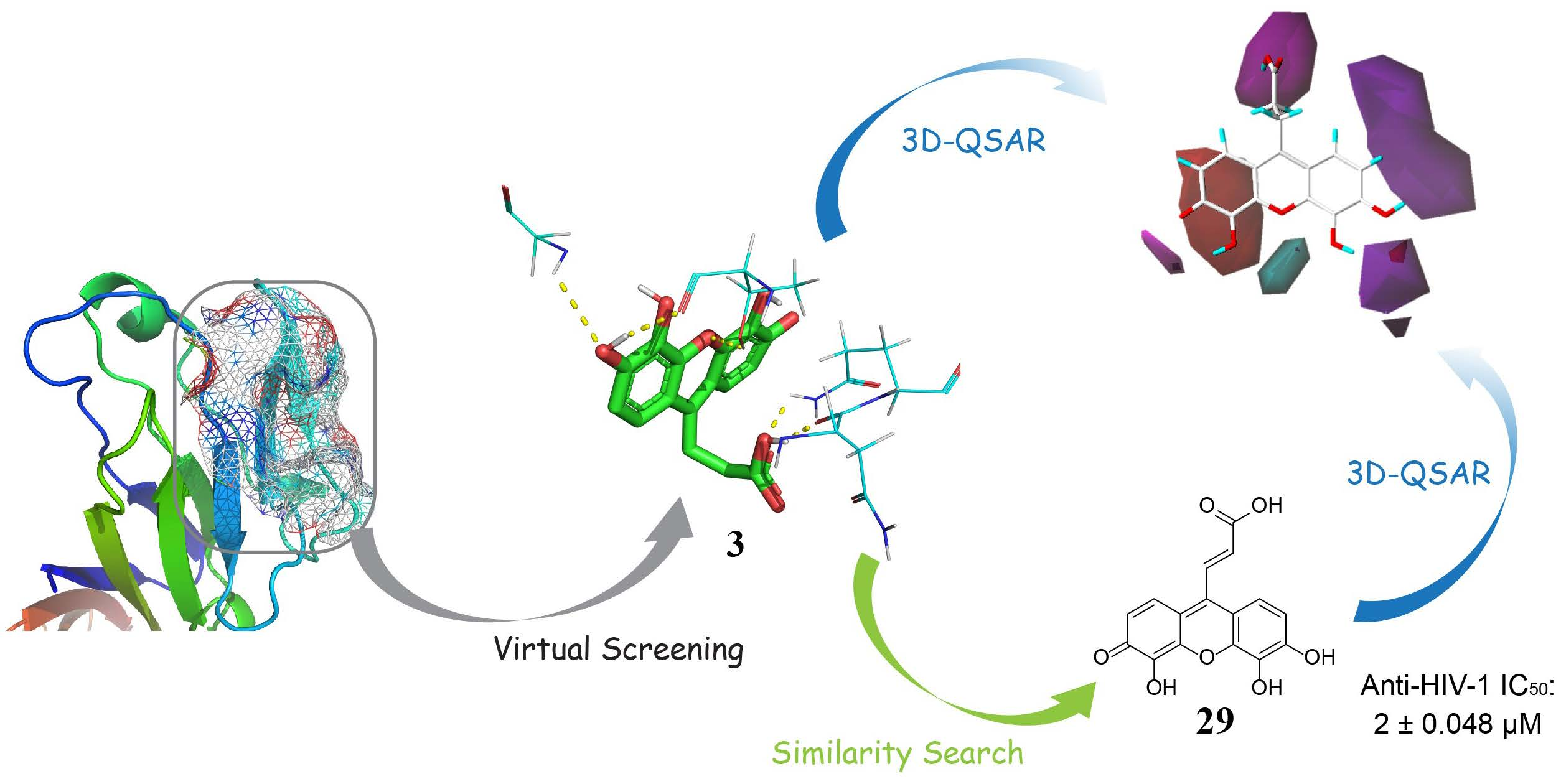
2.1. Virtual Screening
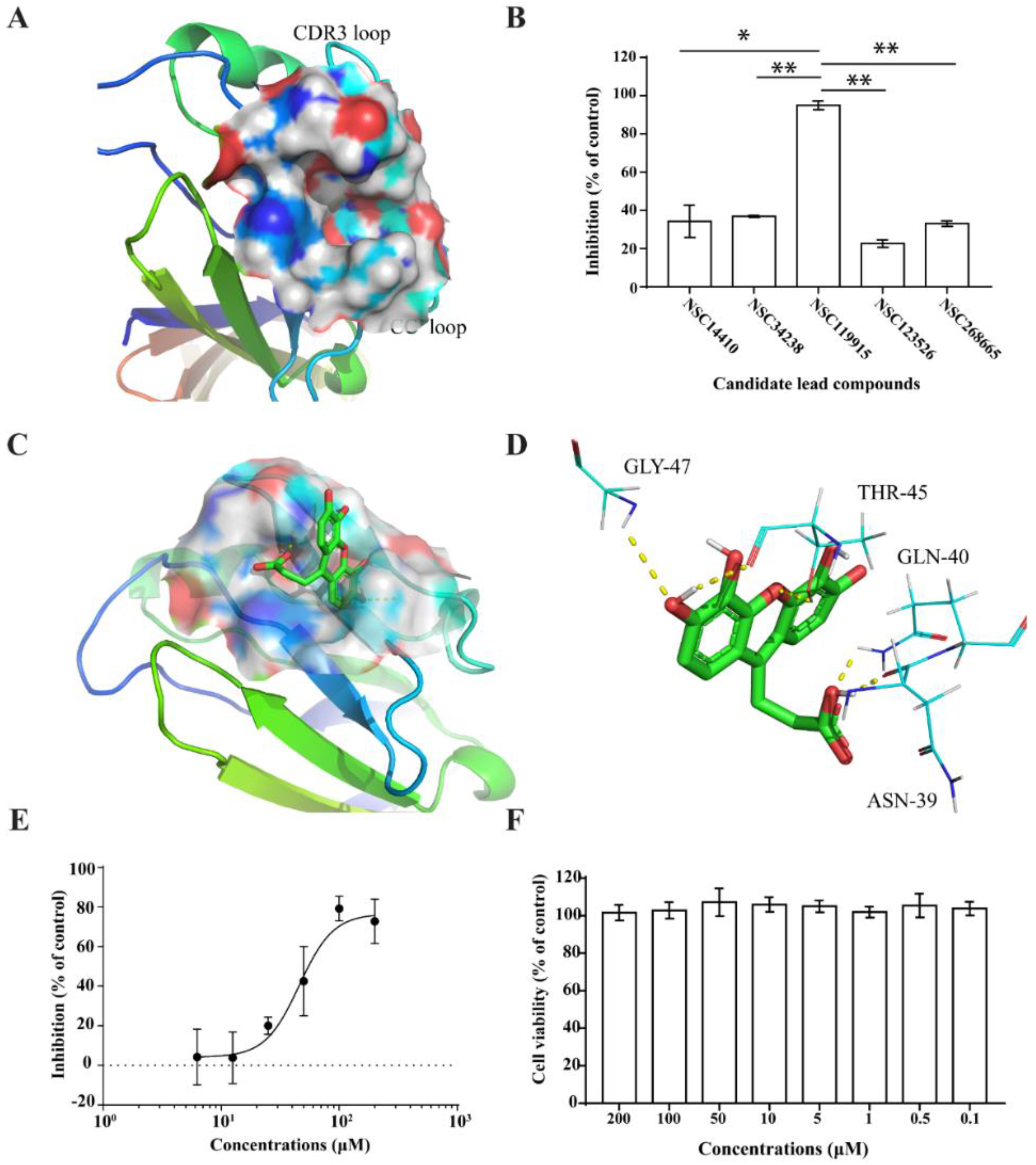
2.2. Competitive CD4-Binding Compounds
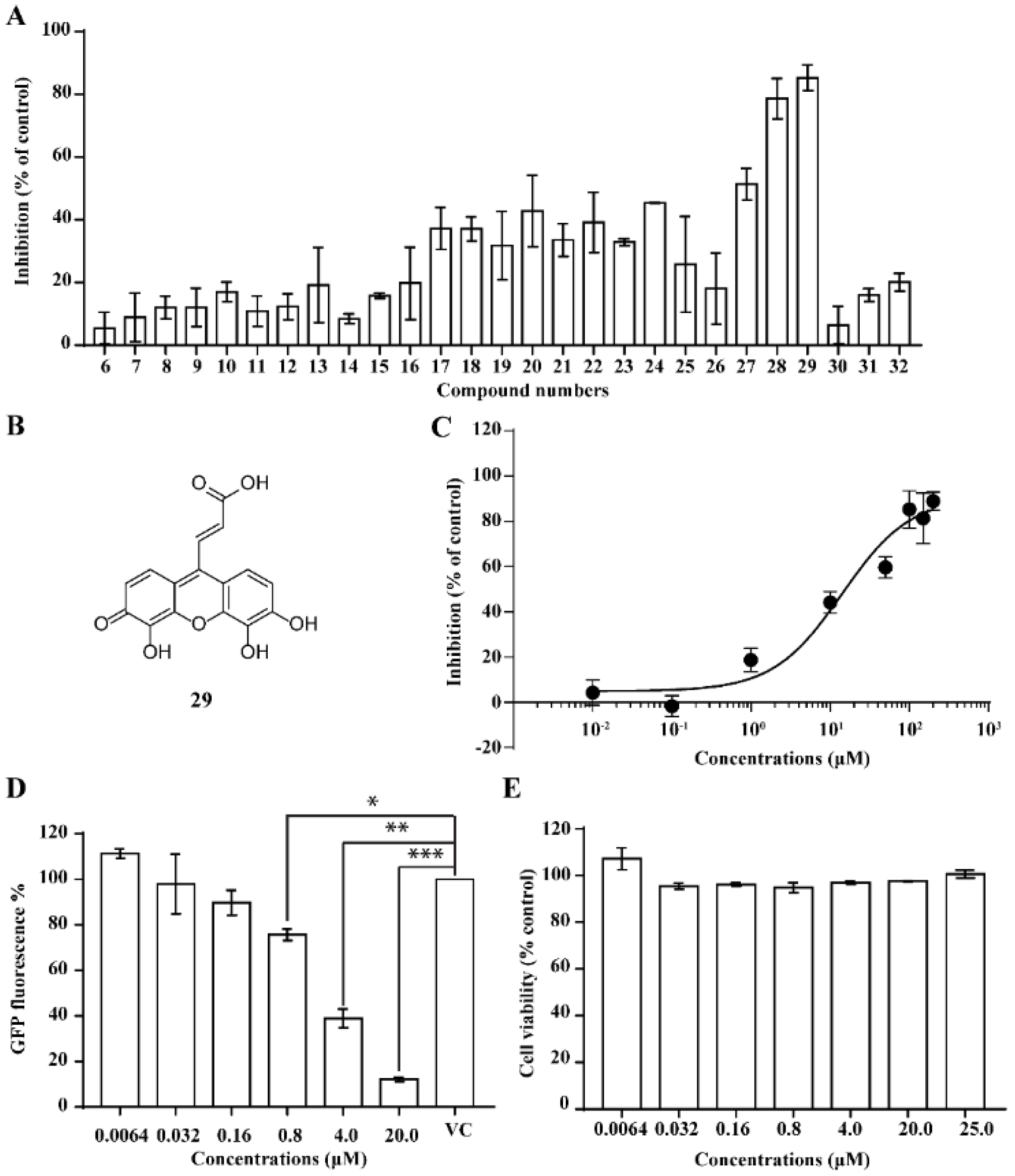
2.3. NSC119915 Displayed Potent Anti-HIV-1 Activity
2.4. NSC119915 Showed No Cytotoxicity in PBMCs
2.5. Bioactivity Evaluations of Compounds Collected from Structural Similarity Screening
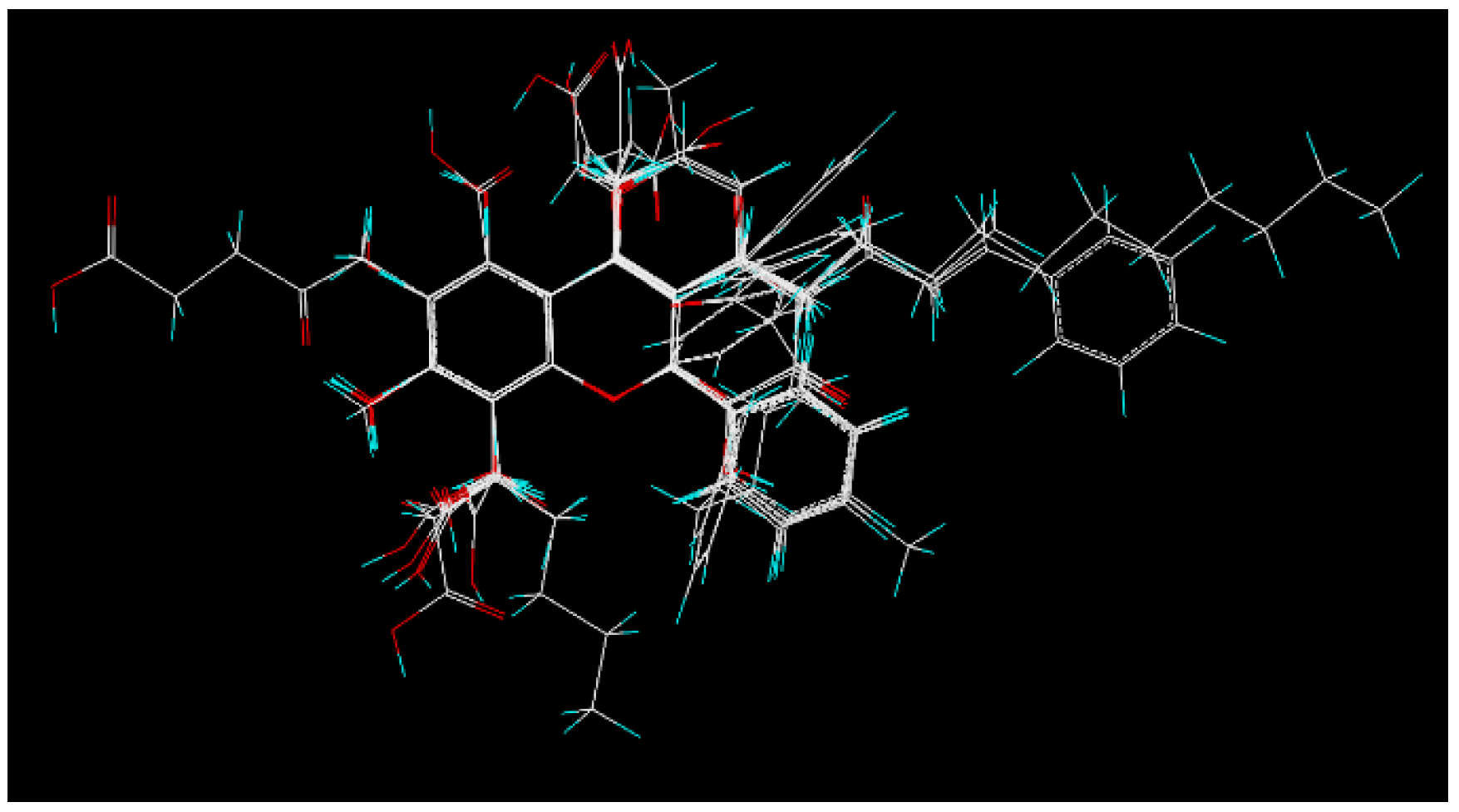
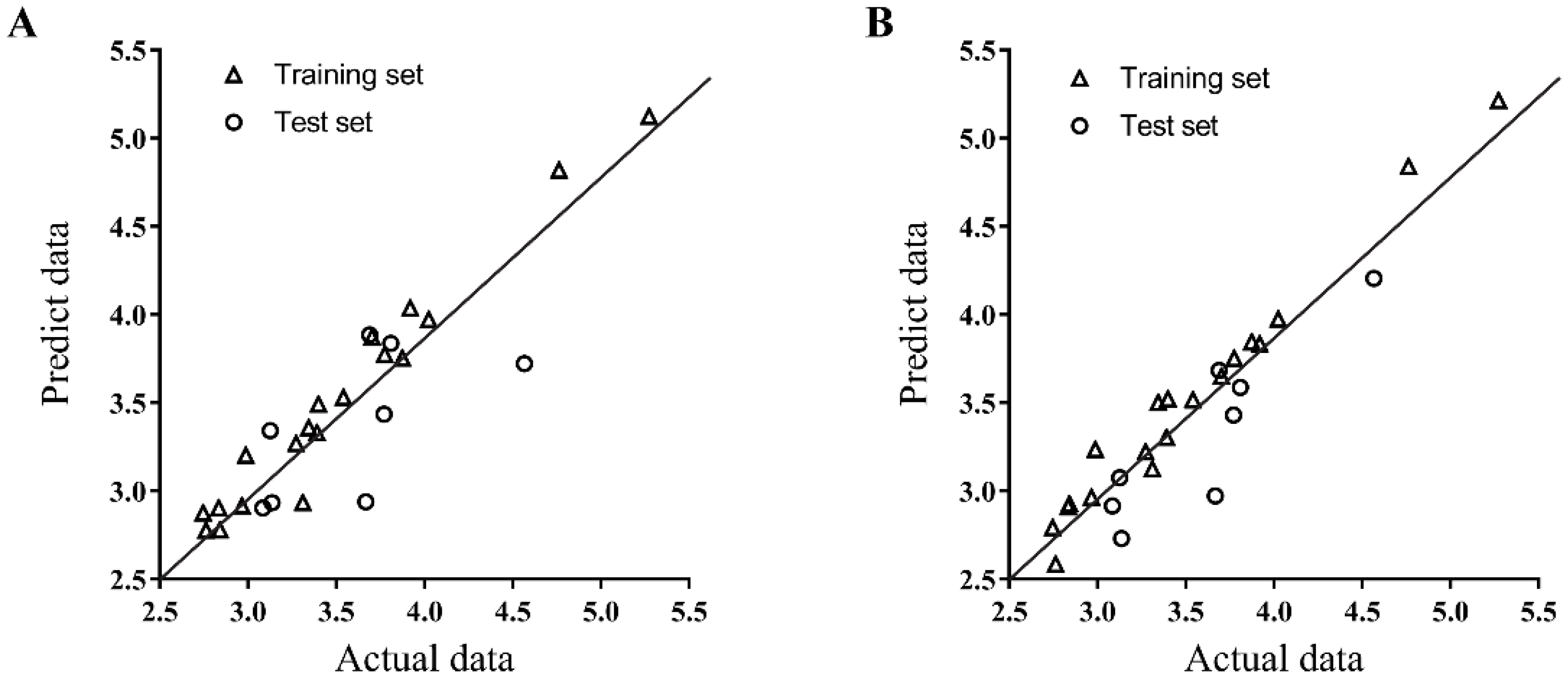
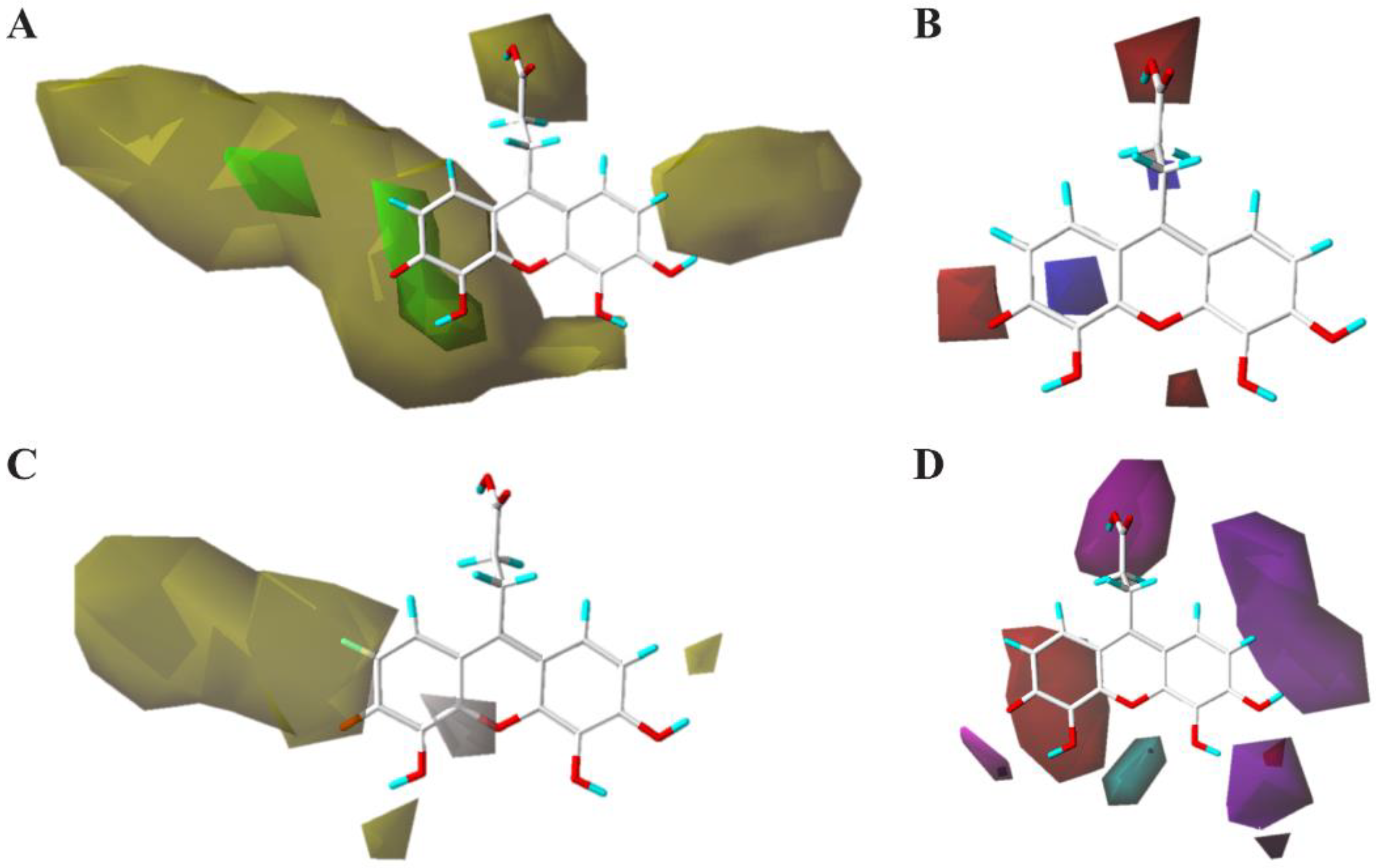
2.6. 3D Quantitative Structure-Activity Relationship (3D-QSAR) Analyses
3. Discussion
4. Materials and Methods
4.1. Molecular Modeling and Virtual Screening
4.2. CD4 Competitive Binding Assay
4.3. Peripheral Blood Mononuclear Cell (PBMC) Isolation
4.4. Anti-HIV-1 Infection Assay
4.5. Anti-HIV-1 Entry Assay
4.6. PBMC Viability Assay
4.7. Compound Structural Similarity Search
4.8. 3D-QSAR Study
4.9. Data and Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barre-Sinoussi, F.; Chermann, J.C.; Rey, F.; Nugeyre, M.T.; Chamaret, S.; Gruest, J.; Dauguet, C.; Axler-Blin, C.; Vezinet-Brun, F.; Rouzioux, C.; et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983, 220, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Hoover, D.R.; Graham, N.M.; Chen, B.; Taylor, J.M.; Phair, J.; Zhou, S.Y.; Munoz, A. Effect of CD4+ cell count measurement variability on staging HIV-1 infection. J. Acq. Immun. Def. Synd. 1992, 5, 794–802. [Google Scholar] [CrossRef]
- Bartlett, J.A.; DeMasi, R.; Quinn, J.; Moxham, C.; Rousseau, F. Overview of the effectiveness of triple combination therapy in antiretroviral-naive HIV-1 infected adults. AIDS 2001, 15, 1369–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palella, F.J., Jr.; Delaney, K.M.; Moorman, A.C.; Loveless, M.O.; Fuhrer, J.; Satten, G.A.; Aschman, D.J.; Holmberg, S.D. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. N. Engl. J. Med. 1998, 338, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Flexner, C. HIV drug development: The next 25 years. Nat. Rev. Drug Discov. 2007, 6, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Clavel, F.; Hance, A.J. HIV drug resistance. N. Engl. J. Med. 2004, 350, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- De Cock, K.M.; Brun-Vezinet, F.; Soro, B. HIV-1 and HIV-2 infections and AIDS in West Africa. AIDS 1991, 5. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Hahn, B.H. Origins of HIV and the AIDS pandemic. CSH Perspect. Med. 2011, 1, a006841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilen, C.B.; Tilton, J.C.; Doms, R.W. HIV: Cell binding and entry. CSH Perspect. Med. 2012, 2, a006866. [Google Scholar] [CrossRef] [PubMed]
- Freeman, M.M.; Seaman, M.S.; Rits-Volloch, S.; Hong, X.G.; Kao, C.Y.; Ho, D.D.; Chen, B. Crystal Structure of HIV-1 Primary Receptor CD4 in Complex with a Potent Antiviral Antibody. Structure 2010, 18, 1632–1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, K.; Liechti, T.; Haas, A.; Rehr, M.; Trkola, A.; Gunthard, H.F.; Oxenius, A. The orientation of HIV-1 gp120 binding to the CD4 receptor differentially modulates CD4+ T cell activation. J. Immunol. 2015, 194, 637–649. [Google Scholar] [CrossRef] [PubMed]
- Sattentau, Q.J.; Moore, J.P. Conformational changes induced in the human immunodeficiency virus envelope glycoprotein by soluble CD4 binding. J. Exp. Med. 1991, 174, 407–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cocchi, F.; DeVico, A.L.; Garzino-Demo, A.; Cara, A.; Gallo, R.C.; Lusso, P. The V3 domain of the HIV-1 gp120 envelope glycoprotein is critical for chemokine-mediated blockade of infection. Nat. Med. 1996, 2, 1244–1247. [Google Scholar] [CrossRef] [PubMed]
- Alkhatib, G.; Combadiere, C.; Broder, C.C.; Feng, Y.; Kennedy, P.E.; Murphy, P.M.; Berger, E.A. CC CKR5: A RANTES, MIP-1α, MIP-1β receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 1996, 272, 1955–1958. [Google Scholar] [CrossRef] [PubMed]
- Berger, E.A.; Doms, R.W.; Fenyo, E.; Korber, B.T.; Littman, D.R.; Moore, J.P.; Sattentau, Q.J.; Schuitemaker, H.; Sodroski, J.; Weiss, R.A. A new classification for HIV-1. Nature 1998, 391, 240. [Google Scholar] [CrossRef] [PubMed]
- Antell, G.C.; Dampier, W.; Aiamkitsumrit, B.; Nonnemacher, M.R.; Jacobson, J.M.; Pirrone, V.; Zhong, W.; Kercher, K.; Passic, S.; Williams, J.W.; et al. Utilization of HIV-1 envelope V3 to identify X4- and R5-specific Tat and LTR sequence signatures. Retrovirology 2016, 13, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, S.; Kaufman, J.D.; Grunwald, M.; Walla, P.J.; Lakomek, N.A.; Wingfield, P.T. HIV-1 gp41 transmembrane oligomerization monitored by FRET and FCS. FEBS Lett. 2018, 592, 939–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamede, J.I.; Cianci, G.C.; Anderson, M.R.; Hope, T.J. Early cytoplasmic uncoating is associated with infectivity of HIV-1. Proc. Natl. Acad. Sci. USA 2017, 114, 7169–7178. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, R.; Sodroski, J. The HIV-1 envelope glycoproteins: Fusogens, antigens, and immunogens. Science 1998, 280, 1884–1888. [Google Scholar] [CrossRef] [PubMed]
- Clayton, L.K.; Sieh, M.; Pious, D.; Reinherz, E.L. Identification of human CD4 residues affecting class II MHC versus HIV-1 gp120 binding. Nature 1989, 339, 548–550. [Google Scholar] [CrossRef] [PubMed]
- Kwong, P.D.; Wyatt, R.; Robinson, J.; Sweet, R.W.; Sodroski, J.; Hendrickson, W.A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998, 393, 648–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capon, D.J.; Ward, R.H. The CD4-gp120 interaction and AIDS pathogenesis. Annu. Rev. Immunol. 1991, 9, 649–678. [Google Scholar] [CrossRef] [PubMed]
- Peterson, A.; Seed, B. Genetic analysis of monoclonal antibody and HIV binding sites on the human lymphocyte antigen CD4. Cell 1988, 54, 65–72. [Google Scholar] [CrossRef]
- Qian, K.; Morris-Natschke, S.L.; Lee, K.H. HIV Entry Inhibitors and Their Potential in HIV Therapy. Med. Res. Rev. 2009, 29, 369–393. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Huang, B.; Zhan, P.; De Clercq, E.; Liu, X. Discovery of small molecular inhibitors targeting HIV-1 gp120-CD4 interaction drived from BMS-378806. Eur. J. Med. Chem. 2014, 86, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhang, Z.; Wallace, O.B.; Deshpande, M.; Fang, H.; Yang, Z.; Zadjura, L.M.; Tweedie, D.L.; Huang, S.; Zhao, F.; et al. Discovery of 4-benzoyl-1-[(4-methoxy-1H-pyrrolo[2,3-b]pyridin-3-yl)oxoacetyl]-2- (R)-methylpiperazine (BMS-378806): A novel HIV-1 attachment inhibitor that interferes with CD4-gp120 interactions. J. Med. Chem. 2003, 46, 4236–4239. [Google Scholar] [CrossRef] [PubMed]
- Lalezari, J.P.; Latiff, G.H.; Brinson, C.; Echevarria, J.; Trevino-Perez, S.; Bogner, J.R.; Thompson, M.; Fourie, J.; Sussmann Pena, O.A.; Mendo Urbina, F.C.; et al. Safety and efficacy of the HIV-1 attachment inhibitor prodrug BMS-663068 in treatment-experienced individuals: 24 week results of AI438011, a phase 2b, randomised controlled trial. Lancet HIV 2015, 2, 427–437. [Google Scholar] [CrossRef]
- Nettles, R.E.; Schurmann, D.; Zhu, L.; Stonier, M.; Huang, S.P.; Chang, I.; Chien, C.; Krystal, M.; Wind-Rotolo, M.; Ray, N.; et al. Pharmacodynamics, safety, and pharmacokinetics of BMS-663068, an oral HIV-1 attachment inhibitor in HIV-1-infected subjects. J. Infect. Dis. 2012, 206, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- Madani, N.; Schon, A.; Princiotto, A.M.; Lalonde, J.M.; Courter, J.R.; Soeta, T.; Ng, D.; Wang, L.; Brower, E.T.; Xiang, S.H.; et al. Small-molecule CD4 mimics interact with a highly conserved pocket on HIV-1 gp120. Structure 2008, 16, 1689–1701. [Google Scholar] [CrossRef] [PubMed]
- Curreli, F.; Kwon, Y.D.; Zhang, H.; Yang, Y.; Scacalossi, D.; Kwong, P.D.; Debnath, A.K. Binding mode characterization of NBD series CD4-mimetic HIV-1 entry inhibitors by X-ray structure and resistance study. Antimicrob. Agents Chemother. 2014, 58, 5478–5491. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Ma, L.; Jiang, S.; Lu, H.; Liu, S.; He, Y.; Strick, N.; Neamati, N.; Debnath, A.K. Identification of N-phenyl-N′-(2,2,6,6-tetramethyl-piperidin-4-yl)-oxalamides as a new class of HIV-1 entry inhibitors that prevent gp120 binding to CD4. Virology 2005, 339, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Curreli, F.; Do Kwon, Y.; Zhang, H.T.; Scacalossi, D.; Belov, D.S.; Tikhonov, A.A.; Andreev, I.A.; Altieri, A.; Kurkin, A.V.; Kwong, P.D.; et al. Structure-Based Design of a Small Molecule CD4-Antagonist with Broad Spectrum Anti-HIV-1 Activity. J. Med. Chem. 2015, 58, 6909–6927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaLonde, J.M.; Elban, M.A.; Courter, J.R.; Sugawara, A.; Soeta, T.; Madani, N.; Princiotto, A.M.; Do Kwon, Y.; Kwong, P.D.; Schon, A.; et al. Design, synthesis and biological evaluation of small molecule inhibitors of CD4-gp120 binding based on virtual screening. Bioorg. Med. Chem. 2011, 19, 91–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melillo, B.; Liang, S.; Park, J.; Schon, A.; Courter, J.R.; LaLonde, J.M.; Wendler, D.J.; Princiotto, A.M.; Seaman, M.S.; Freire, E.; et al. Small-Molecule CD4-Mimics: Structure-Based Optimization of HIV-1 Entry Inhibition. ACS Med. Chem. Lett. 2016, 7, 330–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennings, P.S. HIV Drug Resistance: Problems and Perspectives. Infect. Dis. Rep. 2013, 5, e5. [Google Scholar] [CrossRef] [PubMed]
- Rusconi, S.; Moonis, M.; Merrill, D.P.; Pallai, P.V.; Neidhardt, E.A.; Singh, S.K.; Willis, K.J.; Osburne, M.S.; Profy, A.T.; Jenson, J.C.; et al. Naphthalene sulfonate polymers with CD4-blocking and anti-human immunodeficiency virus type 1 activities. Antimicrob. Agents Chemother. 1996, 40, 234–236. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.E.; Stephen, A.G.; Adelsberger, J.W.; Roberts, P.E.; Zhu, W.M.; Currens, M.J.; Feng, Y.X.; Crise, B.J.; Gorelick, R.J.; Rein, A.R.; et al. Discovery of small-molecule human immunodeficiency virus type 1 entry inhibitors that target the gp120-binding domain of CD4. J. Virol. 2005, 79, 6122–6133. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, J.M.; Kuritzkes, D.R.; Godofsky, E.; DeJesus, E.; Larson, J.A.; Weinheimer, S.P.; Lewis, S.T. Safety, pharmacokinetics, and antiretroviral activity of multiple doses of ibalizumab (formerly TNX-355), an anti-CD4 monoclonal antibody, in human immunodeficiency virus type 1-infected adults. Antimicrob. Agents Chemother. 2009, 53, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Iacob, S.A.; Iacob, D.G. Ibalizumab Targeting CD4 Receptors, an Emerging Molecule in HIV Therapy. Front. Microbiol. 2017, 8, 2323. [Google Scholar] [CrossRef] [PubMed]
- Vermeire, K.; Princen, K.; Hatse, S.; De Clercq, E.; Dey, K.; Bell, T.W.; Schols, D. CADA, a novel CD4-targeted HIV inhibitor, is synergistic with various anti-HIV drugs in vitro. Aids 2004, 18, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Caffrey, M. HIV envelope: Challenges and opportunities for development of entry inhibitors. Trends Microbiol. 2011, 19, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C. The process of structure-based drug design. Chem. Biol. 2003, 10, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W., Jr. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [PubMed]
- Stephen, A.G.; Worthy, K.M.; Towler, E.; Mikovits, J.A.; Sei, S.; Roberts, P.; Yang, Q.E.; Akee, R.K.; Klausmeyer, P.; McCloud, T.G.; et al. Identification of HIV-1 nucleocapsid protein: Nucleic acid antagonists with cellular anti-HIV activity. Biochem. Biophys. Res. Commun. 2002, 296, 1228–1237. [Google Scholar] [CrossRef]
- Rein, A.; Henderson, L.E.; Levin, J.G. Nucleic-acid-chaperone activity of retroviral nucleocapsid proteins: Significance for viral replication. Trends Biochem. Sci. 1998, 23, 297–301. [Google Scholar] [CrossRef]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G.; Abraham, U.; Mietzner, T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem. 1994, 37, 4130–4146. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Satoh, T.; Korngold, R.; Huang, Z. CD4 dimerization and oligomerization: Implications for T-cell function and structure-based drug design. Immunol. Today 1998, 19, 455–462. [Google Scholar] [CrossRef]
- Gervaix, A.; West, D.; Leoni, L.M.; Richman, D.D.; Wong-Staal, F.; Corbeil, J. A new reporter cell line to monitor HIV infection and drug susceptibility in vitro. Proc. Natl. Acad. Sci. USA 1997, 94, 4653–4658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Reynolds, C.H. Performance of similarity measures in 2D fragment-based similarity searching: Comparison of structural descriptors and similarity coefficients. J. Chem. Inf. Comput. Sci. 2002, 42, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Pei, S.N.; Parekh, P.; Salazar, E.; Zu, Y. Blocking interaction of viral gp120 and CD4-expressing T cells by single-stranded DNA aptamers. Int. J. Biochem. Cell Biol. 2014, 51, 10–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, C.A. Lead-and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Capasso, A.; Cerchia, C.; Di Giovanni, C.; Granato, G.; Albano, F.; Romano, S.; De Vendittis, E.; Ruocco, M.R.; Lavecchia, A. Ligand-based chemoinformatic discovery of a novel small molecule inhibitor targeting CDC25 dual specificity phosphatases and displaying in vitro efficacy against melanoma cells. Oncotarget 2015, 6, 40202–40222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavecchia, A.; Di Giovanni, C.; Pesapane, A.; Montuori, N.; Ragno, P.; Martucci, N.M.; Masullo, M.; De Vendittis, E.; Novellino, E. Discovery of new inhibitors of Cdc25B dual specificity phosphatases by structure-based virtual screening. J. Med. Chem. 2012, 55, 4142–4158. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Capuzzi, S.J.; Muratov, E.N.; Tropsha, A. Phantom PAINS: Problems with the Utility of Alerts for Pan-Assay Interference Compounds. J. Chem. Inf. Model. 2017, 57, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ye, L.; Wang, X.; Wang, X.; Liu, H.; Qian, X.; Zhu, Y.; Yu, H. Molecular docking, molecular dynamics simulation, and structure-based 3D-QSAR studies on estrogenic activity of hydroxylated polychlorinated biphenyls. Sci. Total Environ. 2012, 441, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Buzon, V.; Natrajan, G.; Schibli, D.; Campelo, F.; Kozlov, M.M.; Weissenhorn, W. Crystal structure of HIV-1 gp41 including both fusion peptide and membrane proximal external regions. PLoS Pathog. 2010, 6, e1000880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NCI Development Therapeutics Program. Available online: https://dtp.cancer.gov/organization/dscb/obtaining/vialed.htm (accessed on 2 November 2017).
- Enhanced NCI Database Browser 2.2. Available online: https://cactus.nci.nih.gov/ncidb2.2/ (accessed on 2 November 2017).
- Li, X.; Ye, L.; Wang, X.; Wang, X.; Liu, H.; Zhu, Y.; Yu, H. Combined 3D-QSAR, molecular docking and molecular dynamics study on thyroid hormone activity of hydroxylated polybrominated diphenyl ethers to thyroid receptors β. Toxicol. Appl. Pharmacol. 2012, 265, 300–307. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | NSC No. | Structure | Inhibition at 100 μM (%) |
|---|---|---|---|
| 1 | 14410 |  | 34.22 |
| 2 | 34238 |  | 36.86 |
| 3 | 119915 |  | 94.94 |
| 4 | 123526 |  | 22.61 |
| 5 | 268665 |  | 33.09 |
| Class I: | ||||||||
 | ||||||||
| No. | NSC No. | R1 | R2 | R3 | R4 | R5 | R6 | Inhibition at 100 μM (%) |
| 6 | 5302 |  |  |  | | | | 5.64 |
| 7 | 11840 |  | | | | | | 8.87 |
| 8 | 29117 | | | | | |  | 12.04 |
| 9 | 45749 | | | | | | | 6.40 |
| 10 | 65625 | |  | | | | | 16.98 |
| 11 | 72276 | | | | | |  | 10.83 |
| 12 | 100977 |  | | | | | 5.27 | |
| 13 | 118660 |  | | | | | | 11.80 |
| 14 | 251156 |  | | | | | | 8.47 |
| 15 | 289346 |  | | | | | | 15.80 |
| 16 | 372920 | | | | | | | 19.76 |
| 17 | 372922 |  | | | | | | 37.27 |
| 18 | 372923 |  | | | | | | 37.10 |
| 19 | 642907 | |  | | | | | 31.77 |
| 20 | 649799 |  | | | | | | 42.79 |
| Class II: | ||||||||
 | ||||||||
| No. | NSC No. | R1 | R2 | R3 | R4 | Inhibition at 100 μM (%) | ||
| 21 | 347512 |  | | | | 33.51 | ||
| 22 | 354633 |  | | | | 39.17 | ||
| 23 | 358315 | |  | | | 32.87 | ||
| 24 | 361582 |  | | | | 45.42 | ||
| 25 | 361583 |  | | | | 25.79 | ||
| 26 | 362083 | | | | | 18.10 | ||
| 27 | 383452 | |  | | | 51.37 | ||
| Class III: | ||||||||
 | ||||||||
| No. | NSC No. | R1 | Inhibition at 100 μM (%) | |||||
| 28 | 119911 |  | 78.62 | |||||
| 29 | 158917 |  | 85.29 | |||||
| Class IV: | ||||||||
| Other compounds | ||||||||
| No. | NSC No. | Structure | Inhibition at 100 μM (%) | |||||
| 30 | 53584 |  | 6.48 | |||||
| 31 | 107022 |  | 16.02 | |||||
| 32 | 61851 |  | 20.09 | |||||
| PLS Statistics | CoMFA | CoMSIA |
|---|---|---|
| Optimum number of components (ONC) | 3 | 4 |
| q2 | 0.624 | 0.732 |
| r2 | 0.961 | 0.973 |
| SEE | 0.145 | 0.124 |
| F | 123.938 | 127.654 |
| Field contribution% | ||
| Steric | 49.5 | 22.3 |
| Electrostatic | 50.5 | 10.2 |
| Hydrophobic | 23.0 | |
| Hydrogen bond donor | 22.7 | |
| Hydrogen bond acceptor | 21.8 |
| No | PInhibition a (Experimental Data) | CoMFA | CoMSIA | ||
|---|---|---|---|---|---|
| PInhibition a (Predicted Data) | Residual | PInhibition a (Predicted Data) | Residual | ||
| 3 | 5.273 | 5.124 | 0.149 | 5.215 | 0.058 |
| 6 | 2.762 | 2.777 | −0.015 | 2.586 | 0.176 |
| 7 | 2.988 | 3.203 | −0.215 | 3.236 | −0.248 |
| 8b | 3.136 | 2.934 | 0.202 | 2.731 | 0.405 |
| 9 | 2.835 | 2.905 | −0.07 | 2.911 | −0.076 |
| 10 | 3.311 | 2.934 | 0.377 | 3.125 | 0.186 |
| 11b | 3.084 | 2.905 | 0.179 | 2.915 | 0.169 |
| 12 | 2.745 | 2.876 | −0.131 | 2.793 | −0.048 |
| 13b | 3.126 | 3.342 | −0.216 | 3.075 | 0.051 |
| 14 | 2.966 | 2.916 | 0.05 | 2.965 | 0.001 |
| 15 | 3.273 | 3.269 | 0.004 | 3.223 | 0.050 |
| 16 | 3.391 | 3.330 | 0.061 | 3.304 | 0.087 |
| 17 | 3.774 | 3.775 | −0.001 | 3.752 | 0.022 |
| 18b | 3.771 | 3.436 | 0.335 | 3.430 | 0.341 |
| 19b | 3.668 | 2.940 | 0.728 | 2.972 | 0.696 |
| 20 | 3.874 | 3.755 | 0.119 | 3.846 | 0.028 |
| 21 | 3.702 | 3.872 | −0.170 | 3.651 | 0.051 |
| 22b | 3.809 | 3.837 | −0.028 | 3.587 | 0.222 |
| 23b | 3.690 | 3.886 | −0.196 | 3.684 | 0.006 |
| 24 | 3.920 | 4.037 | −0.117 | 3.834 | 0.086 |
| 25 | 3.541 | 3.531 | 0.010 | 3.517 | 0.024 |
| 26 | 3.344 | 3.360 | −0.016 | 3.503 | −0.159 |
| 27 | 4.024 | 3.972 | 0.052 | 3.974 | 0.050 |
| 28b | 4.566 | 3.722 | 0.844 | 4.206 | 0.360 |
| 29 | 4.763 | 4.819 | −0.056 | 4.841 | −0.078 |
| 30 | 2.841 | 2.780 | 0.061 | 2.928 | −0.087 |
| 32 | 3.400 | 3.493 | −0.093 | 3.523 | −0.123 |
| No. | NSC No. | Bioassay (Active) a | PAINS Filter b |
|---|---|---|---|
| 17 | 372922 | 1 (anticancer screen) | PASS |
| 18 | 372923 | 4 (antimicrobial assay; histone lysine methyltransferase G9a inhibitor; SWI/SNF chromatin remodeling complex inhibitor) | PASS |
| 19 | 642907 | 7 (Grb2; HRAR1; p56 lck tyrosine kinase; Fyn protein kinase; phospholipase C gamma | PASS |
| 20 | 649799 | NONE | PASS |
| 21 | 347512 | 5 (all for anti-cancer screens in mice using different models) | PASS |
| 22 | 354633 | 1 (anticancer drug screen. Data for tumor model P388 Leukemia in mice) | PASS |
| 23 | 358315 | NONE | PASS |
| 24 | 361582 | NONE | PASS |
| 25 | 361583 | NONE | PASS |
| 26 | 362083 | NONE | PASS |
| 27 | 383452 | 4 (anticancer drug screen in mice, fructose-bisphosphate aldolase inhibitor) | PASS |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Zhang, H.; Huang, L.S.; Zhu, S.; Xu, Y.; Zhang, X.-Q.; Schooley, R.T.; Yang, X.; Huang, Z.; An, J. Virtual Screening, Biological Evaluation, and 3D-QSAR Studies of New HIV-1 Entry Inhibitors That Function via the CD4 Primary Receptor. Molecules 2018, 23, 3036. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23113036
Zhang C, Zhang H, Huang LS, Zhu S, Xu Y, Zhang X-Q, Schooley RT, Yang X, Huang Z, An J. Virtual Screening, Biological Evaluation, and 3D-QSAR Studies of New HIV-1 Entry Inhibitors That Function via the CD4 Primary Receptor. Molecules. 2018; 23(11):3036. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23113036
Chicago/Turabian StyleZhang, Chaozai, Huijun Zhang, Lina S. Huang, Siyu Zhu, Yan Xu, Xing-Quan Zhang, Robert T. Schooley, Xiaohong Yang, Ziwei Huang, and Jing An. 2018. "Virtual Screening, Biological Evaluation, and 3D-QSAR Studies of New HIV-1 Entry Inhibitors That Function via the CD4 Primary Receptor" Molecules 23, no. 11: 3036. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23113036