On the Mechanism of Action of Anti-Inflammatory Activity of Hypericin: An In Silico Study Pointing to the Relevance of Janus Kinases Inhibition

 , , and
, , and
Abstract
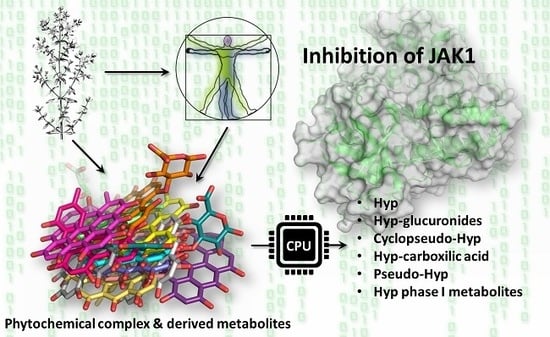
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
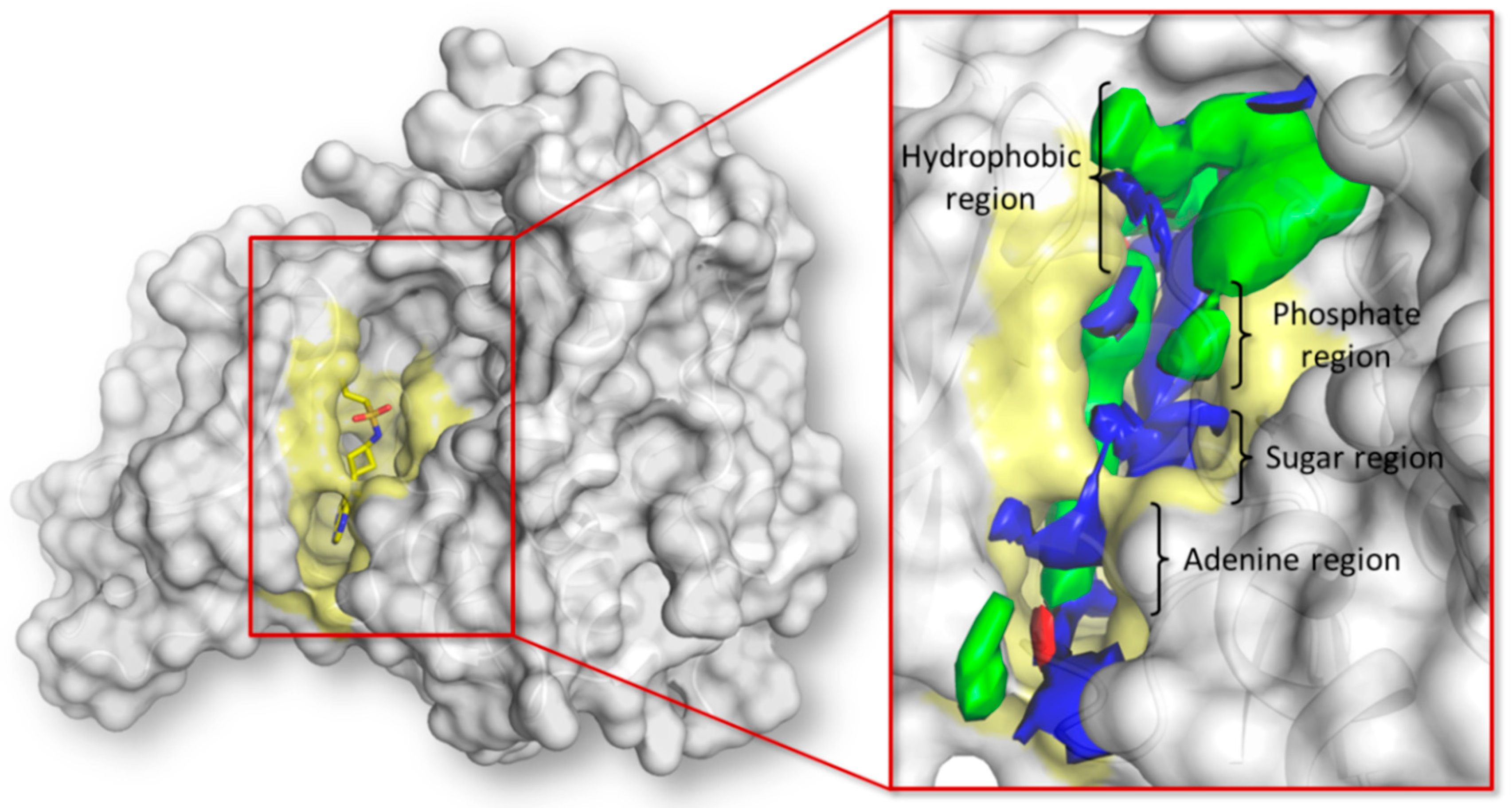
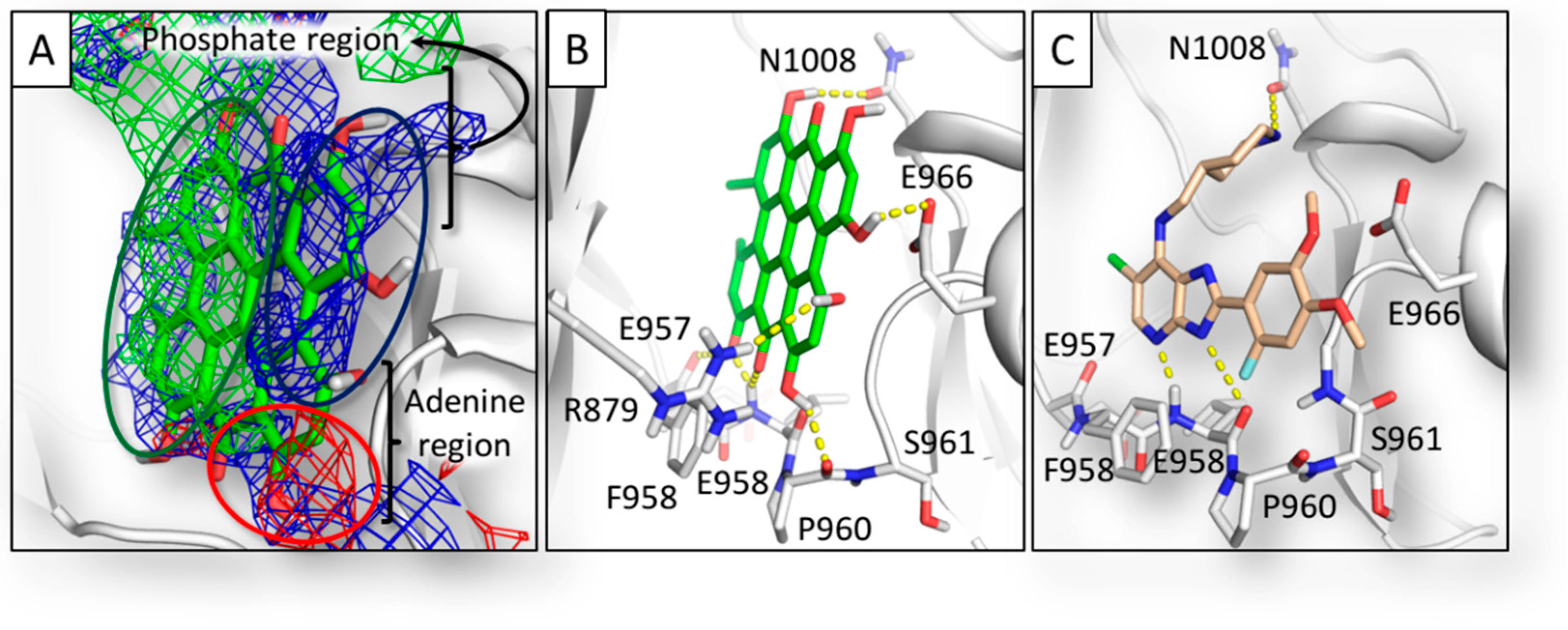
2.1. Pharmacophoric Analysis of the ATP Binding Site
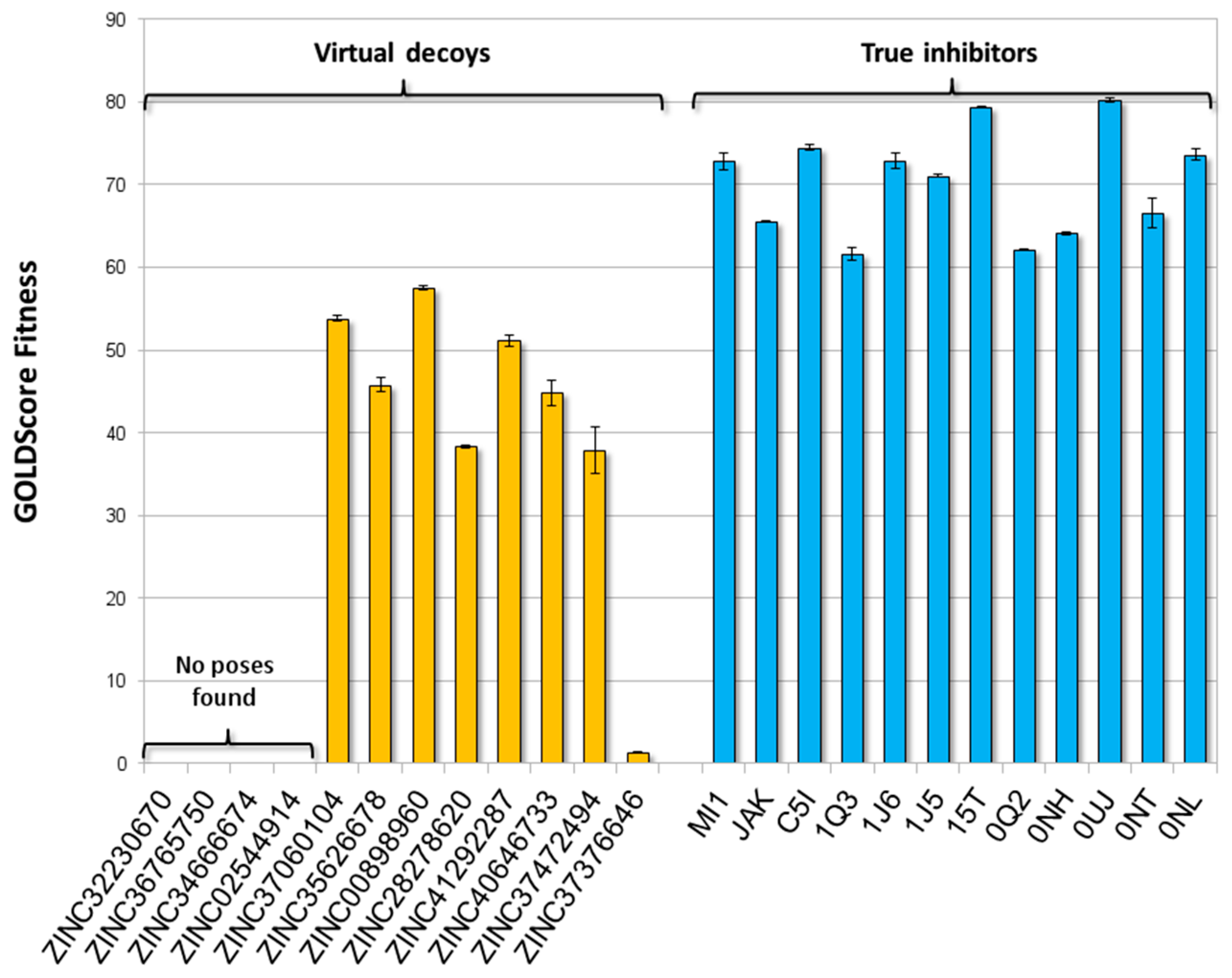
2.2. Training and Validation Procedures
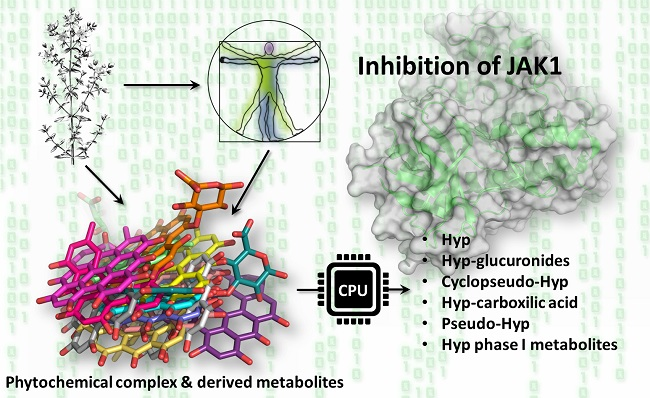
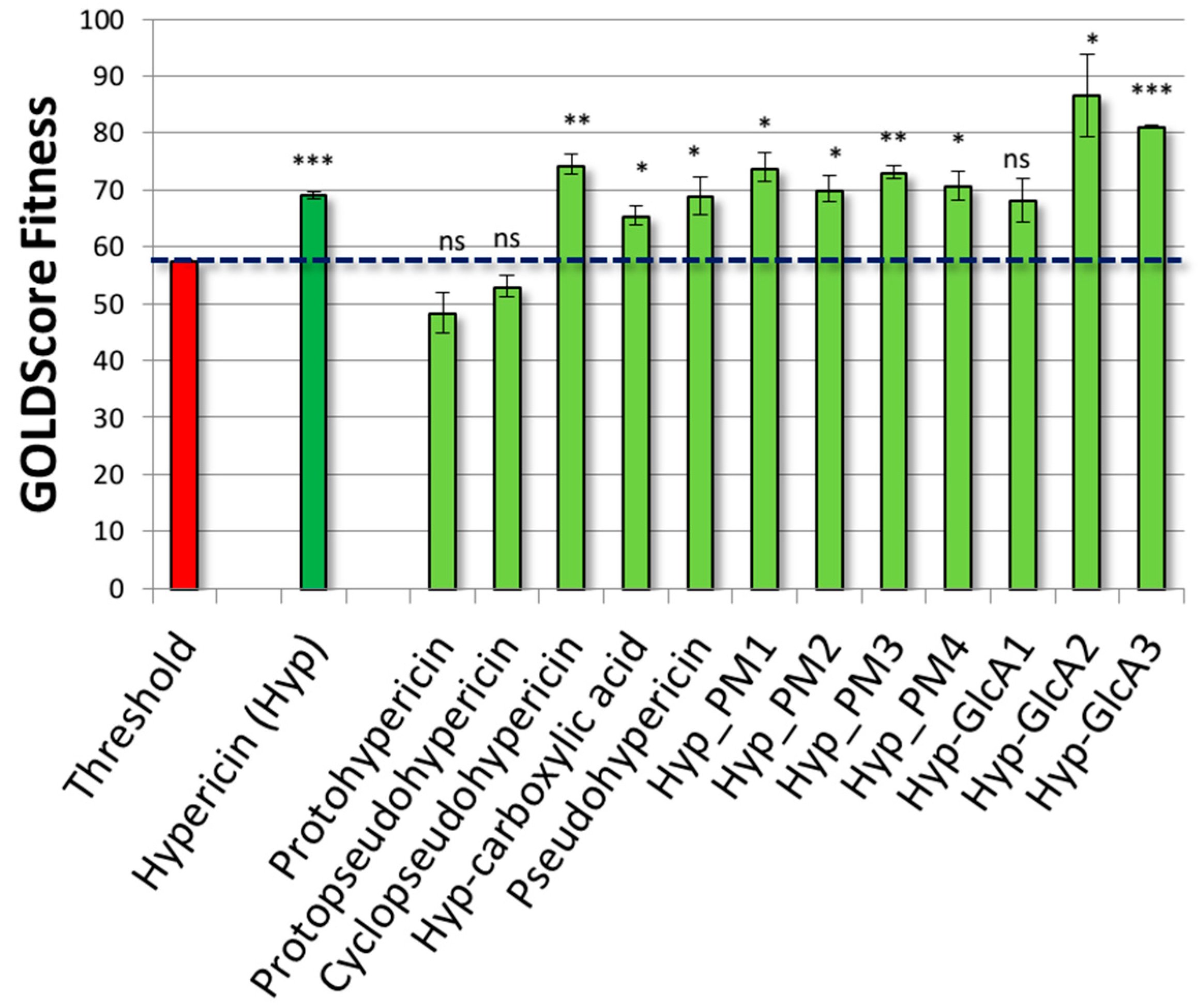
2.3. Results of Hypericin
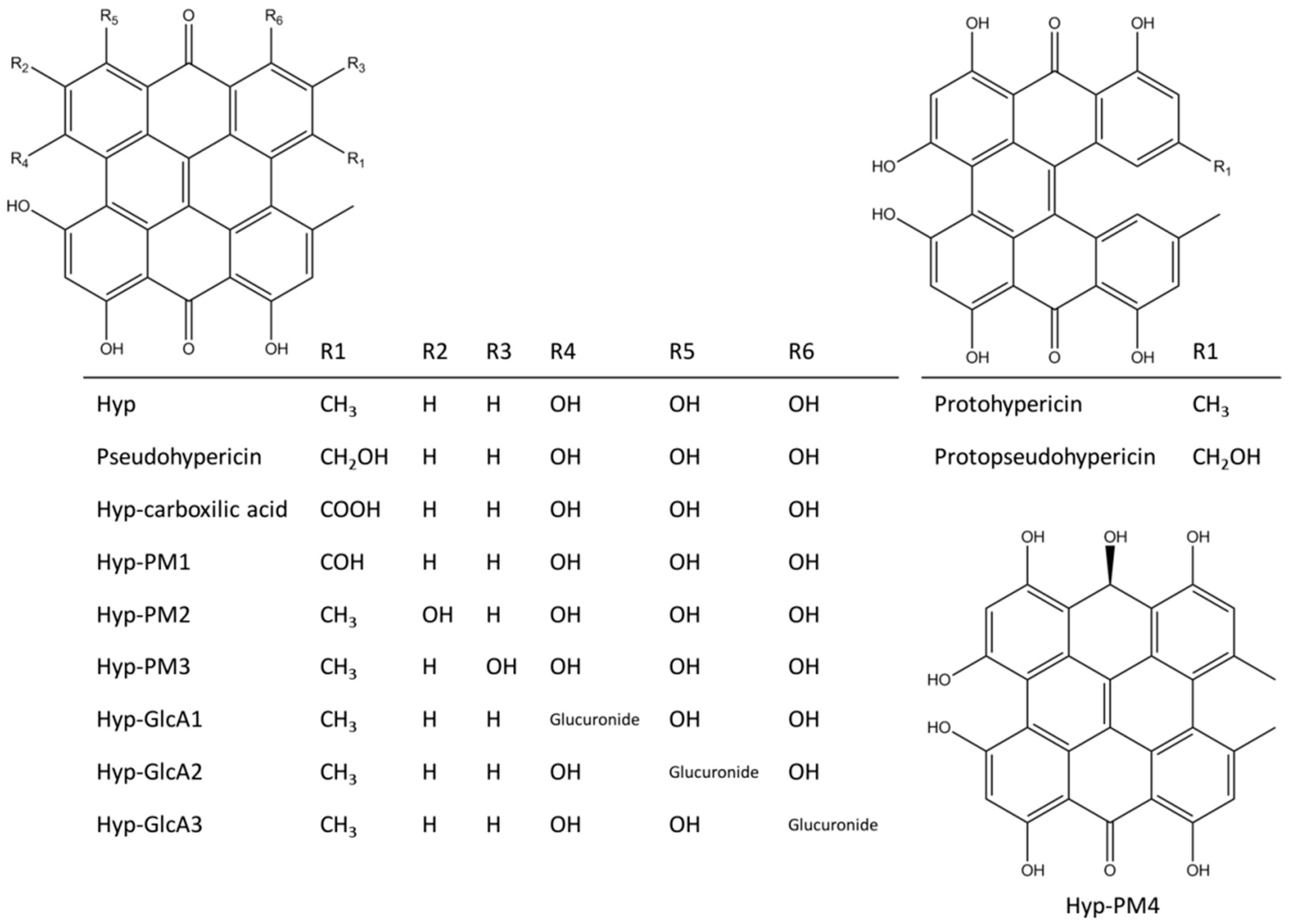
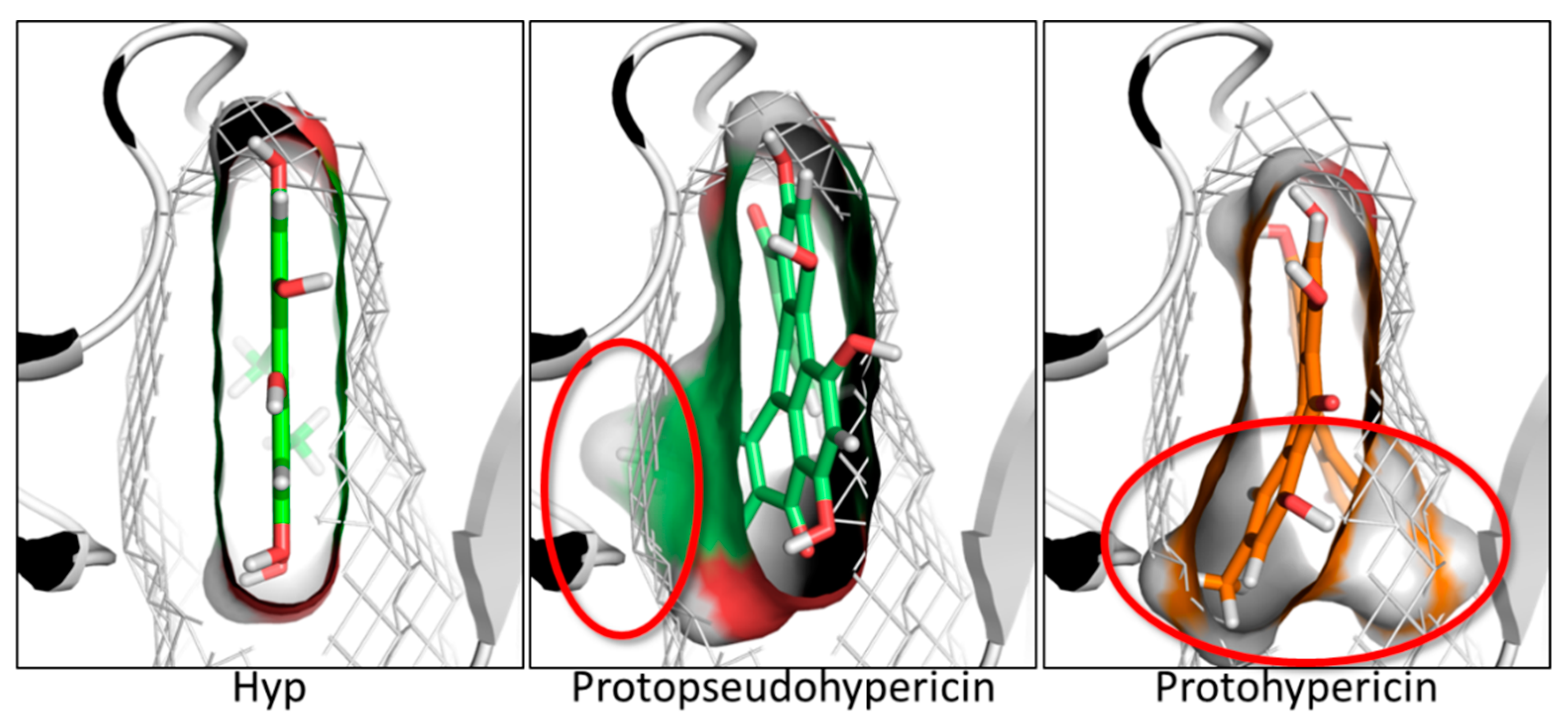
2.4. Results of Hypericin Analogues and Metabolites
3. Materials and Methods
3.1. Design of Training and Validation Sets
3.2. Model Preparation for Docking Simulations
3.3. Pharmacophoric Modeling
3.4. Docking Simulations
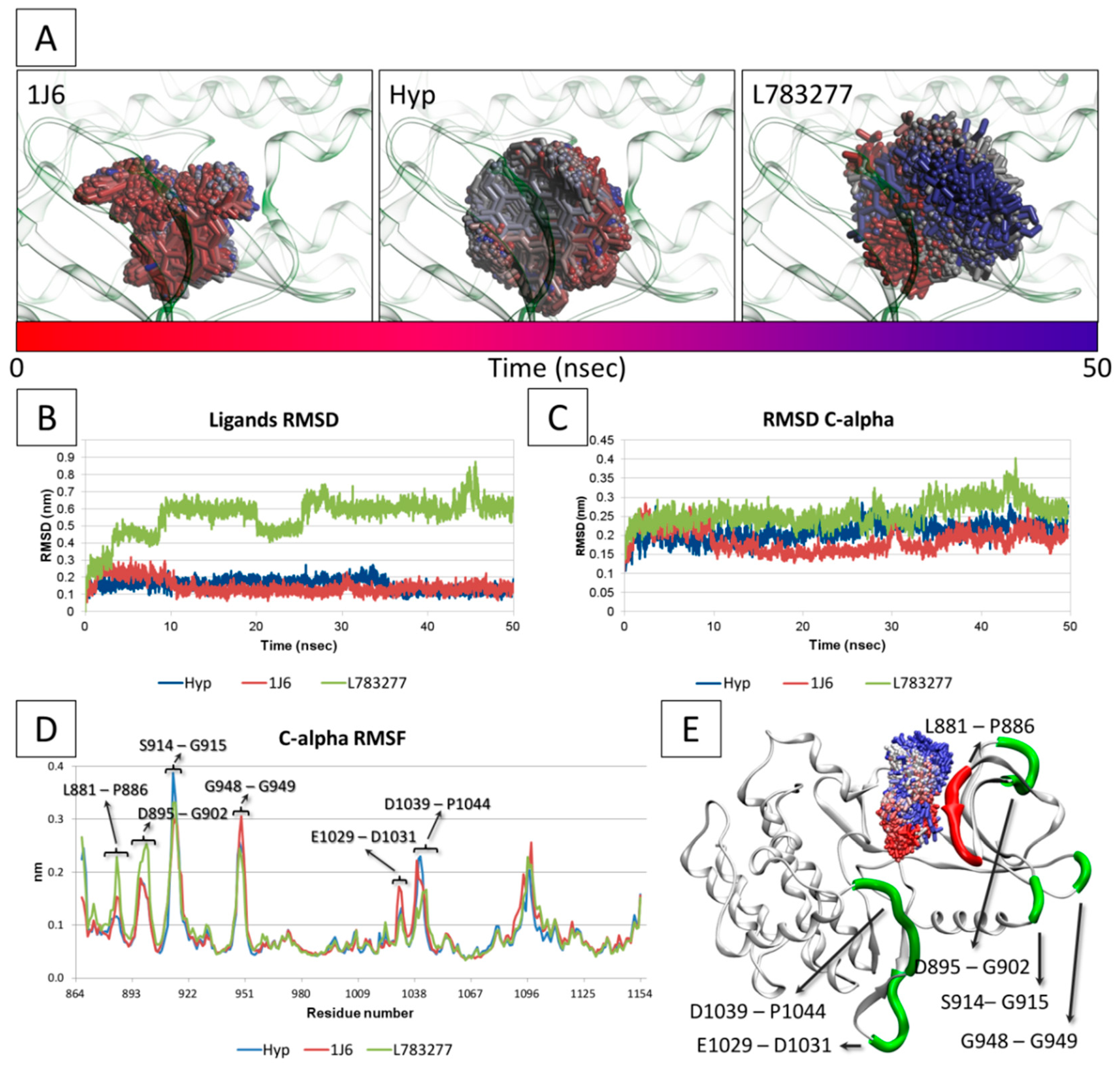
3.5. Molecular Dynamic
3.6. Prediction of Metabolites
3.7. Statistical Analysis
3.8. Graphical Representation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Božin, B.; Kladar, N.; Grujić, N.; Anačkov, G.; Samojlik, I.; Gavarić, N.; Conić, B.S. Impact of origin and biological source on chemical composition, anticholinesterase and antioxidant properties of some St. John’s wort species (Hypericum spp., Hypericaceae) from the Central Balkans. Molecules 2013, 18, 11733–11750. [Google Scholar] [CrossRef] [PubMed]
- EMA. Community Herbal Monograph on Hypericum perforatum L. Herba (Well-Established Medicinal Use); EMA/HMPC/101304/102008; EMA: London, UK, 2009. [Google Scholar]
- EMA. Assessment Report on Hypericum perforatum L., Herba; EMA/HMPC/101303/102008; EMA: London, UK, 2009. [Google Scholar]
- Franz, C.; Chizzola, R.; Novak, J.; Sponza, S. Botanical species being used for manufacturing plant food supplements (PFS) and related products in the EU member states and selected third countries. Food Funct. 2011, 2, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Murthy, H.N.; Kim, Y.S.; Park, S.Y.; Paek, K.Y. Hypericins: Biotechnological production from cell and organ cultures. Appl. Microbiol. Biotechnol. 2014, 98, 9187–9198. [Google Scholar] [CrossRef] [PubMed]
- Wurglics, M.; Schubert-Zsilavecz, M. Hypericum perforatum: A ‘modern’ herbal antidepressant- Pharmacokinetics of active ingredients. Clin. Pharmacokinet. 2006, 45, 449–468. [Google Scholar] [CrossRef] [PubMed]
- Süntar, I.P.; Akkol, E.K.; Yılmazer, D.; Baykal, T.; Kırmızıbekmez, H.; Alper, M.; Yeşilada, E. Investigations on the in vivo wound healing potential of Hypericum perforatum L. J. Ethnopharmacol. 2010, 127, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Rizshsky, M.; Hauck, C.C.; Nikolau, B.J.; Murphy, P.A.; Birt, D.F. The inhibition of lipopolysaccharide-induced macrophage inflammation by 4 compounds in Hypericum perforatum extract is partially dependent on the activation of SOCS3. Phytochemistry 2012, 76, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Paterniti, I.; Briguglio, E.; Mazzon, E.; Galuppo, M.; Oteri, G.; Cordasco, G.; Cuzzocrea, S. Effects of Hypericum perforatum, in a rodent model of periodontitis. BMC Complement. Altern. Med. 2010, 10, 73. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, M.S.; Cardoso, R.D.; Fattori, V.; Arakawa, N.S.; Tomaz, J.C.; Lopes, N.P.; Verri, W.A., Jr. Hypericum perforatum reduces paracetamol-induced hepatotoxicity and lethality in mice by modulating inflammation and oxidative stress. Phytother. Res. 2015, 29, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Raziq, N.; Saeed, M.; Shahid, M.; Muhammad, N.; Khan, H.; Gul, F. Pharmacological basis for the use of Hypericum oblongifolium as a medicinal plant in the management of pain, inflammation and pyrexia. BMC Complement. Altern. Med. 2015, 16, 41. [Google Scholar] [CrossRef] [PubMed]
- Velingkar, V.S.; Gupta, G.L.; Hegde, N.B. A current update on phytochemistry, pharmacology and herb–drug interactions of Hypericum perforatum. Phytochem. Rev. 2017, 16, 725–744. [Google Scholar] [CrossRef]
- Takahashi, I.; Nakanishi, S.; Kobayashi, E.; Nakano, H.; Suzuki, K.; Tamaoki, T. Hypericin and pseudohypericin specifically inhibit protein kinase C: Possible relation to their antiretroviral activity. Biochem. Biophys. Res. Commun. 1989, 165, 1207–1212. [Google Scholar] [CrossRef]
- Yip, L.; Hudson, J.B.; Gruszecka-Kowalik, E.; Zalkow, L.H.; Neil Towers, G.H. Antiviral activity of a derivative of the photosensitive compound Hypericin. Phytomedicine 1996, 3, 185–190. [Google Scholar] [CrossRef]
- Kil, K.S.; Yum, Y.N.; Seo, S.H.; Lee, K.T. Antitumor activities of hypericin as a protein tyrosine kinase blocker. Arch. Pharm. Res. 1996, 19, 490. [Google Scholar] [CrossRef]
- Panossian, A.G.; Gabrielian, E.; Manvelian, V.; Jurcic, K.; Wagner, H. Immunosuppressive effects of hypericin on stimulated human leukocytes: Inhibition of the arachidonic acid release, leukotriene B(4) and Interleukin-Iα production, and activation of nitric oxide formation. Phytomedicine 1996, 3, 19–28. [Google Scholar] [CrossRef]
- Hammer, K.D.P.; Yum, M.Y.; Dixon, P.M.; Birt, D.F. Identification of JAK-STAT pathways as important for the anti-inflammatory activity of a Hypericum perforatum fraction and bioactive constituents in RAW 264.7 mouse macrophages. Phytochemistry 2010, 71, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Babon, J.J.; Lucet, I.S.; Murphy, J.M.; Nicola, N.A.; Varghese, L.N. The molecular regulation of Janus kinase (JAK) activation. Biochem. J. 2014, 462, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, J.D.; Flanagan, M.E.; Telliez, J.B. Discovery and Development of Janus Kinase (JAK) Inhibitors for Inflammatory Diseases. J. Med. Chem. 2014, 57, 5023–5038. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.P.; Yang, X.X.; Chan, S.Y.; Xu, A.L.; Duan, W.; Zhu, Y.Z.; Wang, J.C. St. John’s wort attenuates irinotecan-induced diarrhea via down-regulation of intestinal pro-inflammatory cytokines and inhibition of intestinal epithelial apoptosis. Toxicol. Appl. Pharmacol. 2006, 216, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Dellafiora, L.; Mena, P.; Cozzini, P.; Brighenti, F.; Del Rio, D. Modelling the possible bioactivity of ellagitannin-derived metabolites. In silico tools to evaluate their potential xenoestrogenic behavior. Food Funct. 2013, 4, 1442–1451. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, L.; Karelson, M.; Dobchev, D.A. Identification of Natural Compounds against Neurodegenerative Diseases Using In Silico Techniques. Molecules 2018, 25, 1847. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Yang, F.; Kang, J.; Yang, X.; Lai, X.; Gao, Y. Multi-Layer Identification of Highly-Potent ABCA1 Up-Regulators Targeting LXRβ Using Multiple QSAR Modeling, Structural Similarity Analysis, and Molecular Docking. Molecules 2016, 21, 1639. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Janus kinase (JAK) inhibitors in the treatment of inflammatory and neoplastic diseases. Pharmacol. Res. 2016, 111, 784–803. [Google Scholar] [CrossRef] [PubMed]
- Kubin, A.; Wierrani, F.; Burner, U.; Alth, G.; Grunberger, W. Hypericin—The facts about a controversial agent. Curr. Pharm. Des. 2005, 11, 233–253. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, Y.; Nakaniwa, T.; Kinoshita, T.; Nakanishi, I.; Kitaura, K.; Hirasawa, A.; Tsujimoto, G.; Tada, T. Structural insight into human CK2α in complex with the potent inhibitor ellagic acid. Bioorg. Med. Chem. Lett. 2009, 19, 2920–2923. [Google Scholar] [CrossRef] [PubMed]
- Caspers, N.L.; Han, S.; Rajamohan, F.; Hoth, L.R.; Geoghegan, K.F.; Subashi, T.A.; Vazquez, M.L.; Kaila, N.; Cronin, C.N.; Johnson, E.; et al. Development of a high-throughput crystal structure-determination platform for JAK1 using a novel metal-chelator soaking system. Acta Crystallogr. Sect. F-Struct. Biol. Commun. 2016, 72, 840–845. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, M.L.; Kaila, N.; Strohbach, J.W.; Trzupek, J.D.; Brown, M.F.; Flanagan, M.E.; Mitton-Fry, M.J.; Johnson, T.A.; TenBrink, R.E.; Arnold, E.P.; et al. Identification of N-{cis-3-(Methyl(7H-pyrrolo 2,3-d pyrimidin-4-yl)aminolcyclobutyl}propan e-1-sulfonamide (PF-04965842): A Selective JAK1 Clinical Candidate for the Treatment of Autoimmune Diseases. J. Med. Chem. 2018, 61, 1130–1152. [Google Scholar] [CrossRef] [PubMed]
- Vasbinder, M.M.; Alimzhanov, M.; Augustin, M.; Bebernitz, G.; Bell, K.; Chuaqui, C.; Deegan, T.; Ferguson, A.D.; Goodwin, K.; Huszar, D.; et al. Identification of azabenzimidazoles as potent JAK1 selective inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Bruni, R.; Sacchetti, G. Factors affecting polyphenol biosynthesis in wild and field grown St. John’s Wort (Hypericum perforatum L. Hypericaceae/Guttiferae). Molecules 2009, 14, 682–725. [Google Scholar] [CrossRef] [PubMed]
- Dellafiora, L.; Dall’Asta, C.; Galaverna, G. Toxicodynamics of Mycotoxins in the Framework of Food Risk Assessment-An In Silico Perspective. Toxins 2018, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.; D’Avolio, A.; Moyle, G.; Bonora, S.; Di Perri, G.; Else, L.; Simiele, M.; Singh, G.J.; Back, D.; Boffito, M. Pharmacokinetics of the co-administration of boceprevir and St John’s wort to male and female healthy volunteers. J. Antimicrob. Chemother. 2014, 69, 1911–1915. [Google Scholar] [CrossRef] [PubMed]
- Dellafiora, L.; Galaverna, G.; Dall’Asta, C. In silico analysis sheds light on the structural basis underlying the ribotoxicity of trichothecenes—A tool for supporting the hazard identification process. Toxicol. Lett. 2017, 270, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dellafiora, L.; Dall’Asta, C.; Cozzini, P. Ergot alkaloids: From witchcraft till in silico analysis. Multi-receptor analysis of ergotamine metabolites. Toxicol. Rep. 2015, 2, 535–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Cereto-Massague, A.; Guasch, L.; Valls, C.; Mulero, M.; Pujadas, G.; Garcia-Vallve, S. DecoyFinder: An easy-to-use python GUI application for building target-specific decoy sets. Bioinformatics 2012, 28, 1661–1662. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Berdini, V.; Hartshorn, M.J.; Mooij, W.T.; Murray, C.W.; Taylor, R.D.; Watson, P. Virtual screening using protein-ligand docking: Avoiding artificial enrichment. J. Chem. Inf. Model. 2004, 44, 793–806. [Google Scholar]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.Y.; He, J.E.; He, S.Q.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Dellafiora, L.; Galaverna, G.; Dall’Asta, C.; Cozzini, P. Hazard identification of cis/trans-zearalenone through the looking-glass. Food Chem. Toxicol. 2015, 86, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Baroni, M.; Cruciani, G.; Sciabola, S.; Perruccio, F.; Mason, J.S. A common reference framework for analyzing/comparing proteins and ligands. Fingerprints for Ligands and Proteins (FLAP): Theory and application. J. Chem. Inf. Model. 2007, 47, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Carosati, E.; Sciabola, S.; Cruciani, G. Hydrogen bonding interactions of covalently bonded fluorine atoms: From crystallographic data to a new angular function in the GRID force field. J. Med. Chem. 2004, 47, 5114–5125. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-Rojas, W.; Olivero-Verbel, J. Potential interaction of natural dietary bioactive compounds with COX-2. J. Mol. Graph. Model. 2011, 30, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Nongonierma, A.B.; Dellafiora, L.; Paolella, S.; Galaverna, G.; Cozzini, P.; FitzGerald, R.J. In Silico Approaches Applied to the Study of Peptide Analogs of Ile Pro-Ile in Relation to Their Dipeptidyl Peptidase IV Inhibitory Properties. Front. Endocrinol. 2018, 9, 329. [Google Scholar] [CrossRef] [PubMed]
- Rollinger, J.M.; Schuster, D.; Baier, E.; Ellmerer, E.P.; Langer, T.; Stuppner, H. Taspine: Bioactivity-guided isolation and molecular ligand-target insight of a potent acetylcholinesterase inhibitor from Magnolia x soulangiana. J. Nat. Prod. 2006, 69, 1341–1346. [Google Scholar] [CrossRef] [PubMed]
- Dellafiora, L.; Paolella, S.; Dall’Asta, C.; Dossena, A.; Cozzini, P.; Galaverna, G. Hybrid in Silico/in Vitro Approach for the Identification of Angiotensin I Converting Enzyme Inhibitory Peptides from Parma Dry-Cured Ham. J. Agric. Food Chem. 2015, 22, 6366–6375. [Google Scholar] [CrossRef] [PubMed]
- Dellafiora, L.; Aichinger, G.; Geib, E.; Sánchez-Barrionuevo, L.; Brock, M.; Cánovas, D.; Dall’Asta, C.; Marko, D. Hybrid in silico/in vitro target fishing to assign function to “orphan” compounds of food origin–The case of the fungal metabolite atromentin. Food Chem. 2019, 270, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Liniger, M.; Neuhaus, C.; Hofmann, T.; Fransioli-Ignazio, L.; Jordi, M.; Drueckes, P.; Trappe, J.; Fabbro, D.; Altmann, K.H. Kinase Inhibition by Deoxy Analogues of the Resorcylic Lactone L-783277. ACS Med. Chem. Lett. 2011, 2, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using Modeller. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; Mackerell, A.D.J. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory. Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A Fast Force Field Generation Tool for Small Organic Molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Cruciani, G.; Carosati, E.; De Boeck, B.; Ethirajulu, K.; Mackie, C.; Howe, T.; Vianello, R. MetaSite: Understanding Metabolism in Human Cytochromes from the Perspective of the Chemist. J. Med. Chem. 2005, 48, 6970–6979. [Google Scholar] [CrossRef] [PubMed]
- Rudik, A.V.; Dmitriev, A.V.; Lagunin, A.A.; Filimonov, D.A.; Poroikov, V.V. Prediction of reacting atoms for the major biotransformation reactions of organic xenobiotics. J. Cheminform. 2016, 8, 68. [Google Scholar] [CrossRef] [PubMed]
- Pulga, A.; Porte, Y.; Morel, J.L. Changes in C57BL6 Mouse Hippocampal Transcriptome Induced by Hypergravity Mimic Acute Corticosterone-Induced Stress. Front. Mol. Neurosci. 2016, 9, 153. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Birt, D.F.; Widrlechner, M.P.; Hammer, K.D.; Hillwig, M.L.; Wei, J.; Kraus, G.A.; Murphy, P.A.; McCoy, J.; Wurtele, E.S.; Neighbors, J.D.; et al. Hypericum in infection: Identification of anti-viral and anti-inflammatory constituents. Pharm. Biol. 2009, 47, 774–782. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dellafiora, L.; Galaverna, G.; Cruciani, G.; Dall’Asta, C.; Bruni, R. On the Mechanism of Action of Anti-Inflammatory Activity of Hypericin: An In Silico Study Pointing to the Relevance of Janus Kinases Inhibition. Molecules 2018, 23, 3058. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23123058
Dellafiora L, Galaverna G, Cruciani G, Dall’Asta C, Bruni R. On the Mechanism of Action of Anti-Inflammatory Activity of Hypericin: An In Silico Study Pointing to the Relevance of Janus Kinases Inhibition. Molecules. 2018; 23(12):3058. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23123058
Chicago/Turabian StyleDellafiora, Luca, Gianni Galaverna, Gabriele Cruciani, Chiara Dall’Asta, and Renato Bruni. 2018. "On the Mechanism of Action of Anti-Inflammatory Activity of Hypericin: An In Silico Study Pointing to the Relevance of Janus Kinases Inhibition" Molecules 23, no. 12: 3058. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23123058