Molecular Modeling and Structural Stability of Wild-Type and Mutant CYP51 from Leishmania major: In Vitro and In Silico Analysis of a Laboratory Strain

 ,
,
Abstract
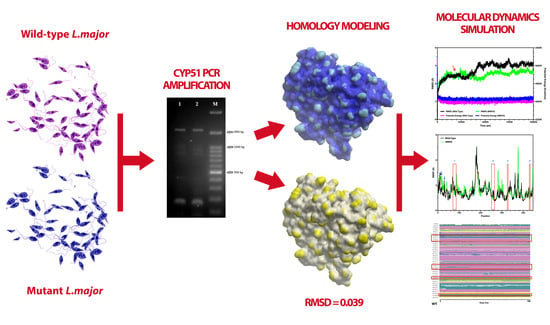
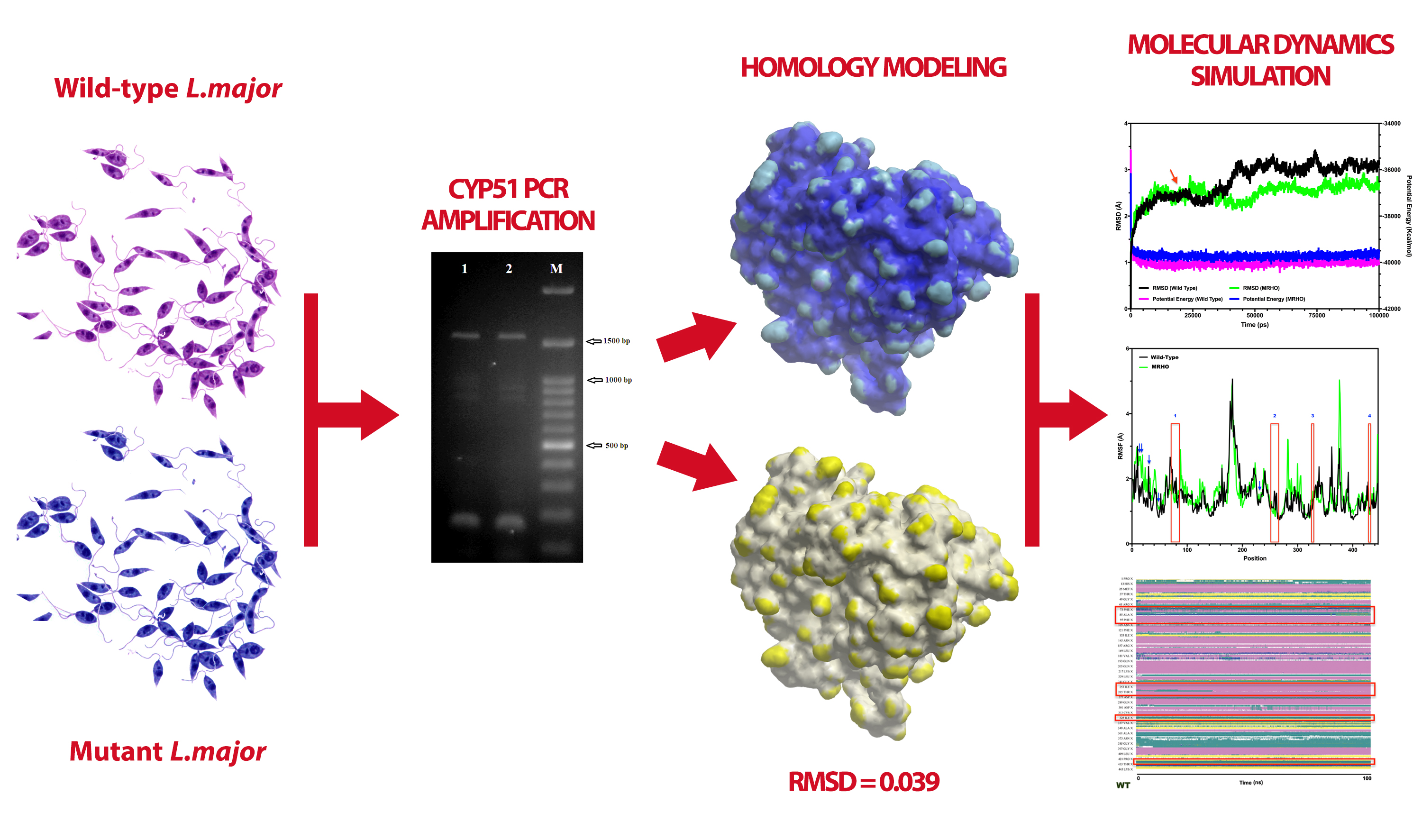
:
1. Introduction
2. Results and Discussion
2.1. CYP51 Expression and Sequence Analysis
2.2. Homology Modeling and Structural Analysis of CYP51
2.3. In Silico Analysis of Protein Stability Upon Mutation
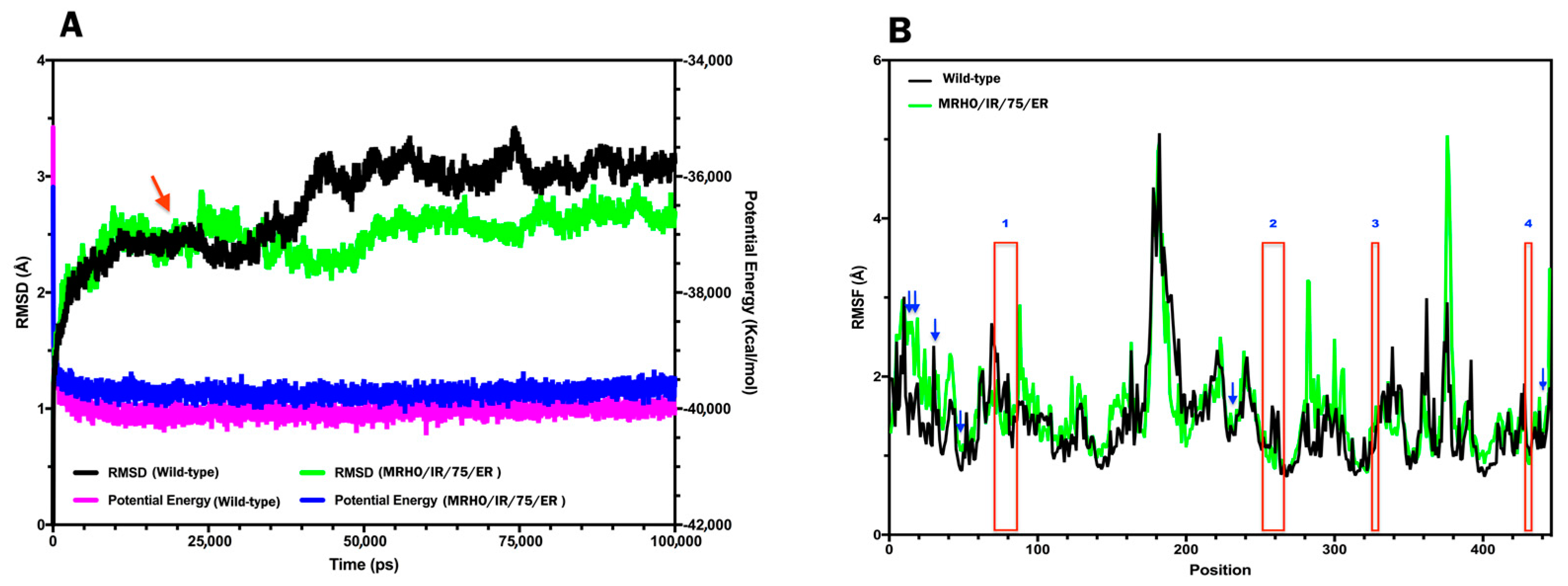
2.4. Molecular Dynamics (MD) Simulations and Interpretations
3. Material and Methods
3.1. Parasite Culture
3.2. Genomic DNA Extraction and RNA Extraction
3.3. cDNA Synthesis and Polymerase Chain Reaction (PCR)
3.4. DNA Sequencing and Sequence Analysis
3.5. Homology Modeling and Sequence Analysis
3.6. In Silico Site-Directed Mutagenesis
3.7. Molecular Dynamics Simulation
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Alvar, J.; Velez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; den Boer, M.; WHO Leishmaniasis Control Team. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 2012, 7, e35671. [Google Scholar] [CrossRef] [PubMed]
- Ready, P.D. Biology of phlebotomine sand flies as vectors of disease agents. Annu. Rev. Entomol. 2013, 58, 227–250. [Google Scholar] [CrossRef] [PubMed]
- Murray, H.W.; Berman, J.D.; Davies, C.R.; Saravia, N.G. Advances in leishmaniasis. Lancet 2005, 366, 1561–1577. [Google Scholar] [CrossRef]
- Croft, S.L.; Sundar, S.; Fairlamb, A.H. Drug resistance in leishmaniasis. Clin. Microbiol. Rev. 2006, 19, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Hargrove, T.Y.; Wawrzak, Z.; Liu, J.; Waterman, M.R.; Nes, W.D.; Lepesheva, G.I. Structural complex of sterol 14α-demethylase (cyp51) with 14α-methylenecyclopropyl-Δ7-24, 25-dihydrolanosterol. J. Lipid Res. 2012, 53, 311–320. [Google Scholar] [CrossRef] [PubMed]
- McCall, L.I.; El Aroussi, A.; Choi, J.Y.; Vieira, D.F.; De Muylder, G.; Johnston, J.B.; Chen, S.; Kellar, D.; Siqueira-Neto, J.L.; Roush, W.R.; et al. Targeting ergosterol biosynthesis in leishmania donovani: Essentiality of sterol 14α-demethylase. PLoS Negl. Trop. Dis. 2015, 9, e0003588. [Google Scholar] [CrossRef] [PubMed]
- Lepesheva, G.I.; Waterman, M.R. Sterol 14α-demethylase cytochrome p450 (cyp51), a p450 in all biological kingdoms. Biochim. Biophys. Acta 2007, 1770, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Alrajhi, A.A.; Ibrahim, E.A.; De Vol, E.B.; Khairat, M.; Faris, R.M.; Maguire, J.H. Fluconazole for the treatment of cutaneous leishmaniasis caused by leishmania major. N. Engl. J. Med. 2002, 346, 891–895. [Google Scholar] [CrossRef] [PubMed]
- Lepesheva, G.I.; Waterman, M.R. Sterol 14α-demethylase (cyp51) as a therapeutic target for human trypanosomiasis and leishmaniasis. Curr. Top. Med. Chem. 2011, 11, 2060–2071. [Google Scholar] [CrossRef] [PubMed]
- Melesina, J.; Robaa, D.; Pierce, R.J.; Romier, C.; Sippl, W. Homology modeling of parasite histone deacetylases to guide the structure-based design of selective inhibitors. J. Mol. Graph. Model. 2015, 62, 342–361. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, M.; Balasubramanian, S.; Ramaswamy, A. Structural dynamics of wild type and mutated forms of human L1 endonuclease and insights into its sequence specific nucleic acid binding mechanism: A molecular dynamics study. J. Mol. Graph. Model. 2017, 76, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Hargrove, T.Y.; Wawrzak, Z.; Liu, J.; Nes, W.D.; Waterman, M.R.; Lepesheva, G.I. Substrate preferences and catalytic parameters determined by structural characteristics of sterol 14α-demethylase (cyp51) from Leishmania infantum. J. Biol. Chem. 2011, 286, 26838–26848. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Mehta, A.; Shaha, C. Cyp5122a1, a novel cytochrome p450 is essential for survival of Leishmania donovani. PLoS ONE 2011, 6, e25273. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.K.; Leung, S.S.; Guilbert, C.; Jacobson, M.P.; McKerrow, J.H.; Podust, L.M. Structural characterization of cyp51 from Trypanosoma cruzi and Trypanosoma brucei bound to the antifungal drugs posaconazole and fluconazole. PLoS Negl. Trop. Dis. 2010, 4, e651. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Madison, V.; Chau, A.S.; Loebenberg, D.; Palermo, R.E.; McNicholas, P.M. Three-dimensional models of wild-type and mutated forms of cytochrome p450 14α-sterol demethylases from Aspergillus fumigatus and Candida albicans provide insights into posaconazole binding. Antimicrob. Agents Chemother. 2004, 48, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, E.; Fariselli, P.; Rossi, I.; Casadio, R. A three-state prediction of single point mutations on protein stability changes. BMC Bioinform. 2008, 9 (Suppl. 2), S6. [Google Scholar] [CrossRef] [PubMed]
- Lepesheva, G.I.; Podust, L.M.; Bellamine, A.; Waterman, M.R. Folding requirements are different between sterol 14α-demethylase (cyp51) from Mycobacterium tuberculosis and human or fungal orthologs. J. Biol. Chem. 2001, 276, 28413–28420. [Google Scholar] [CrossRef] [PubMed]
- Betts, M.J.; Russell, R.B. Amino acid properties and consequences of substitutions. In Bioinformatics for Geneticists; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2003; pp. 289–316. [Google Scholar]
- Nitahara, Y.; Kishimoto, K.; Yabusaki, Y.; Gotoh, O.; Yoshida, Y.; Horiuchi, T.; Aoyama, Y. The amino acid residues affecting the activity and azole susceptibility of rat cyp51 (sterol 14-demethylase p450). J. Biochem. 2001, 129, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Snelders, E.; Karawajczyk, A.; Schaftenaar, G.; Verweij, P.E.; Melchers, W.J. Azole resistance profile of amino acid changes in Aspergillus fumigatus cyp51a based on protein homology modeling. Antimicrob. Agents Chemother. 2010, 54, 2425–2430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellado, E.; Garcia-Effron, G.; Alcazar-Fuoli, L.; Melchers, W.J.; Verweij, P.E.; Cuenca-Estrella, M.; Rodriguez-Tudela, J.L. A new Aspergillus fumigatus resistance mechanism conferring in vitro cross-resistance to azole antifungals involves a combination of cyp51a alterations. Antimicrob. Agents Chemother. 2007, 51, 1897–1904. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.E.; Warrilow, A.G.; Price, C.L.; Mullins, J.G.; Kelly, D.E.; Kelly, S.L. Resistance to antifungals that target cyp51. J. Chem. Biol. 2014, 7, 143–161. [Google Scholar] [CrossRef] [PubMed]
- Fukuyoshi, S.; Kometani, M.; Watanabe, Y.; Hiratsuka, M.; Yamaotsu, N.; Hirono, S.; Manabe, N.; Takahashi, O.; Oda, A. Molecular dynamics simulations to investigate the influences of amino acid mutations on protein three-dimensional structures of cytochrome P450 2D6.1, 2, 10, 14A, 51, and 62. PLoS ONE 2016, 11, e0152946. [Google Scholar] [CrossRef] [PubMed]
- Paul, R.A.; Rudramurthy, S.M.; Meis, J.F.; Mouton, J.W.; Chakrabarti, A. A novel Y319H substitution in CYP51C associated with azole resistance in Aspergillus flavus. Antimicrob. Agents Chemother. 2015, 59, 6615–6619. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Arnautova, Y.A.; Abagyan, R.A.; Totrov, M. Development of a new physics-based internal coordinate mechanics force field and its application to protein loop modeling. Proteins 2011, 79, 477–498. [Google Scholar] [CrossRef] [PubMed]
- Abagyan, R.; Totrov, M. Biased probability monte carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 1994, 235, 983–1002. [Google Scholar] [CrossRef] [PubMed]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Bordner, A.J.; Abagyan, R.A. large scale prediction of protein geometry and stability changes for arbitrary single point mutations. Proteins 2004, 57, 400–413. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; Mackerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain X1 and X2 dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with namd. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds DNA samples of wild-type and mutant strains of L. major are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nucleotide/Codon Position * | Substitution Type | Nucleotide and Amino Acid Changes ** | Nucleotide/Codon Position * | Substitution Type | Nucleotide and Amino Acid Changes ** |
|---|---|---|---|---|---|
| 13/5 | Nonsynonymous | T/C; Phe/Leu | 915/305 | Synonymous | T/C |
| 112/38 | Nonsynonymous | G/A; Ala/Thr | 924/308 | Synonymous | T/C |
| 116/39 | Nonsynonymous | T/C; Met/Thr | 927/309 | Synonymous | A/G |
| 161/54 | Nonsynonymous | A/G; Asp/Gly | 1020/340 | Synonymous | C/T |
| 226/76 | Nonsynonymous | G/A; Val/Ile | 1053/351 | Synonymous | T/C |
| 312/104 | Synonymous | C/T | 1296/432 *** | Synonymous | C/G |
| 729/243 | Synonymous | A/C | 1323/441 | Synonymous | C/T |
| 759/253 | Synonymous | A/G | 1353/451 | Synonymous | A/G |
| 777/259 | Synonymous | T/C | 1395/465 *** | Synonymous | C/G |
| 787/263 | Nonsynonymous | A/G; Ser/Gly | 1421/474 | Nonsynonymous | G/A; Arg/Lys |
| 882/294 | Synonymous | G/C |
| Amino Acid Position | Amino Acid Changes | ΔΔG (kcal/mol) |
|---|---|---|
| 5 * | Phe/Leu | - |
| 38 | Ala/Thr | 0.20 |
| 39 | Met/Thr | 0.50 |
| 54 | Asp/Gly | 0.53 |
| 76 | Val/Ile | 0.44 |
| 263 | Ser/Gly | −1.02 |
| 474 | Arg/Lys | 1.17 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keighobadi, M.; Emami, S.; Lagzian, M.; Fakhar, M.; Rafiei, A.; Valadan, R. Molecular Modeling and Structural Stability of Wild-Type and Mutant CYP51 from Leishmania major: In Vitro and In Silico Analysis of a Laboratory Strain. Molecules 2018, 23, 696. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23030696
Keighobadi M, Emami S, Lagzian M, Fakhar M, Rafiei A, Valadan R. Molecular Modeling and Structural Stability of Wild-Type and Mutant CYP51 from Leishmania major: In Vitro and In Silico Analysis of a Laboratory Strain. Molecules. 2018; 23(3):696. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23030696
Chicago/Turabian StyleKeighobadi, Masoud, Saeed Emami, Milad Lagzian, Mahdi Fakhar, Alireza Rafiei, and Reza Valadan. 2018. "Molecular Modeling and Structural Stability of Wild-Type and Mutant CYP51 from Leishmania major: In Vitro and In Silico Analysis of a Laboratory Strain" Molecules 23, no. 3: 696. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23030696