In Vitro Anti-Toxoplasma gondii and Antimicrobial Activity of Amides Derived from Cinnamic Acid

Abstract
:1. Introduction
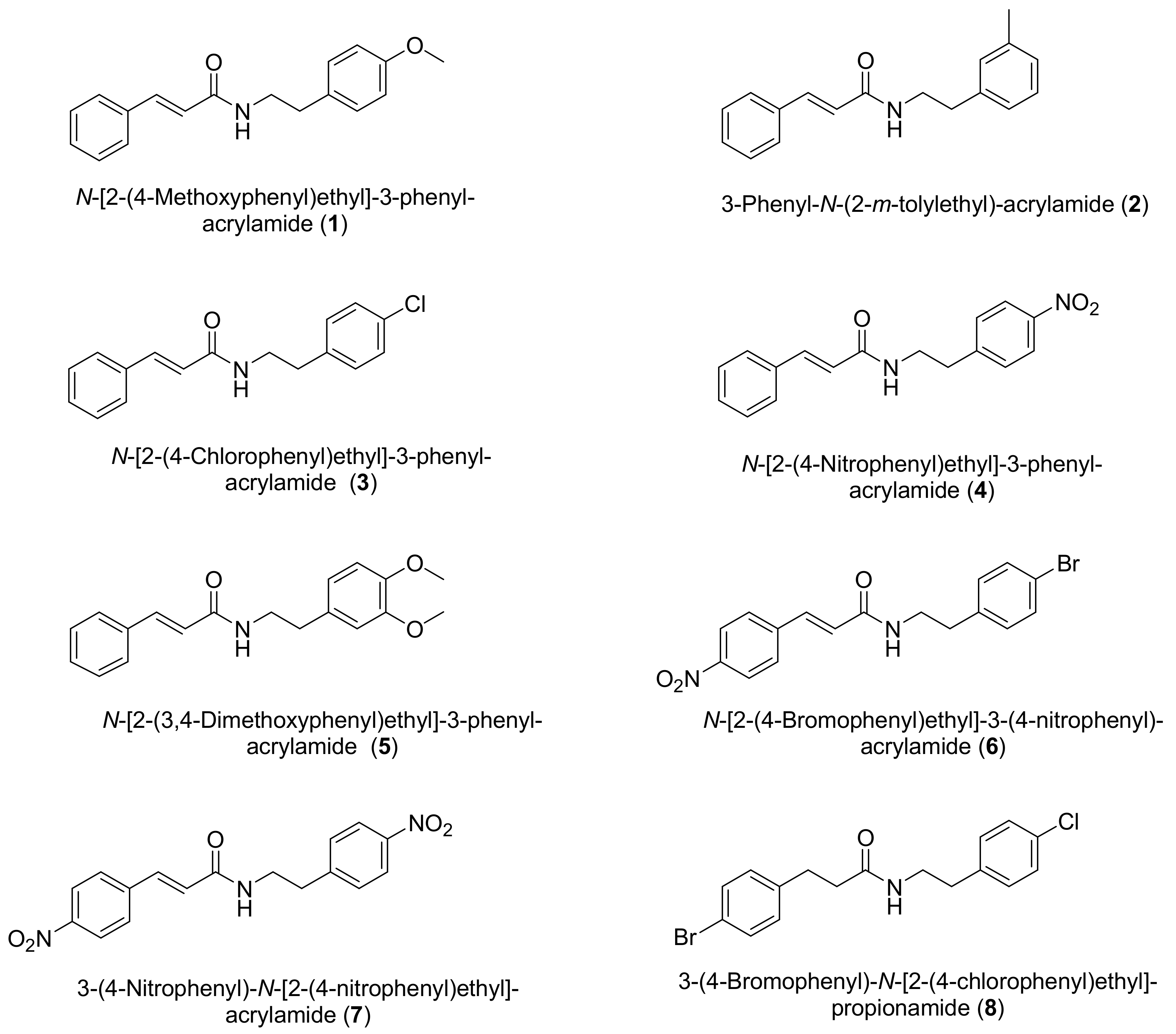
2. Results and Discussion
2.1. Chemistry
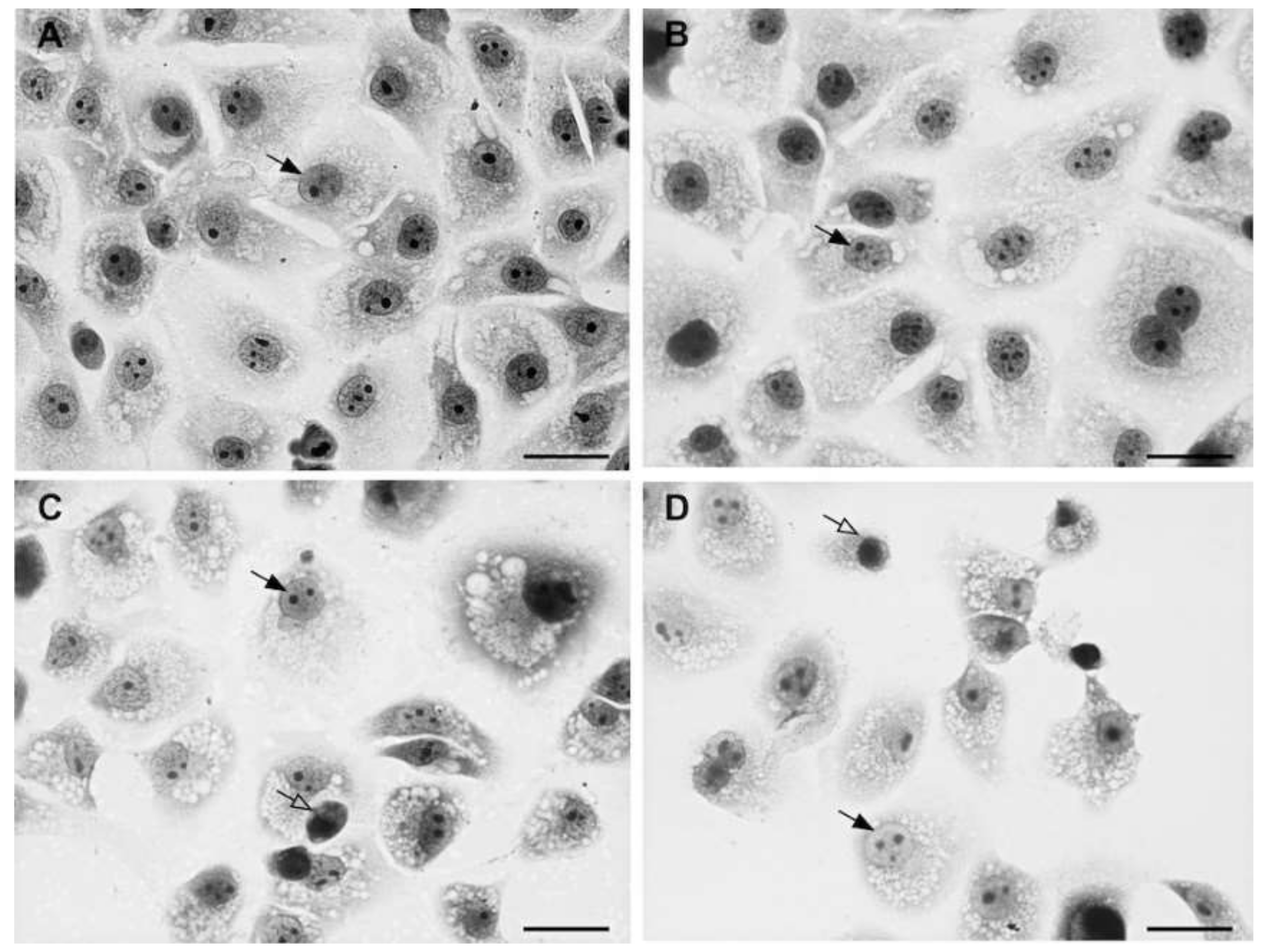
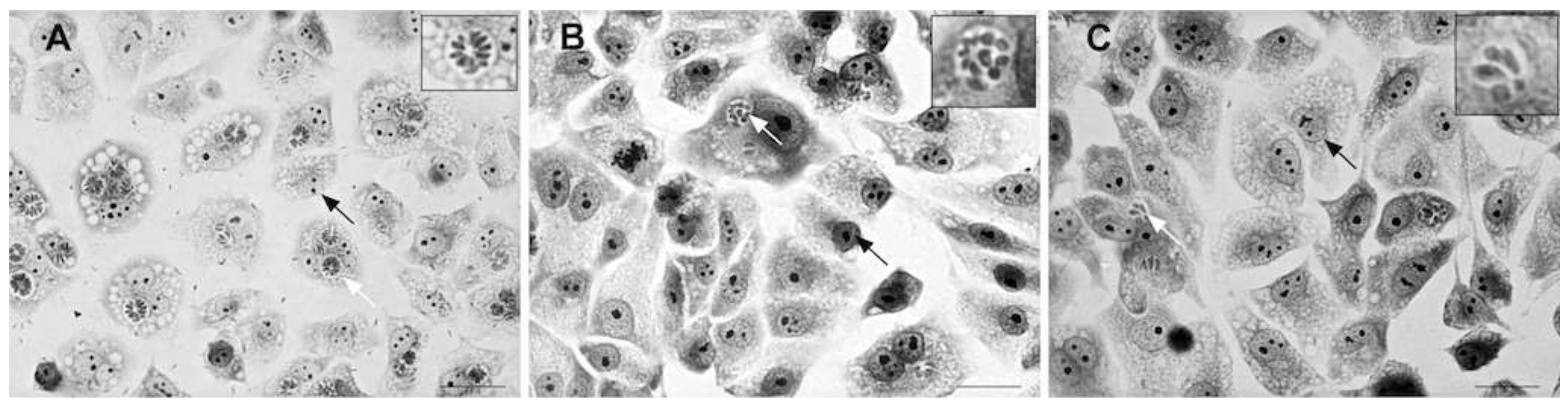
2.2. Assay for Anti-Toxoplasma Gondii Activity
2.3. Assay for Antimicrobial Activity
3. Materials and Methods
3.1. General Information
3.2. General Synthetic Procedure
3.2.1. General Synthetic Procedure for Compounds 1–5 and 8
3.2.2. Synthetic Procedure for Compounds 6 and 7
3.3. Anti-Toxoplasma gondii Activity
3.4. Antimicrobial Activity
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pittman, B.S. Cinnamon: It’s Not Just For Making Cinnamon Rolls. Ethnobot. Leafl. 2010, 2010, 11. [Google Scholar]
- Guzman, J.D. Natural Cinnamic Acids, Synthetic Derivatives and Hybrids with Antimicrobial Activity. Molecules 2014, 19, 19292–19349. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, L.H.; Maillet, J.; LeBlanc, L.M.; Jean-François, J.; Touaibia, M.; Flamand, N.; Surette, M.E. Caffeic acid phenethyl ester and its amide analogue are potent inhibitors of leukotriene biosynthesis in human polymorphonuclear leukocytes. PLoS ONE 2012, 7, 31833. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.H.; Hwang, T.L.; Thang, T.; Leu, Y.L.; Kuo, P.C.; Nguyet, B.M.; Dai, D.N.; Wu, T.S. Isolation and Synthesis of Melodamide A, a New Anti-inflammatory Phenolic Amide from the Leaves of Melodorum fruticosum. Planta Med. 2013, 79, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.-H.; Li, N.-G.; Shi, Q.-P.; Tang, H.; Tang, Y.-P.; Li, W.; Yin, L.; Yang, J.-P.; Duan, J.-A. Synthesis and structure-activity relationship analysis of caffeic acid amides as selective matrix metalloproteinase inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 1206–1211. [Google Scholar] [CrossRef] [PubMed]
- Ferraz, R.; Pinheiro, M.; Gomes, A.; Teixeira, C.; Prudêncio, C.; Reis, S.; Gomes, P. Effects of novel triple-stage antimalarial ionic liquids on lipid membrane models. Bioorg. Med. Chem. Lett. 2017, 27, 4190–4193. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, J.; Mitsch, A.; Wissner, P.; Jomaa, H.; Schlitzer, M. Structure-activity relationships of novel anti-malarial agents. Part 2: Cinnamic acid derivatives. Bioorg. Med. Chem. Lett. 2001, 11, 423–424. [Google Scholar] [CrossRef]
- Carvalho, S.A.; Feitosa, L.O.; Soares, M.; Costa, T.E.M.M.; Henriques, M.G.; Salomão, K.; De Castro, S.L.; Kaiser, M.; Brun, R.; Wardell, J.L.; et al. Design and synthesis of new (E)-cinnamic N-acylhydrazones as potent antitrypanosomal agents. Eur. J. Med. Chem. 2012, 54, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Zang, C.; Tian, S.; Liu, W.; Tan, S.; Cai, Z.; Ni, T.; An, M.; Li, R.; Gao, Y.; et al. Design, synthesis, and evaluation of caffeic acid amides as synergists to sensitize fluconazole-resistant Candida albicans to fluconazole. Bioorg. Med. Chem. Lett. 2015, 25, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Montes, R.C.; Perez, A.L.; Medeiros, C.I.S.; Araújo, M.O.; Lima, E.D.; Scotti, M.T.; Sousa, D.P. Synthesis, Antifungal Evaluation and In Silico Study of N-(4-Halobenzyl)amides. Molecules 2016, 21, 1716. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, B.; Belsare, D.; Pharande, D.; Mourya, V.; Dhake, A. Esters, amides and substituted derivatives of cinnamic acid: Synthesis, antimicrobial activity and QSAR investigations. Eur. J. Med. Chem. 2004, 39, 827–834. [Google Scholar] [CrossRef] [PubMed]
- William, J.; Sullivan, V.J., Jr. Mechanisms of Toxoplasma gondii persistence and latency. NIH Public Access 2013, 36, 717–733. [Google Scholar] [CrossRef]
- Tenter, A.M.; Heckeroth, A.R.; Weiss, L.M. Toxoplasma gondii: From animals to humans. Int. J. Parasitol. 2000, 30, 1217–1258. [Google Scholar] [CrossRef]
- Galván-Ramírez, M.D.L.; Gutiérrez-Maldonado, A.F.; Verduzco-Grijalva, F.; Marcela, J.; Jiménez, D. The role of hormones on Toxoplasma gondii infection: A systematic review. Front. Microbiol. 2014, 5, 503. [Google Scholar] [CrossRef]
- Galvani, A.T. Toxoplasma gondii: Um novo desafio. Núcleo Pesquisas em Avaliação Riscos Ambientais 2014, 1, 1–2. [Google Scholar]
- Wei, H.; Wei, S.; Lindsay, D.S.; Peng, H. A Systematic Review and Meta-Analysis of the Efficacy of Anti-Toxoplasma gondii Medicines in Humans. PLoS ONE 2015, 10, 0138204. [Google Scholar] [CrossRef] [PubMed]
- Tenório, R.P.; Carvalho, C.S.; Pessanha, C.S.; Lima, J.G.; Faria, A.R.; Alves, A.J.; Melo, E.J.T.; Góes, A.J.S. Synthesis of thiosemicarbazone and 4-thiazolidinone derivatives and their in vitro anti-Toxoplasma gondii activity. Bioorg. Med. Chem. Lett. 2005, 15, 2575–2578. [Google Scholar] [CrossRef] [PubMed]
- Liesen, A.P.; Aquino, T.M.; Carvalho, C.S.; Lima, V.T.; Araújo, J.M.; Lima, J.G.; Faria, A.R.; Melo, E.J.T.; Alves, A.J.; Alves, E.W.; et al. Synthesis and evaluation of anti-Toxoplasma gondii and antimicrobial activities of thiosemicarbazides, 4-thiazolidinones and 1,3,4-thiadiazoles. Eur. J. Med. Chem. 2010, 45, 3685–3691. [Google Scholar] [CrossRef] [PubMed]
- Aquino, T.M.; Liesen, A.P.; Silva, R.E.A.; Lima, V.T.; Carvalho, C.S.; Faria, A.R.; Araujo, J.M.; Lima, J.G.; Alves, A.J.; Melo, E.J.T.; et al. Synthesis, anti-Toxoplasma gondii and antimicrobial activities of benzaldehyde 4-phenyl-3-thiosemicarbazones 5-thiazolidineacetic acids. Bioorg. Med. Chem. 2008, 16, 446–456. [Google Scholar] [CrossRef] [PubMed]
- De Almeida Carlos, L.; da Silva Amaral, K.A.; Curcino Vieira, I.J.; Mathias, L.; Braz-Filho, R.; Silva Samarão, S.; Vieira-da-Motta, O. Rauvolfia Grandiflora (Apocynaceae) Extract Interferes with Staphylococcal Density, Enterotoxin Production and Antimicrobial Activity. Braz. J. Microbiol. 2010, 41, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, J.H. Manual of Clinical Microbiology; Murray, P.R., Baro, E.J., Pfaller, M.A., Tonover, F.C., Eds.; ASM Press: Washington, DC, USA, 1995. [Google Scholar]
- Georgiev, L.; Chochkova, M.; Ivanova, G.; Najdenski, H.; Ninova, M.; Milkova, T. Radical scavenging and antimicrobial activities of cinnamoyl amides of biogenic monoamines. Riv. Ital. Sost. Grasse 2012, 89, 91–102. [Google Scholar]
- CLSI. Performance Standards for Antimicrobial Disk Susceptibility Tests; Approved Standard, 12th ed.; CLSI: Wayne, PA, USA, 2015; Volume 35, ISBN 1562387812. [Google Scholar]
- Sheikh, M.C.; Takagi, S.; Yoshimura, T.; Morita, H. Mechanistic studies of DCC/HOBt-mediated reaction of 3-phenylpropionic acid with benzyl alcohol and studies on the reactivities of “active ester” and the related derivatives with nucleophiles. Tetrahedron 2010, 66, 7272–7278. [Google Scholar] [CrossRef]
- Vieira-Da-Motta, O.; Folly, M.M.; Sakyiama, C.C.H. Detection of different Staphylococcus aureus strains in bovine milk from subclinical mastitis using PCR and routine techniques. Braz. J. Microbiol. 2001, 32, 27–31. [Google Scholar] [CrossRef]
- Silva, F.A.S.; Azevedo, C.A.V. The Assistat Software Version 7.7 and its use in the analysis of experimental data. Afr. J. Agric. Res. 2016, 11, 3733–3740. [Google Scholar]
Sample Availability: Samples of all the compounds are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
| Cytotoxicity (Uninfected Culture) a | |||||
|---|---|---|---|---|---|
| Concentration (mM) | |||||
| Drug | Untreated | 0.17 | 0.35 | 1.69 | 2.92 |
| Compound 1 | 226 ± 09 | - | 229 ± 13 | 112 ± 14 | 0 |
| Compound 3 | 196 ± 12 | - | 194 ± 11 | - | 0 |
| Compound 4 | 196 ± 12 | 200 ± 21 | - | 0 | 0 |
| Compound 5 | 226 ± 09 | - | - | 217 ± 30 | 71 ± 10 |
| Compound 6 | 206 ± 10 | - | - | - | 209 ± 12 |
| Compound 7 | 206 ± 10 | - | - | - | 209 ± 17 |
| Cytotoxicity (Infected Culture) a | |||||
|---|---|---|---|---|---|
| Drugs | Concentration (mM) | Uninfected Cells | Infected Cells | Total of Cells | Parasites |
| Control | 0 | 83 ± 8 | 28 ± 4 | 111 ± 12 | 179 ± 29 |
| Compound 1 | 0.18 | 129 ± 6 | 24 ± 4 | 153 ± 10 | 166 ± 15 |
| Control | 0 | 108 ± 19 | 51 ± 6 | 159 ± 25 | 254 ± 38 |
| Compound 3 | 0.077 | 147 ± 3 | 23 ± 5 | 170 ± 8 | 127 ± 32 |
| Control | 0 | 108 ± 19 | 51 ± 6 | 159 ± 25 | 254 ± 38 |
| Compound 4 | 0.038 | 149 ± 24 | 35 ± 10 | 184 ± 34 | 134 ± 32 |
| Control | 0 | 83 ± 8 | 28 ± 4 | 111 ± 12 | 179 ± 29 |
| Compound 5 | 0.16 | 114 ± 3 | 12 ± 1 | 126 ± 4 | 82 ± 19 |
| Control | 0 | 79 ± 7 | 37 ± 3 | 116 ± 10 | 174 ± 16 |
| Compound 6 | 1.33 | 89 ± 12 | 31 ± 5 | 120 ± 17 | 83 ± 23 |
| Control | 0 | 79 ± 7 | 37 ± 3 | 116 ± 10 | 174 ± 16 |
| Compound 7 | 1.46 | 104 ± 9 | 28 ± 3 | 132 ± 12 | 98 ± 12 |
| Compound | Minimum Inhibitory Concentration (MIC) in µg mL−1 a | |||||
|---|---|---|---|---|---|---|
| ATCC 15442 | LSA 88 | ATCC 33591 | ATCC 25923 | ATCC 25922 | ATCC 12228 | |
| 1 | >250 | − | − | − | − | − |
| 2 | >250 | >250 | >250 | 250 | − | >250 |
| 3 | >250 | − | − | − | − | − |
| 4 | >250 | − | − | − | − | − |
| 5 | >250 | 250 | − | − | − | − |
| 6 | − | − | − | − | >250 | >250 |
| 7 | − | − | − | − | >250 | >250 |
| 8 | − | − | − | − | − | − |
| Strains | ||||
|---|---|---|---|---|
| ATCC 25923 | LSA 88 | |||
| Antibiotic | Control | 2 | Control | 5 |
| VAN | 16.74Bg | 23.98Ad | 17.50Ae | 17.45Ai |
| AMP | 27.05Ba | 36.99Aa | 33.17Aab | 34.64Ab |
| SUT | 25.00Bbc | 37.00Aa | 28.05Bc | 29.68Ade |
| CLI | 21.40Be | 34.42Aab | 26.76Ac | 27.17Ag |
| CFX | 23.70Bcd | 34.86Aab | 28.09Bc | 28.90Aef |
| ERI | 20.87Be | 32.42Aabc | 25.73Bc | 27.99Afg |
| AMO | 24.95Bbc | 32.88Aab | 33.98Aab | 31.36Bc |
| PEN | 25.69Bab | 33.40Aab | 36.11Aa | 36.21Aa |
| GEN | 21.37Be | 30.24Abc | 26.72Ac | 26.99Ag |
| AMC | 26.81Ba | 35.52Aab | 35.06Aab | 35.62Aab |
| OXA | 19.02Bf | 26.61Acd | 21.31Ad | 21.32Ah |
| TET | 22.40Bde | 33.33Aab | 32.49Ab | 30.91Acd |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silveira, G.R.; Campelo, K.A.; Lima, G.R.S.; Carvalho, L.P.; Samarão, S.S.; Vieira-da-Motta, O.; Mathias, L.; Matos, C.R.R.; Vieira, I.J.C.; Melo, E.J.T.d.; et al. In Vitro Anti-Toxoplasma gondii and Antimicrobial Activity of Amides Derived from Cinnamic Acid. Molecules 2018, 23, 774. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23040774
Silveira GR, Campelo KA, Lima GRS, Carvalho LP, Samarão SS, Vieira-da-Motta O, Mathias L, Matos CRR, Vieira IJC, Melo EJTd, et al. In Vitro Anti-Toxoplasma gondii and Antimicrobial Activity of Amides Derived from Cinnamic Acid. Molecules. 2018; 23(4):774. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23040774
Chicago/Turabian StyleSilveira, Graziela Rangel, Karoline Azerêdo Campelo, Gleice Rangel Silveira Lima, Lais Pessanha Carvalho, Solange Silva Samarão, Olney Vieira-da-Motta, Leda Mathias, Carlos Roberto Ribeiro Matos, Ivo José Curcino Vieira, Edesio José Tenório de Melo, and et al. 2018. "In Vitro Anti-Toxoplasma gondii and Antimicrobial Activity of Amides Derived from Cinnamic Acid" Molecules 23, no. 4: 774. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23040774