ω-Methylsulfanylalkyl Glucosinolates: A General Synthetic Pathway



1
Institut de Chimie Organique et Analytique (ICOA), Université d’Orléans et CNRS, UMR 7311, BP 6759, F-45067 Orléans CEDEX 2, France
2
Laboratoire IMRCP, UMR CNRS 5623, Université P. Sabatier Toulouse III, 118 route de Narbonne, 31062 Toulouse CEDEX 9, France
3
Department of Chemistry and Biochemistry, Biomolecular Sciences Programme, Laurentian University, Sudbury, ON P3E 2C6, Canada
*
Author to whom correspondence should be addressed.
Molecules 2018, 23(4), 786; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23040786
Submission received: 9 March 2018
/
Revised: 20 March 2018
/
Accepted: 21 March 2018
/
Published: 28 March 2018
(This article belongs to the Special Issue Focusing on Sulfur in Medicinal Chemistry)
Abstract
:A general pathway was devised to synthesize ω-methylsulfanylalkyl glucosinolates, which represent an important class of structurally homogeneous plant secondary metabolites. The required thiofunctionalized hydroximoyl chlorides were obtained from the corresponding α,ω-nitroalkyl methylsulfide precursors, involving as the key-step, a nitronate chlorination strategy. A coupling reaction with 1-thio-beta-d-glucopyranose, followed by O-sulfation of the intermediate thiohydroximate and final deprotection of the sugar moiety afforded the target compounds.
1. Introduction
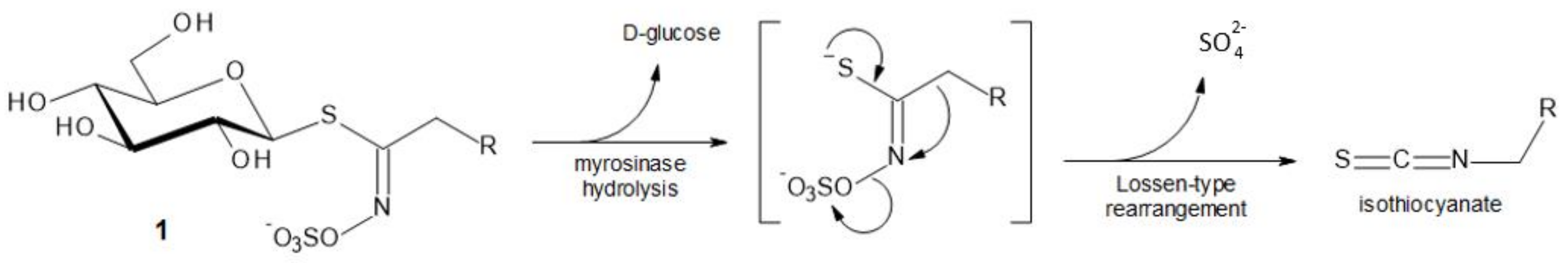
Glucosinolates (GLs) 1 stand as molecular tags of the plant order Brassicales [1,2]. Those strikingly bio-relevant thiosaccharidic secondary metabolites are found without exception in all 17 families of this order. All of the known GLs (ca. 130 molecules to date) display a remarkable structural homogeneity based on a hydrophilic beta-d-glucopyrano unit, which bears a O-sulfated anomeric (Z)-thiohydroximate function connected to a rather hydrophobic side chain R. Depending on plant species, the constitution of R is the sole structural variant, in which diversified aliphatic, arylaliphatic, or indolic arrangements can be found. Associated in plants with an atypical glycohydrolase, myrosinase (E.C. 3.2.1.147), GLs can undergo hydrolytic cleavage of the anomeric C–S bond. After d-glucose release, the detached aglycon undergoes a Lossen-type rearrangement to produce biologically active isothiocyanates (Figure 1) [1,2,3]. GLs can thus be regarded as isothiocyanate bio-precursors.
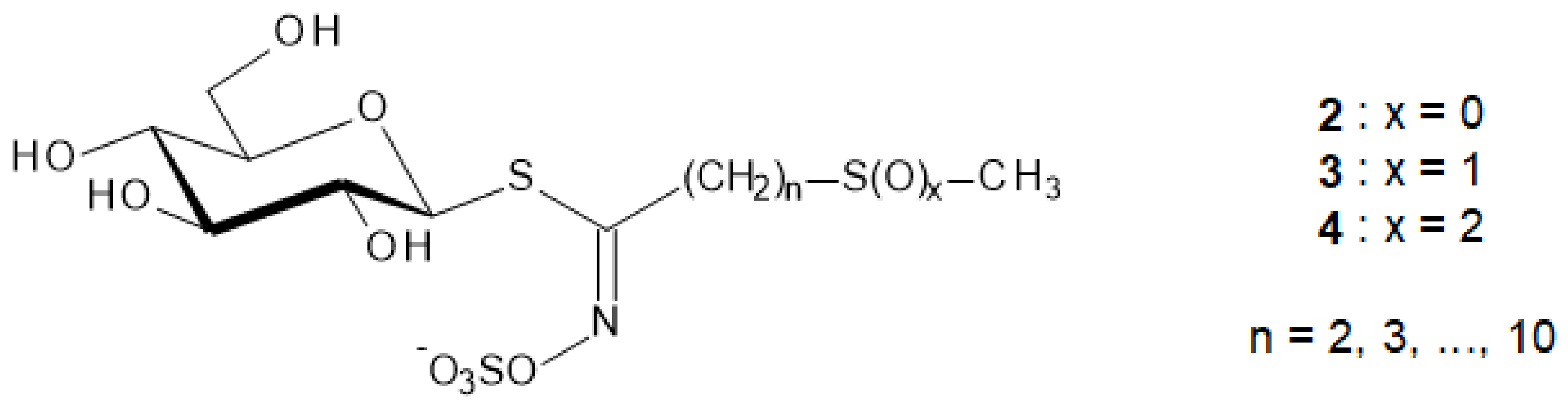
It is remarkable that more than one-third of the actually registered GL structures display an additional thio-function—namely sulfide (2), sulfoxide (3) or sulfone (4)—in ω-position of their aglycon chain, as represented in Figure 2 [4,5].
Mainly found in the Brassicaceae and Capparaceae families, such thiofunctionalized GLs have been investigated over the last decade for their redox properties in various in vitro free radical-scavenging assays, with a view to assessing their antioxidant potential [6,7,8].
Notwithstanding the large number of known GL-containing plant species, accessing these compounds from appropriate vegetable sources via extractive processes is generally not straightforward [9,10]. “Specialist” plants, which can be considered for convenient isolation of one specific GL in reasonable amount, are in very limited number [11,12,13]. This is particularly the case for thiofunctionalized GLs, for which the synthetic approach should be privileged to make available attractive substrates for biological studies [14,15]. Nevertheless, since our pioneering work [16], only a small number of syntheses have focused on GLs bearing an external thio-function in the aglycon part. Starting from a sulfoxide-oxime precursor, isotopically labelled 4-methylsulfinylbutyl GL (glucoraphanin) was synthesized by Botting et al. for analysis and metabolic studies [17,18]. 4-Methylsulfanylbut-3-enyl GL (glucoraphasatin) and 13C-labelled 4-methylsulfanylbutyl GL (glucoerucin) were also synthesized [19,20].
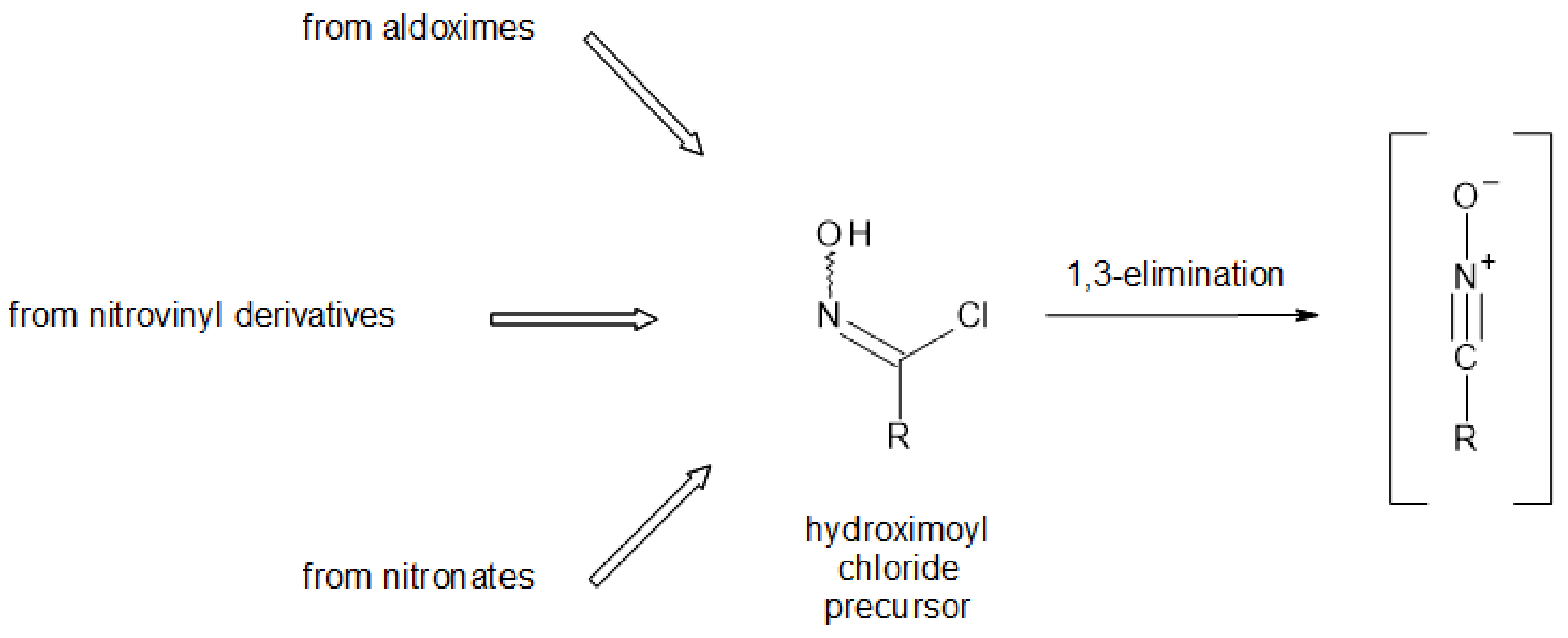
For most synthetic methods that have been developed for GLs over the years, the key step was the stereospecific formation of a thiohydroximate function, usually resulting from the 1,3-addition of a glucosyl mercaptan onto a transient nitrile oxide [15]. As they are generally unstable molecular species, nitrile oxides have to be generated in situ through base-induced conversion of hydroximoyl chlorides, (Figure 3), which in turn currently result from electrophilic chlorination of aldoximes [21].
In this particular case, however, the presence on the carbon chain of a methylsulfanyl moiety a priori precludes the use of reagents like chlorine or N-chlorosuccinimide which are prone to oxidize a sulfide function [22].
2. Results and Discussion
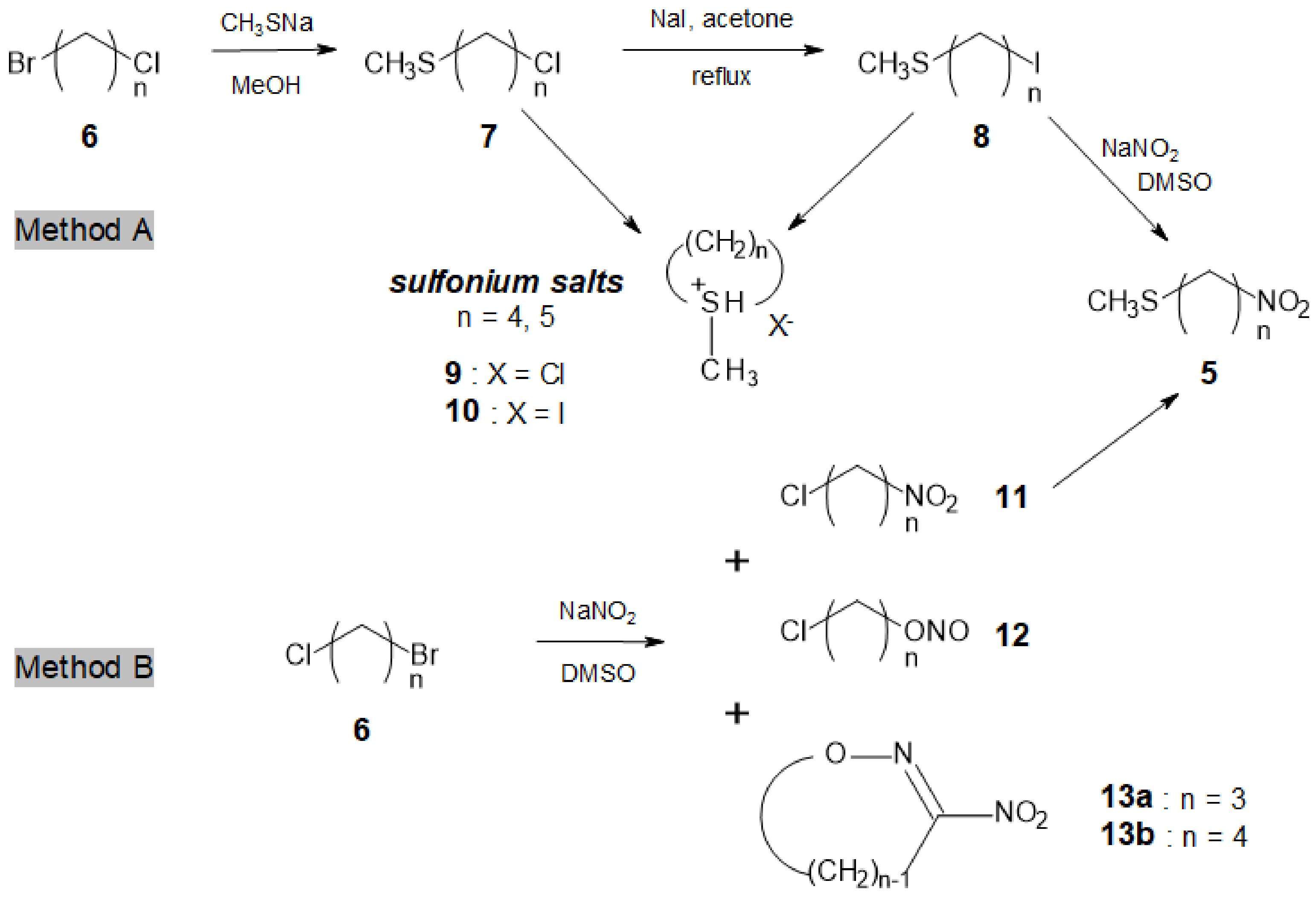
The preparation of a range of linear ω-nitroalkyl methyl sulfides 5 of variable length (n = 3–12) from commercially available ω-bromochloroalkanes 6 was expected to be straightforward. However, in the case of small sized polymethylene chain (n = 3–6), undesirable side-reactions took place. Hence, distinct strategies had to be devised to provide the ω-nitroalkyl methyl sulfide precursors 5.
Method A (Scheme 2) is suggested for dihaloalkanes 6a (n = 3) or 6d (n = 6). In the cases of 6b and 6c (n = 4 and 5), the reaction sequence was almost disrupted because of the formation of cyclic sulfonium salts 9 and 10 [23,24].
Compounds 7 and 8 have a tendency to undergo cyclization during their formation, on standing or/and during their purification by distillation. Sulfonium salts formation rate is carbon chain length-, temperature-, and halogen type-dependent. Thus, rapid cyclization occurred in the case of ω-methylsulfanyl-iodobutane 8b and -pentane 8c. Slightly slower, but still significant, cyclization took place with the corresponding chloro-analogues, ω-methylsulfanyl-chlorobutane 7b and -pentane 7c while in the case of 6-methylsulfanyl-1-chlorohexane 7d, the cyclization was minimized (Table 1). Halogen-exchange reaction of compounds 7 with iodide ion was very slow, even at an elevated temperature, thus explaining the result reported in Table 1 for n = 4 or 5. In cases where n = 3 or 6, chloro- and iodo-compounds were more stable—enough at least for completing the method A reaction sequence and afford the desired α,ω-nitro-sulfides 5.
α,ω-Bis-methylsulfanylalkanes can be simultaneously formed [25] during the preparation of compounds 7. Their occurrence can be minimized by controlling the temperature, the reaction time, and the molar ratio of CH3SNa. Nevertheless, even if they are not separated from the reactive intermediates, the presence of such side-products is not likely to disturb the subsequent reaction steps.
According to method B (Scheme 2), reaction of NaNO2 with ω-bromo-chloroalkanes 6 afforded the ω-chloro-nitroalkanes 11 together with the isomeric nitrites 12 [27,28]. The reaction needed to be time-controlled for each dihaloalkane 6 in order to optimize the nitro-compound to nitrite ratio (Table 2), and at the same time to prevent destruction of the nitro-derivatives 11 due to the simultaneous presence of the nitrite ion and an alkyl nitrite [29].
In the case of 6a (n = 3), notably, the formation in ca. 40% yield of the known 3-nitro-2-isoxazoline 13a occurred, because of the interaction of the nitro-derivative 11a with the nitrite 12a [30]. In fact, a prolonged reaction of 1,3-bromochloropropane with NaNO2 in the presence of 1-propyl nitrite has been patented [31] as a preparative method for 13a [32]. In contrast in the case of 5b (n = 4), no side-formation of the corresponding oxazine 13b was observed.
The ω-chloro-nitroalkanes 11a–d were isolated by flash chromatography and were characterized by GC-MS and NMR analysis. Subsequent conversion of 11a–d into the expected ω-methylsulfanyl-nitroalkanes 5a–d was carried out by reaction with freshly prepared sodium methanethiolate, obtained by passing CH3SH gas in a 1 M methanolic solution of sodium methoxide. Indeed, commercial powdered CH3SNa inevitably gave poor conversion yields and produced a number of unexpected side products (acids, esters, lactones, thiosulfonates...), e.g., a high yield of γ-butyrolactone in the case of 11b (n = 4).
After their formation, ω-methylsulfanyl-nitroalkanes 5 had to be recovered from the basic reaction medium by the addition of a protic acid. However the regeneration of aliphatic nitro-compounds from their salts is not a routine matter [33]. Depending on the experimental conditions, acidic treatment of primary nitronates often results in the production of aldehydes and/or hydroxamic acids via Nef and Meyer reactions [34]. Optimization of nitroalkane recovery is dependent on resorting to mild acidification protocols, such as hydroxylamine hydrochloride in 10% aqueous acetic acid at low temperature. Applying such conditions in the knotty case of 11b (n = 4) allowed to satisfactorily recover (1-methylsulfanyl)-4-nitrobutane 5b, without the formation of γ-butyrolactone.
Access to longer chain ω-methylsulfanyl-nitroalkanes 5e-g (n = 8, 10, 12) was more straightforward (Method C, Scheme 3). Commercially available α,ω-dibromoalkanes 14 were reacted with CH3SNa to afford ω-methylsulfanyl bromoalkanes 15e-g (#60% yield), together with minor amounts of the corresponding α,ω-bis-methylsulfanylalkanes [25]. Finally, compounds 15e-g were converted, as described above, into the desired ω-methylsulfanyl nitroalkanes 5e-g in 65–70% yields.
Following a procedure previously established by Kjaer [35,36], all of the nitro-compounds 5 were converted into their moisture-sensitive sodium salts, which were isolated by brief filtration under dry atmosphere, then reacted in chloroform with SOCl2 (in our hands, the oxalyl chloride variant [37] did not bring substantial improvement) at low temperature (−60 °C) to afford the hydroximoyl chlorides 16. Those were subsequently used without further purification in the next steps of the synthesis (Scheme 4).
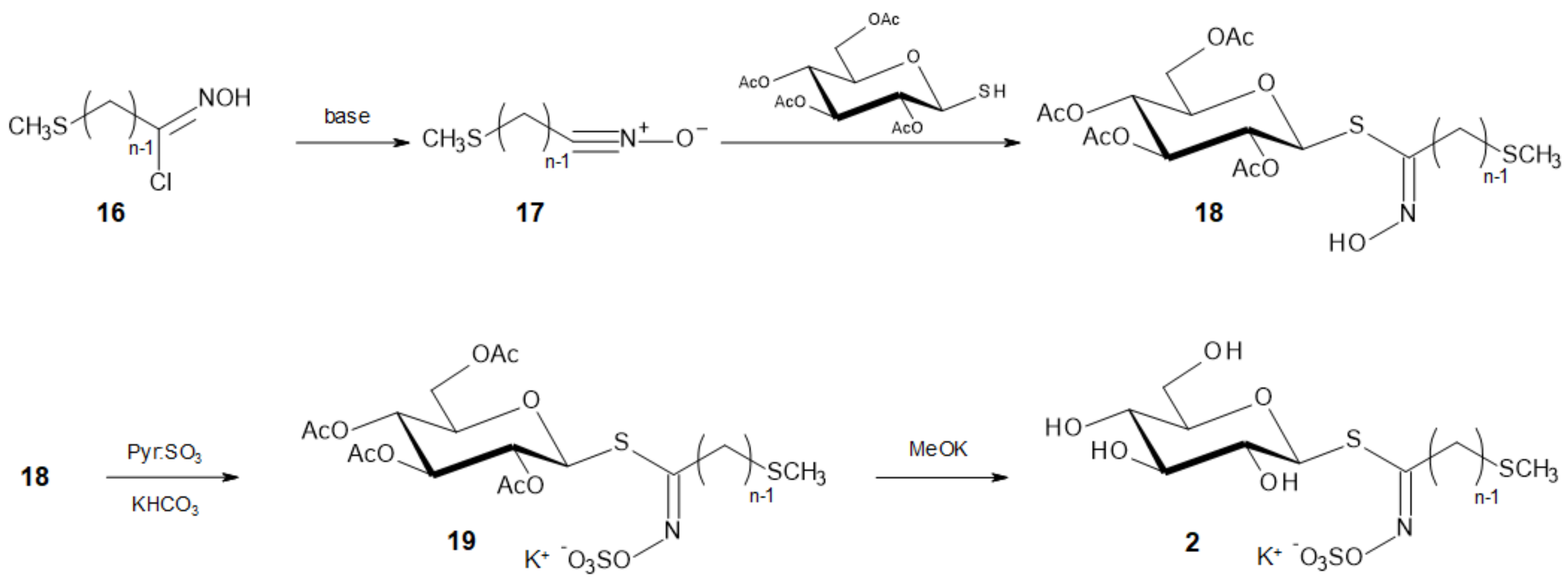
The intermediate nitrile oxides 17 were generated in situ by base-catalyzed (NEt3) 1,3-elimination on hydroximoyl chlorides 16 to react [15,38] with 2,3,4,6-tetra-O-acetyl-1-thio-β-d-glucopyranose [39] to afford the corresponding ω-methylthioalkyl (Z)-thiohydroximates 18.
Final conversion of the thiohydroximates 18a–g into the target GLs 2a–g was performed according to a previously described protocol [15]. O-Sulfation of the hydroximino moiety using sulfur trioxide pyridine complex, followed by quenching with aqueous potassium hydrogenocarbonate afforded the per-O-acetylated GLs 19a–g, the carbohydrate part of which was readily deprotected under basic conditions to afford the corresponding GLs.
3. Experimental Section
3.1. General Information
α,ω-Bromochloroalkanes 6a–d Br-(CH2)n-Cl (n = 3–6) and α,ω-dibromoalkanes 14 Br-(CH2)n-Br (n = 8, 10, 12) were obtained from commercial sources (Sigma-Aldrich, Saint-Quentin-Fallavier, France) and were used without further purification. Anhydrous reactions were performed under argon atmosphere in pre-dried flasks, using distilled anhydrous solvents. Unless otherwise stated, all of the chemicals obtained from Sigma-Aldrich were used without further purification. TLC on precoated aluminum-back plates (Merck Kieselgel 60F254, E. Merck, Darmstadt, Germany) were generally visualized by UV light (254 nm) and by charring after exposure to a 5% H2SO4 solution in ethanol. Flash column chromatography was carried out using silica gel (E. Merck, 36–63 mesh, unless otherwise indicated). Melting points (°C) were obtained using a Büchi 510 apparatus (Büchi Sarl, Rungis, France) and are uncorrected. Optical rotations were measured at 20 °C with a Perkin Elmer 341 polarimeter (Perkin Elmer France, Villebon s/Yvette, France) with a path length of 1 dm. 1H and 13C-NMR spectra were recorded on 250 MHz Bruker Avance DPX250 or 400 MHz Bruker Avance2 spectrometers (Bruker France, Wissembourg, France). Chemical shifts are expressed in parts per million (ppm) downfield from TMS internal standard and coupling constants are given in Hz. NMR peak assignments were established using NOESY, COSY, HSQC, and HMBC methods for all the reported compounds.
GC monitoring was carried out using a Varian 3700 gas chromatograph (Agilent Technologies, Les Ulis, France) equipped with a split/splitess injector at 240 °C, a DB-5 (Agilent) capillary column (30 m × 0.25 mm, 0.25 µm film thickness—carrier gas nitrogen 1 mL·min−1) and a flame ionization detector at 280 °C. Depending on the solute analyzed, two temperature programs were used: (1) initial temperature 100 °C, held for 2 min, and then ramped to 220 °C with a 10 °C·min−1 rate maintained for 10 min. (2) initial temperature 100 °C, held for 2 min, then ramped to 250 °C with a 15 °C·min−1 rate maintained for 15 min.
GC-MS analyses were performed using a HP-5890 (Agilent) chromatograph hyphenated to a HP-5970 mass spectrometer (Agilent quadrupole). Spectra were obtained in EI mode at 70 eV. The gas chromatograph was equipped with a HP-1 capillary column (Agilent, 100% dimethylsiloxane, 30 m × 0. 25 mm, 0.33 µm film thickness). The injector temperature was held at 200 °C and a 1 µL sample was injected with a 1/100 split ratio under a constant helium flow (1 mL·min−1). The oven temperature was initially held at 60 °C for 3 min, and then ramped with a 3 °C·min−1 rate to 80 °C. This temperature was held for 1 min, then ramped with a 3 °C·min−1 rate to 150 °C. A 5 °C·min−1 rate was used to bring the oven temperature to 200 °C. The final temperature of 230 °C was reached with a 10 °C·min−1 rate and maintained for 20 min.
Mass spectra (ion spray; IS) were recorded on a Perkin–Elmer Sciex API-300 spectrometer (Sciex Sarl, Les Ulis, France).
High resolution mass spectra (HRMS) were recorded with a MicrOTOF-QII spectrometer (Bruker France S.A.S., Champs-sur-Marne, France) in the electrospray ionisation (ESI) mode.
3.2. Synthesis
3.2.1. General Procedure for the Synthesis of Small-Sized Chloroalkyl Methyl Sulfides 7a–d
A 2.6 M methanolic solution of MeSNa (23 mL, 1.2 equiv.) was added dropwise to a stirred solution of ω-bromochloroalkane 6 (50 mmol) in dry methanol (80 mL) at room temperature (r.t.). Stirring was continued until complete consumption of the dihalide (2–3 h, GC monitoring). The mixture was concentrated in vacuo and the resulting white slurry was taken in cold water and extracted with dichloromethane. After drying the organic phase over MgSO4, filtration and concentration in vacuo, the oily residue was purified by flash chromatography using silica gel (230–400 mesh) with hexane as eluent to afford 7a–d as light yellow oils.
3.2.2. General Procedure for the Synthesis of Long-Sized Bromoalkyl Methylsulfides 15e–g
Similarly, the α,ω-dibromoalkane 14 (50 mmol) dissolved in a stirred methanol (200 mL)-dichloromethane (50 mL) mixture was treated dropwise (over 30 min) by a 2.6 M methanolic solution of MeSNa (20 mL, 1.04 equiv.), while cooling at r.t. After complete consumption of the dihalide (1–2 h, TLC monitoring), the mixture was concentrated in vacuo and the resulting white slurry was taken in cold water and extracted with dichloromethane. After drying the organic phase over MgSO4, filtration and concentration in vacuo, the oily residue was purified by flash chromatography using silica gel (230–400 mesh) with hexane as eluent to afford 15e–g [25].
8-Bromo-1-methylsulfanyloctane [64053-04-9] 15e [43]. Light yellow viscous oil (57% yield). 1H-NMR (CDCl3) δ 1.25–1.44 (m, 8H, CH2), 1.57 (qt, 2H, H-2), 1.84 (qt, 2H, H-7), 2.08 (s, 3H, MeS), 2.48 (t, 2H, Jvic = 7.3, H-1), 3.39 (t, 2H, Jvic = 6.8, H-8). 13C-NMR δ 15.3 (MeS), 27.9, 28.5, 28.9 (C-2-C-6), 32.6 (C-7), 33.8 (C-8), 34.1 (C-1). ESI-MS m/z 240.2 [M + H]+.
10-Bromo-1-methylsulfanyldecane 15f. Light yellow solid (55% yield). 1H-NMR (CDCl3) δ 1.22–1.47 (m, 12H, CH2), 1.58 (qt, 2H, H-2), 1.85 (qt, 2H, H-9), 2.08 (s, 3H, MeS), 2.47 (t, 2H, Jvic = 7.3, H-1), 3.39 (t, 2H, Jvic = 6.8, H-10). 13C-NMR δ 15.3 (MeS), 28.0, 28.6, 29.0, 29.2 (C-2-C-8), 32.7 (C-9), 33.8 (C-10), 34.1 (C-1). ESI-MS m/z 268.3 [M + H]+.
12-Bromo-1-methylsulfanyldodecane 15g. Light yellow solid (59% yield). 1H-NMR (CDCl3) δ 1.19–1.48 (m, 16H, CH2), 1.58 (qt, 2H, H-2), 1.84 (qt, 2H, H-11), 2.08 (s, 3H, MeS), 2.48 (t, 2H, Jvic = 7.3, H-1), 3.39 (t, 2H, Jvic = 6.8, H-12). 13C-NMR δ 15.4 (MeS), 28.0, 28.6, 28.7, 29.0, 29.1, 29.3, 29.4 (C-2-C-10), 32.7 (C-11), 33.9 (C-12), 34.2 (C-1). ESI MS m/z 296.3 [M + H]+.
3.2.3. Procedures for the Synthesis of Nitroalkyl Methylsulfides 5
Method A. Synthesis of 5a (n = 3) and 5d (n = 6)
The chloroalkyl methyl sulfide 7 (42 mmol) was refluxed in dry acetone with dry NaI (13.5 g, 90 mmol) for 2–3 days until the complete conversion of the starting material (GC monitoring). After concentration in vacuo, the resulting slurry was taken in a 0.6 M aqueous sodium thiosulfate solution, and extracted with CH2Cl2. The combined organic phases were dried over MgSO4, filtered and concentrated in vacuo to afford compound 8 as fragrant pale orange oil, which was used as such in the next step.
To a solution of dry sodium nitrite (2.0 g, 29 mmol) in DMSO (80 mL), a solution of 8 (24 mmol) in DMSO (15 mL) was added dropwise, while keeping the temperature at 20–25 °C. After 2–3 h, the reaction was quenched by adding ice-water (300 mL) and repeatedly extracted with diethyl ether. The combined organic phases were dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (petroleum ether–Et2O 4:1) to afford 5a and 5d as colourless oils.
(1-Methylsulfanyl)-3-nitropropane [182258-93-1] 5a. Colourless oil (2.27 g, 40% overall yield). 1H-NMR (CDCl3) δ 2.09 (s, 3H, MeS), 2.27 (qt, 2H, H-2), 2.58 (t, 2H, Jvic = 6.8, H-1), 4.51 (t, 2H, Jvic = 6.8, H-3). 13C-NMR δ 15.1 (MeS), 26.1 (C-2), 30.4 (C-1), 73.7 (C-3). MS IS m/z 136.2 [M + H]+.
(1-Methylsulfanyl)-6-nitrohexane 5d. Colourless oil (3.13 g, 42% overall yield). 1H-NMR (CDCl3) δ 1.32–1.48 (m, 4H, H-3, H-4), 1.54–1.63 (m, 2H, H-2), 1.94–2.04, m, 2H, H-5), 2.06 (s, 3H, MeS), 2.46 (t, 2H, Jvic = 7.3, H-1), 4.36 (t, 2H, Jvic = 7.3, H-6). 13C-NMR δ 15.2 (MeS), 25.6 (C-4), 27.0 (C-5), 27.7 (C-3), 28.4 (C-2), 33.8 (C-1), 75.5 (C-6). MS IS m/z 178.3 [M + H]+.
Method B. Synthesis of 5a–d (n = 3–6) from ω-bromochloroalkanes 6a–d
To a solution of dry sodium nitrite (1.24 g, 18 mmol) in DMSO (100 mL), a solution of ω-bromochloroalkane 6 (15 mmol) in DMSO (20 mL) was added dropwise while keeping the temperature at 20–25 °C. After 2–5 h, the reaction was quenched by adding ice-water (400 mL) and was repeatedly extracted with diethyl ether. The combined organic phases were dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by column chromatography (petroleum ether–Et2O 5:1) to afford ω-chloronitroalkanes 11 as colourless oils.
1-Chloro-3-nitropropane [16694-52-3] 11a. Colourless oil (0.8 g, 42% yield) 1H-NMR (CDCl3) δ 2.45 (qt, 2H, H-2), 3.65 (t, 2H, Jvic = 6.3, H-1), 4.58 (t, 2H, Jvic = 6.3, H-3). 13C-NMR δ 29.6 (C-2), 40.7 (C-1), 72.1 (C-3) [44].
1-Chloro-4-nitrobutane [41168-66-5] 11b. Colourless oil (1.2 g, 60% yield) 1H-NMR (CDCl3) δ 1.83–1.92 (m, 2H, H-2), 2.13–2.23 (m, 2H, H-3), 3.58 (t, 2H, Jvic = 6.1, H-1), 4.43 (t, 2H, Jvic = 6.8, H-4). 13C-NMR δ 24.4 (C-2), 28.8 (C-3), 43.5 (C-1), 74.6 (C-4) [45].
1-Chloro-5-nitropentane [1173694-33-1] 11c. Colourless oil (1.5 g, 66% yield) 1H-NMR (CDCl3) δ 1.51–1.60 (m, 2H, H-3), 1.77–1.87 (m, 2H, H-2), 1.98–2.08 (m, 2H, H-4), 3.54 (t, 2H, Jvic = 6.3, H-1), 4.39 (t, 2H, Jvic = 7.0, H-5). 13C-NMR δ 23.4 (C-3), 26.4 (C-4), 31.5 (C-2), 44.2 (C-1), 75.3 (C-5).
1-Chloro-6-nitrohexane [898543-32-3] 11d. Colourless oil (1.9 g, 76% yield) 1H-NMR (CDCl3) δ 1.38–1.55 (m, 4H, H-3, H-4), 1.73–1.83 (m, 2H, H-2), 1.97–2.07 (m, 2H, H-5), 3.53 (t, 2H, Jvic = 6.6, H-1), 4.38 (t, 2H, Jvic = 7.0, H-6). 13C-NMR δ 25.4 (C-4), 25.9 (C-3), 27.0 (C-5), 31.9 (C-2), 44.6 (C-1), 75.4 (C-6) [46].
A solution of chloronitro derivative 11 (25 mmol) in dry methanol (50 mL) was reacted with a 2.6 M methanolic solution of MeSNa (20 mL, 2.08 equiv.) under the conditions that are reported in Table 3. After concentration in vacuo, the resulting slurry was poured into ice water (200 mL) under vigorous stirring and treated dropwise at 0 °C by a cooled solution of hydroxylamine hydrochloride (4.0 g, 57 mmol) in 20% aqueous acetic acid (17 mL) [28]. The two-phase system obtained was extracted three times with CH2Cl2, and the combined organic phases were dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by column chromatography (petroleum ether–Et2O 4:1) to afford 5a–d as colourless oils (5a and 5d were described above).
(1-Methylsulfanyl)-4-nitrobutane [182258-94-2] 5b. Colourless oil (1.6 g, 43% yield). 1H-NMR (CDCl3) δ 1.63–1.73 (m, 2H, H-2), 2.03–2.16 (m, 5H, MeS, H-3), 2.52 (t, 2H, Jvic = 7.0, H-1), 4.40 (t, 2H, Jvic = 7.0, H-4). 13C-NMR δ 15.1 (MeS), 25.2 (C-2), 25.9 (C-3), 33.0 (C-1), 75.0 (C-4). MS IS m/z 150.2 [M + H]+.
(1-Methylsulfanyl)-5-nitropentane [182258-95-3] 5c. Colourless oil (3.5 g, 86% yield). 1H-NMR (CDCl3) d 1.42–1.52 (m, 2H, H-3), 1.58–1.68 (m, 2H, H-2), 1.96–2.06 (m, 2H, H-4), 2.07 (s, 3H, MeS), 2.48 (t, 2H, Jvic = 7.1, H-1), 4.37 (t, 2H, Jvic = 7.1, H-5). 13C-NMR δ 15.2 (MeS), 25.0 (C-3), 26.7 (C-4), 28.00 (C-2), 33.5 (C-1), 75.3 (C-5). MS IS m/z 164.2 [M + H]+.
Method C. Synthesis of Long-Sized Nitroalkyl Methylsulfides 5e–g (n = 8, 10, 12)
To a solution of dry sodium nitrite (2.0 g, 29 mmol) in DMSO (80 mL), a solution of 15 (24 mmol) in DMSO (15 mL) was added dropwise, while keeping the temperature at 20–25 °C. After 2–3 h, the reaction was quenched by adding ice-water (300 mL) and repeatedly extracted with diethyl ether. The combined organic phases were dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by column chromatography (petroleum ether-Et2O 19:1) to afford 5e–g as colourless oils.
(1-Methylsulfanyl)-8-nitrooctane 5e. Colourless oil (3.2 g, 65% yield). 1H-NMR (CDCl3) δ 1.23–1.42 (m, 8H, H-3-6), 1.57 (qt, 2H, H-2), 1.98 (qt, 2H, H-7), 2.07 (s, 3H, MeS), 2.46 (t, 2H, Jvic = 7.3, H-1), 4.36 (t, 2H, Jvic = 7.3, H-8). 13C-NMR δ 15.3 (MeS), 25.9 (C-4), 27.1 (C-7), 28.3, 28.5, 28.6, (C-3, C-5, C-6), 28.8 (C-2), 34.0 (C-1), 75.6 (C-8). MS IS m/z 206.3 [M + H]+.
(1-Methylsulfanyl)-10-nitrodecane 5f. Colourless oil (3.8 g, 68% yield). 1H-NMR (CDCl3) δ 1.21–1.41 (m, 12H, H-3-8), 1.57 (qt, 2H, H-2), 1.99 (qt, 2H, H-9), 2.07 (s, 3H, MeS), 2.47 (t, 2H, Jvic = 7.3, H-1), 4.36 (t, 2H, Jvic = 7.3, H-10). 13C-NMR δ 15.3 (MeS), 26.0, 27.2, 28.5, 28.6, 28.9, 29.0 (C-3-9), 29.1 (C-2), 34.1 (C-1), 75.6 (C-10). MS IS m/z 234.4 [M + H]+.
(1-Methylsulfanyl)-12-nitrododecane 5g. Colourless oil (4.2 g, 67% yield). 1H-NMR (CDCl3) δ 1.21–1.42 (m, 16H, H-3-10), 1.58 (qt, 2H, H-2), 2.0 (qt, 2H, H-11), 2.09 (s, 3H, MeS), 2.48 (t, 2H, Jvic = 7.3, H-1), 4.37 (t, 2H, Jvic = 7.3, H-12). 13C-NMR δ 15.3 (MeS), 26.0, 27.2, 28.6, 28.9, 29.2 (C-3-11), 29.3 (C-2), 34.1 (C-1), 75.6 (C-12). MS IS m/z 262.4 [M + H]+.
3.2.4. General Procedure for the Nitronate Chlorination and Coupling with the Thioglucose Unit
Nitronate formation. To a stirred freshly prepared solution of sodium (0.3 g) in 2-butanol (17 mL), under argon atmosphere, a solution of nitro derivative 5 (13 mmol) in anhydrous ether (20 mL) is added dropwise under exclusion of moisture. The slow addition of anhydrous ether (200 mL) while stirring caused precipitation of the nitronate as a white solid. After 10 min more stirring, the suspension was rapidly filtered off on sintered glass, and, after rinsing with anhydrous ether (20–30 mL), the nitronate cake was dried in vacuo for 30 min (preparation of nitronates can be more confortably realized in a glovebox).
Conversion of nitronate into hydroximoyl chloride. The powdered nitronate obtained was suspended in dry chloroform (40 mL), the stirred mixture was cooled at −60 °C, and a chloroform solution of freshly distilled thionyl chloride (1 mL in 5 mL) was added dropwise. After 20 min more stirring at −60°C, the reaction was quenched by pouring the mixture into ice water; the chloroform solution was separated and the aqueous phase was extracted twice with chloroform. The combined organic phases were dried over MgSO4, filtered and concentrated in vacuo to afford the raw hydroximoyl chloride as a greenish oil.
Coupling with the thioglucose unit. This oily residue was immediately dissolved in 70 mL of a dry dichloromethane–diethyl ether mixture (2:1 v/v), 2,3,4,6-tetra-O-acetyl-1-thio-β-d-glucopyranose (3.64 g, 10 mmol) was added, then dry argon was bubbled for 15 min in the solution cooled to −10 °C. A solution of triethylamine (4.2 mL, 30 mmol) in dichloromethane (10 mL) was added in one portion and the mixture was stirred during 45 min more. After washing with ice-cold 1N sulfuric acid, the organic phase was separated and the aqueous phase extracted twice with dichloromethane. The combined organic phases were dried over MgSO4, filtered and concentrated in vacuo. The syrupy residue was purified by column chromatography (petroleum ether–EtOAc 3:2) to afford the glycosyl thiohydroximates 18 as amorphous solids.
S-(2,3,4,6-Tetra-O-acetyl-β-d-glucopyranosyl) (Z)-(3′-methylsulfanyl)propanethiohydroximate 18a. White amorphous powder (32% yield), [α]D −20 (c = 1.0, CHCl3). 1H-NMR (CDCl3) δ 2.00, 2.03, 2.05, 2.10, (4s, 12H, CH3CO), 2.14 (s, 3H, MeS), 2.78–2.81 (m, 4H, H-2′, H-3′), 3.78 (dt, 1H, J4–5 = 10.0, H-5), 4.17 (d, 2H, J5–6a = J5–6b = 3.9, H-6a, H-6b), 5.05–5.12 (m, 3H, H-1, H-2, H-4), 5.24–5.30 (m, 1H, H-3), 8.04 (brs, NOH). 13C-NMR δ 15.5 (MeS), 20.3, 20.4, 20.6 (CH3CO), 31.1 (C-2′), 32.7 (C-1′), 62.0 (C-6), 67.9 (C-4), 70.0 (C-2), 73.6 (C-3), 75.9 (C-5), 79.9 (C-1), 150.8 (C=N), 169.4, 169.5, 170.3, 170.8 (C=O). HR-ESI-MS: C18H27NO10S2: calcd. 481.1076; found 481.1061.
S-(2,3,4,6-Tetra-O-acetyl-β-d-glucopyranosyl) (Z)-(4′-methylsulfanyl)butanethiohydroximate 18b. White amorphous powder (34% yield), [α]D −16 (c = 1.0, CHCl3). 1H-NMR (CDCl3) δ 1.90–1.95 (m, 2H, H-3′), 1.99, 2.01, 2.03, 2.06 (4s, 12H, CH3CO), 2.10 (s, 3H, MeS), 2.52–2.67 (m, 4H, H-2′, H-4′), 3.82 (ddd, 1H, J4–5 = 9.9, H-5), 4.13 (dd, 1H, J5–6b = 2.4, Jgem = 11.8, H-6b), 4.20 (dd, 1H, J5–6a = 5.0, H-6a), 5.00–5.12 (m, 3H, H-1, H-2, H-4), 5.24 (t, 1H, J3–4 = 9.0, H-3), 8.80 (brs, NOH). 13C-NMR δ 15.4 (MeS), 20.4, 20.5, 20.6 (CH3CO), 26.1 (C-3′), 30.9 (C-4′), 33.4 (C-2′), 62.0 (C-6), 67.9 (C-4), 70.1 (C-2), 73.8 (C-3), 75.8 (C-5), 79.8 (C-1), 151.7 (C=N), 169.3, 169.5, 170.4, 170.8 (C=O). HR-ESI-MS: C19H29NO10S2: calcd. 495.1233; found 495.1230.
S-(2,3,4,6-Tetra-O-acetyl-β-d-glucopyranosyl) (Z)-(5′-methylsulfanyl)pentanethiohydroximate 18c. White amorphous powder (40% yield), [α]D −17 (c = 1.0, CHCl3). 1H-NMR (CDCl3) δ 1.60–1.85 (m, 4H, H-3′, H-4′), 2.02, 2.05, 2.07, 2.09 (4s, 12H, CH3CO), 2.11 (s, 3H, MeS), 2.45–2.55 (m, 4H, H-2′, H-5′), 3.79 (ddd, 1H, J4–5 = 9.7, H-5), 4.14 (dd, 1H, J5–6b = 2.5, Jgem = 12.6, H-6b), 4.22 (dd, 1H, J5–6a = 5.4, H-6a), 5.05–5.15 (m, 3H, H-1, H-2, H-4), 5.25 (dd, 1H, J3–4 = 9.3, H-3), 8.20 (brs, NOH). 13C-NMR δ 15.3 (MeS), 20.4, 20.5 (CH3CO), 25.8 (C-4′), 28.0 (C-3′), 31.9 (C-5′), 33.5 (C-2′), 62.1 (C-6), 68.0 (C-4), 70.1 (C-2), 73.7 (C-3), 75.8 (C-5), 79.8 (C-1), 152.0 (C=N), 169.3, 169.5, 170.3, 170.8 (C=O). HR-ESI-MS: C20H31NO10S2: calcd. 509.1389; found 509.1378.
S-(2,3,4,6-Tetra-O-acetyl-β-d-glucopyranosyl) (Z)-(6′-methylsulfanyl)hexanethiohydroximate 18d. White amorphous powder (48% yield), [α]D −11 (c = 1.0, CHCl3). 1H-NMR (CDCl3) δ 1.40–1.51 (m, 2H, H-4′), 1.58–1.72 (m, 4H, H-3′, H-5′), 2.01, 2.02, 2.04, 2.08 (4s, 12H, CH3CO), 2.10 (s, 3H, MeS), 2.50 (t, 4H, Jvic = 7.1, H-2′, H-6′), 3.75 (ddd, 1H, J4–5 = 10.1, H-5), 4.11 (dd, 1H, J5–6b = 2.4, Jgem = 12.3, H-6b), 4.20 (dd, 1H, J5–6a = 5.6, H-6a), 5.04–5.12 (m, 3H, H-1, H-2, H-4), 5.27 (dd, 1H, J3–4 = 9.3, H-3), 8.20 (brs, NOH). 13C-NMR δ 15.3 (MeS), 20.4, 20.5 (CH3CO), 26.4 (C-4′), 28.0 (C-5′), 28.5 (C-3′), 32.3 (C-6′), 33.8 (C-2′), 62.1 (C-6), 68.0 (C-4), 70.0 (C-2), 73.7 (C-3), 75.9 (C-5), 79.8 (C-1), 152.1 (C=N), 169.3, 169.5, 170.5, 170.7 (C=O). HR-ESI-MS: C21H33NO10S2: calcd. 523.1546; found 523.1541.
S-(2,3,4,6-Tetra-O-acetyl-β-d-glucopyranosyl) (Z)-(8′-methylsulfanyl)octanethiohydroximate 18e. White amorphous powder (47% yield), [α]D −11 (c = 1.0, CHCl3). 1H-NMR (CDCl3) δ 1.32–1.40 (m, 6H, H-4′, H-5′, H-6′), 1.47–1.64 (m, 4H, H-3′, H-7′), 1.99, 2.01, 2.02, 2.05 (4s, 12H, CH3CO), 2.07 (s, 3H, MeS), 2.41–2.46 (m, 4H, H-2′, H-8′), 3.73 (ddd, 1H, J4–5 = 10.0, H-5), 4.09 (dd, 1H, J5–6b = 2.1, Jgem = 12.3, H-6b), 4.18 (dd, 1H, J5–6a = 5.4, H-6a), 5.02–5.10 (m, 3H, H-1, H-2, H-4), 5.25 (dd, 1H, J3–4 = 9.4, H-3), 8.83 (brs, NOH). 13C-NMR δ 15.3 (MeS), 20.4, 20.5 (CH3CO), 26.8, 28.5, 28.7 (C-4′, C-5′, C-6′), 28.8 (C-7′), 28.9 (C-3′), 32.3 (C-8′), 34.5 (C-2′), 62.1 (C-6), 68.0 (C-4), 70.0 (C-2), 73.7 (C-3), 75.9 (C-5), 79.8 (C-1), 152.2 (C=N), 169.3, 169.5, 170.4, 170.7 (C=O). HR-ESI-MS: C23H37NO10S2: calcd. 551.1859; found 551.1851.
S-(2,3,4,6-Tetra-O-acetyl-β-d-glucopyranosyl) (Z)-(10′-methylsulfanyl)decanethiohydroximate 18f. White amorphous powder (51% yield), [α]D −12 (c = 1.0, CHCl3). 1H-NMR (CDCl3) δ 1.28–1.34 (m, 10H, H-4′ to H-8′), 1.51–1.63 (m, 4H, H-3′, H-9′), 1.98, 2.01, 2.02, 2.04 (4s, 12H, CH3CO), 2.06 (s, 3H, MeS), 2.38–2.47 (m, 4H, H-2′, H-10′),3.71 (ddd, 1H, J4–5 = 10.0, H-5), 4.08 (dd, 1H, J5–6b = 2.4, Jgem = 12.1, H-6b), 4.19 (dd, 1H, J5–6a = 5.2, H-6a), 5.00–5.09 (m, 3H, H-1, H-2, H-4), 5.22 (dd, 1H, J3–4 = 9.2, H-3), 8.86 (brs, NOH). 13C-NMR δ 15.3 (MeS), 20.4, 20.5 (CH3CO), 26.9, 28.6, 28.9, (C-4′ to C-8′), 29.0 (C-9′), 29.2 (C-3′), 32.4 (C-10′), 34.1 (C-2′), 62.1 (C-6), 68.0 (C-4), 70.0 (C-2), 73.7 (C-3), 75.9 (C-5), 79.8 (C-1), 152.3 (C=N), 169.3, 169.5, 170.4, 170.7 (C=O). HR-ESI-MS: C25H41NO10S2: calcd. 579.2172; found 579.2163.
S-(2,3,4,6-Tetra-O-acetyl-β-d-glucopyranosyl) (Z)-(12′-methylsulfanyl)dodecanethiohydroximate 18g. White amorphous powder (56% yield), [α]D −12 (c = 1.0, CHCl3). 1H-NMR (CDCl3) δ 1.31–1.38 (m, 14H, H-4′ to H-10′), 1.53–1.66 (m, 4H, H-3′, H-11′), 1.98, 2.01, 2.02, 2.05 (4s, 12H, CH3CO), 2.07 (s, 3H, MeS), 2.40–2.45 (m, 4H, H-2′, H-12′), 3.72 (ddd, 1H, J4–5 = 9.8, H-5), 4.09 (dd, 1H, J5–6b = 2.5, Jgem = 12.5, H-6b), 4.19 (dd, 1H, J5–6a = 5.5, H-6a), 5.01–5.12 (m, 3H, H-1, H-2, H-4), 5.24 (dd, 1H, J3–4 = 9.5, H-3), 8.88 (brs, NOH). 13C-NMR δ 15.3 (MeS), 20.4, 20.5 (CH3CO), 26.7, 28.9, 29.0 (C-4′ to C-10′), 29.1 (C-11′), 29.2 (C-3′), 32.4 (C-12′), 34.3 (C-2′), 62.1 (C-6), 67.9 (C-4), 70.0 (C-2), 73.6 (C-3), 75.8 (C-5), 79.8 (C-1), 152.3 (C=N), 169.2, 169.5, 170.3, 170.6 (C=O). HR-ESI-MS: C27H45NO10S2: calcd. 607.2485; found 607.2478.
3.2.5. General Procedure for NO-Sulfation of the Glucosyl Thiohydroximates 18
Sulfur trioxide pyridine complex (400 mg, 5 eq.) was added to a solution of compound 18 (0.5 mmol) in dimethylformamide (7 mL). After 24 h stirring at rt, the reaction mixture was treated with a 0.2 M aqueous solution of KHCO3 (12 mL) then the solvents were evaporated in vacuo. Chromatographic purification (CH2Cl2/MeOH 17:3) provided compounds 19.
Per-O-acetylated 2-methylsulfanylethyl glucosinolate 19a. White amorphous powder (61% yield), [α]D −20 (c = 0.9, MeOH). 1H-NMR (DMSO-d6) δ 1.96, 1.99, 2.01, 2.03 (4s, 12H, CH3CO), 2.23 (s, 3H, MeS), 2.46–2.53 (m, 4H, H-1′, H-2′), 4.03–4.15 (m, 3H, H-5, H-6a, H-6b), 4.88 (t, 1H, J2–3 = 9.4, H-2), 4.94 (t, 1H, J4–5 = 8.6, H-4), 5.39 (dd, 1H, J3–4 = 9.6, H-3), 5.56 (d, 1H, J1–2 = 10.1, H-1). 13C-NMR δ 14.6 (MeS), 20.2, 20.3, 20.5 (CH3CO), 26.5 (C-2′), 32.9 (C-1′), 62.2 (C-6), 68.1 (C-4), 69.8 (C-2), 72.9 (C-3), 74.0 (C-5), 78.3 (C-1), 153.2 (C=N), 169.4, 169.7, 170.0, 170.3 (C=O). HR-ESI-MS: C18H26NO13S3: calcd. 560.0566; found 560.0571.
Per-O-acetylated 3-methylsulfanylpropyl glucosinolate 19b. White amorphous powder (68% yield), [α]D −22 (c = 1.1, MeOH). 1H-NMR (DMSO-d6) δ 1.79–1.90 (m, 2H, H-2′), 1.94, 1.97, 1.99, 2.00 (4s, 12H, CH3CO), 2.05 (s, 3H, MeS), 2.53 (t, 2H, Jvic = 7.3, H-3′), 2.63 (t, 2H, Jvic = 7.3, H-1′), 4.00–4.18 (m, 3H, H-5, H-6a, H-6b), 4.86 (t, 1H, J2–3 = 9.0, H-2), 4.89 (t, 1H, J4–5 = 8.8, H-4), 5.43 (dd, 1H, J3–4 = 9.3, H-3), 5.50 (d, 1H, J1–2 = 10.2, H-1). 13C-NMR δ 14.7 (MeS), 20.2, 20.3, 20.5 (CH3CO), 26.4 (C-3′), 30.3, 32.5 (C-1′, C-2′), 62.1 (C-6), 68.0 (C-4), 69.6 (C-2), 72.8 (C-3), 74.4 (C-5), 78.2 (C-1), 153.5 (C=N), 169.4, 169.6, 169.9, 170.3 (C=O). HR-ESI-MS: C19H28NO13S3: calcd. 574.0723; found 574.0730.
Per-O-acetylated 4-methylsulfanylbutyl glucosinolate 19c. White amorphous powder (86% yield), [α]D −18 (c = 1.0, MeOH). 1H-NMR (DMSO-d6) δ 1.52–1.68 (m, 4H, H-2′, H-3′), 1.94, 1.97, 1.99, 2.00 (4s, 12H, CH3CO), 2.02 (s, 3H, MeS), 2.48–2.59 (m, 4H, H-1′, H-4′), 4.00–4.15 (m, 3H, H-5, H-6a, H-6b), 4.86 (t, 1H, J2–3 = 9.5, H-2), 4.91 (t, 1H, J4–5 = 8.5, H-4), 5.43 (dd, 1H, J3–4 = 9.5, H-3), 5.50 (d, 1H, J1–2 = 10.2, H-1). 13C-NMR δ 14.5 (MeS), 20.2, 20.3, 20.5 (CH3CO), 25.7 (C-4′), 27.9, 31.0, 32.8 (C-1′, C-2′, C-3′), 62.2 (C-6), 68.1 (C-4), 69.6 (C-2), 72.8 (C-3), 74.4 (C-5), 78.2 (C-1), 154.0 (C=N), 169.4, 169.6, 169.9, 170.3 (C=O). HR-ESI-MS: C20H30NO13S3: calcd. 588.0879; found 588.0887.
Per-O-acetylated 5-methylsulfanylpentyl glucosinolate 19d. White amorphous powder (84% yield), [α]D −15 (c = 1.1, MeOH). 1H-NMR (DMSO-d6) δ 1.38–1.68 (m, 6H, H-2′, H-3′, H-4′), 1.94, 1.98, 1.99, 2.00 (4s, 12H, CH3CO), 2.02 (s, 3H, MeS), 2.43–2.57 (m, 4H, H-1′, H-5′), 3.98–4.15 (m, 3H, H-5, H-6a, H-6b), 4.86 (t, 1H, J2–3 = 9.8, H-2), 4.91 (t, 1H, J4–5 = 8.8, H-4), 5.45 (dd, 1H, J3–4 = 9.7, H-3), 5.51 (d, 1H, J1–2 = 10.2, H-1). 13C-NMR δ 15.6 (MeS), 20.2, 20.4, 20.5 (CH3CO), 26.4 (C-5′), 27.8, 28.2, 31.4, 33.2 (C-1′ to C-4′), 62.2 (C-6), 68.2 (C-4), 69.6 (C-2), 72.8 (C-3), 74.4 (C-5), 78.2 (C-1), 154.1 (C=N), 169.4, 169.6, 169.9, 170.3 (C=O). HR-ESI-MS: C21H32NO13S3: calcd. 602.1036; found 602.1041.
Per-O-acetylated 7-methylsulfanylheptyl glucosinolate 19e. White amorphous powder (83% yield), [α]D −17 (c = 1.0, MeOH). 1H-NMR (DMSO-d6) δ 1.24–1.39 (m, 6H, H-3′, H-4′, H-5′), 1.44–1.65 (m, 4H, H-2′, H-6′), 1.94, 1.97, 1.99, 2.01 (4s, 12H, CH3CO), 2.01 (s, 3H, MeS), 2.40–2.54 (m, 4H, H-1′, H-7′), 3.98–4.15 (m, 3H, H-5, H-6a, H-6b), 4.85 (t, 1H, J2–3 = 9.2, H-2), 4.91 (t, 1H, J4–5 = 8.8, H-4), 5.46 (dd, 1H, J3–4 = 9.4, H-3), 5.49 (d, 1H, J1–2 = 10.3, H-1). 13C-NMR δ 14.7 (MeS), 20.2, 20.3, 20.4 (CH3CO), 26.8 (C-7′), 28.2, 28.3, 28.6, 31.4, 33.2 (C-1′ to C-6′), 62.2 (C-6), 68.2 (C-4), 69.6 (C-2), 72.8 (C-3), 74.4 (C-5), 78.2 (C-1), 154.1 (C=N), 169.4, 169.6, 169.9, 170.2 (C=O). HR-ESI-MS: C23H36NO13S3: calcd. 630.1049; found 630.1060.
Per-O-acetylated 9-methylsulfanylnonyl glucosinolate 19f. White amorphous powder (85% yield), [α]D −16 (c = 1.0, MeOH). 1H-NMR (DMSO-d6) δ 1.21–1.40 (m, 10H, H-3′ to H-7′), 1.44–1.59 (m, 4H, H-2′, H-8′), 1.95, 1.97, 1.98, 2.00 (4s, 12H, CH3CO), 2.01 (s, 3H, MeS), 2.40–2.54 (m, 4H, H-1′, H-9′), 3.98–4.14 (m, 3H, H-5, H-6a, H-6b), 4.84 (t, 1H, J2–3 = 9.7, H-2), 4.90 (t, 1H, J4–5 = 8.5, H-4), 5.46 (dd, 1H, J3–4 = 9.7, H-3), 5.50 (d, 1H, J1–2 = 10.0, H-1). 13C-NMR δ 15.1 (MeS), 20.2, 20.3, 20.4 (CH3CO), 26.9 (C-9′), 28.2, 28.4, 28.5, 28.8, 29.1, 31.6, 34.3 (C-1′ to C-8′), 62.2 (C-6), 68.4 (C-4), 70.0 (C-2), 73.1 (C-3), 74.7 (C-5), 78.7 (C-1), 154.5 (C=N), 169.3, 169.5, 170.0, 170.3 (C=O). HR-ESI-MS: C25H40NO13S3: calcd. 658.1662; found 658.1665.
Per-O-acetylated 11-methylsulfanylundecyl glucosinolate 19g. White amorphous powder (84% yield), [α]D −16 (c = 1.0, MeOH). 1H-NMR (DMSO-d6) δ 1.24–1.40 (m, 14H, H-3′ to H-9′), 1.42–1.63 (m, 4H, H-2′, H-10′), 1.96, 1.98, 2.00, 2.02 (4s, 12H, CH3CO), 2.01 (s, 3H, MeS), 2.47–2.56 (m, 4H, H-1′, H-11′), 3.93–4.13 (m, 3H, H-5, H-6a, H-6b), 4.86 (t, 1H, J2–3 = 9.6, H-2), 4.93 (t, 1H, J4–5 = 8.8, H-4), 5.47 (dd, 1H, J3–4 = 9.5, H-3), 5.51 (d, 1H, J1–2 = 10.0, H-1). 13C-NMR δ 14.71 (MeS), 20.2, 20.3, 20.4 (CH3CO), 26.5 (C-11′), 28.8, 29.0, 29.3, 29.5, 29.9, 31.5, 32.4, 34.8 (C-1′ to C-10′), 62.1 (C-6), 68.4 (C-4), 69.8 (C-2), 73.2 (C-3), 75.1 (C-5), 78.3 (C-1), 154.5 (C=N), 169.4, 169.6, 170.0, 170.3 (C=O). HR-ESI-MS: C27H44NO13S3: calcd. 686.1975; found 686.1987.
3.2.6. General Procedure for Deprotection of the Glucopyranosyl Moiety
To a suspension of peracetylated glucosinolate 19 (0.1 mmol) in dry methanol (5 mL) under argon, a freshly prepared 1 M solution of potassium methoxide was added dropwise until the pH reached 9. After 4 h standing at rt, the reaction mixture was neutralized by the addition of Dowex H+ resin. After filtration and evaporation in vacuo, the resulting crude was purified by C-18 silica gel column chromatography (eluent: water) and freeze-drying to provide glucosinolates 2.
2-Methylsulfanylethyl glucosinolate [27303-30-6] 2a. White amorphous powder (80% yield), [α]D −27 (c = 0.95, H2O). 1H-NMR (D2O) δ 2.19 (s, 3H, MeS), 2.92–2.98 (m, 2H, H-2′), 3.04–3.10 (m, 2H, H-1′), 3.44–3.53 (m, 2H, H-2, H-4), 3.58–3.69 (m, 2H, H-3, H-5), 3.76 (dd, 1H, J5–6b = 5.8, Jgem = 13.0, H-6b), 3.95 (dd, 1H, J5–6a = 2.6, H-6a), 5.09 (d, 1H, J1–2 = 10.1, H-1). 13C-NMR δ 15.3 (MeS), 30.5 (C-2′), 33.0 (C-1′), 61.5 (C-6), 70.2 (C-4), 73.5 (C-2), 78.3 (C-3), 81.5 (C-5), 83.1 (C-1), 164.9 (C=N). HR-ESI-MS: C10H18NO9S3: calcd. 392.0144; found 392.0133.
3-Methylsulfanylpropyl glucosinolate [26888-03-9] 2b. Glucoibervirin. White amorphous powder (84% yield), [α]D −21 (c = 1.05, H2O). 1H-NMR (D2O) δ 2.05 (qt, 2H, H-2′), 2.16 (s, 3H, MeS), 2.67 (t, 2H, Jvic = 7.5, H-3′), 2.63 (t, 2H, Jvic = 7.5, H-1′), 3.46–3.52 (m, 2H, H-2, H-4), 3.55–3.66 (m, 2H, H-3, H-5), 3.73 (dd, 1H, J5–6b = 5.8, Jgem = 13.2, H-6b), 3.93 (dd, 1H, J5–6a = 2.7, H-6a), 5.09 (d, 1H, J1–2 = 10.1, H-1). 13C-NMR δ 15.3 (MeS), 26.7 (C-2′), 30.9 (C-3′), 34.1 (C-1′), 61.4 (C-6), 70.7 (C-4), 73.2 (C-2), 78.2 (C-3), 82.5 (C-5), 83.8 (C-1), 165.1 (C=N). HR-ESI-MS: C11H20NO9S3: calcd. 406.0300; found 406.0291.
4-Methylsulfanylbutyl glucosinolate [21973-56-8] 2c. Glucoerucin. White amorphous powder (90% yield), [α]D −20 (c = 1.00, H2O). 1H-NMR (D2O) δ 1.67–1.89 (m, 4H, CH2), 2.12 (s, 3H, MeS), 2.61 (t, 2H, Jvic = 7.5, H-4′), 2.76 (t, 2H, Jvic = 7.5, H-1′), 3.42–3.51 (m, 2H, H-2, H-4), 3.57–3.63 (m, 2H, H-3, H-5), 3.72 (dd, 1H, J5–6b = 5.8, Jgem = 13.1, H-6b), 3.91 (dd, 1H, J5–6a = 2.7, H-6a), 5.06 (d, 1H, J1–2 = 10.1, H-1). 13C-NMR δ 15.2 (MeS), 25.7 (C-2′), 28.2 (C-3′), 31.6 (C-4′), 33.0 (C-1′), 62.2 (C-6), 71.2 (C-4), 73.5 (C-2), 78.7 (C-3), 81.8 (C-5), 83.0 (C-1), 165.4 (C=N) [47]. HR-ESI-MS: C12H22NO9S3: calcd. 420.0457; found 420.0453.
5-Methylsulfanylpentyl glucosinolate [29611-01-6] 2d. Glucoberteroin. White amorphous powder (90% yield), [α]D −21 (c = 0.95, H2O). 1H-NMR (D2O) δ 1.44 (qt, 2H, Jvic = 7.4, H-3′), 1.61 (qt, 2H, Jvic = 7.3, H-4′) 1.70 (qt, 2H, Jvic = 7.3, H-2′), 2.06 (s, 3H, MeS), 2.53 (t, 2H, Jvic = 7.3, H-5′), 2.68 (t, 2H, Jvic = 7.3, H-1′), 3.38–3.45 (m, 2H, H-2, H-4), 3.50–3.56 (m, 2H, H-3, H-5), 3.68 (dd, 1H, J5–6b = 5.8, Jgem = 13.1, H-6b), 3.85 (dd, 1H, J5–6a = 2.6, H-6a), 5.00 (d, 1H, J1–2 = 10.0, H-1). 13C-NMR δ 15.1 (MeS), 27.5 (C-3′), 28.5 (C-2′), 29.0 (C-4′), 33.0 (C-5′), 34.2 (C-1′), 61.8 (C-6), 70.3 (C-4), 73.3 (C-2), 78.4 (C-3), 81.4 (C-5), 83.0 (C-1), 164.5 (C=N) [48].HR-ESI-MS: C13H24NO9S3: calcd. 434.0613; found 434.0610.
7-Methylsulfanylheptyl glucosinolate [80667-67-0] 2e. White amorphous powder (90% yield), [α]D −22 (c = 1.00, H2O). 1H-NMR (D2O) δ 1.24–1.39 (m, 6H, CH2), 1.44–1.65 (m, 4H, CH2), 2.01 (s, 3H, MeS), 2.38–2.54 (m, 4H, CH2), 3.45–3.52 (m, 2H, H-2, H-4), 3.55–3.63 (m, 2H, H-3, H-5), 3.68 (dd, 1H, J5–6b = 5.7, Jgem = 12.8, H-6b), 3.89 (dd, 1H, J5–6a = 2.6, H-6a), 5.06 (d, 1H, J1–2 = 10.0, H-1). 13C-NMR δ 15.1(MeS), 27.8, 28.7, 28.9 (C-3′-C-5′), 29.0 (C-2′), 29.2 (C-6′), 33.0 (C-1′), 34.3 (C-7′), 61.6 (C-6), 70.1 (C-4), 73.0 (C-2), 78.2 (C-3), 81.2 (C-5), 82.9 (C-1), 165.4 (C=N). HR-ESI--MS: C15H28NO9S3: calcd. 462.0926; found 462.0932 [48]. HR-ESI-MS: C15H28NO9S3: calcd. 462.0926; found 462.0919.
9-Methylsulfanylnonyl glucosinolate [81149-01-1] 2f. White amorphous powder (93% yield), [α]D −19 (c = 0.84, H2O). 1H-NMR (D2O) δ 1.21–1.40 (m, 10H, CH2), 1.44–1.59 (m, 4H, CH2), 2.00 (s, 3H, MeS), 2.43–2.58 (m, 4H, (CH2), 3.40–3.51 (m, 2H, H-2, H-4), 3.57–3.62 (m, 2H, H-3, H-5), 3.71 (dd, 1H, J5–6b = 5.7, Jgem = 13.0, H-6b), 3.91 (dd, 1H, J5–6a = 2.7, H-6a), 5.06 (d, 1H, J1–2 = 10.1, H-1). 13C-NMR δ 15.4 (MeS), 26.9, 28.2, 28.4, 28.6 (C-3′-C-7′), 28.9 (C-2′), 29.2 (C-8′), 31.9 (C-1′), 34.6 (C-9′), 61.4 (C-6), 70.5 (C-4), 73.4 (C-2), 78.3 (C-3), 82.2 (C-5), 83.5 (C-1), 164.8 (C=N). HR-ESI-MS: C17H32NO9S3: calcd. 490.1239; found 490.1231.
11-Methylsulfanylundecyl glucosinolate 2g. Colourless gum (91% yield), [α]D −21 (c = 0.92, H2O). 1H-NMR δ (D2O) 1.26–1.44 (m, 10H, CH2), 1.42–1.63 (m, 4H, (CH2), 2.02 (s, 3H, MeS), 2.44–2.56 (m, 4H, (CH2), 3.39–3.50 (m, 2H, H-2, H-4), 3.54–3.61 (m, 2H, H-3, H-5), 3.74 (dd, 1H, J5–6b = 5.8, Jgem = 13.0, H-6b), 3.93 (dd, 1H, J5–6a = 2.6,, H-6a), 5.06 (d, 1H, , J1–2 = 10.1, H-1). 13C-NMR δ 15.2 (MeS), 26.5, 28.9, 29.0, 29.5 (C3′-C-9′), 29.7 (C-2′), 30.0 (C-10′), 32.2 (C-1′), 34.8 (C-11′), 61.6 (C-6), 70.2 (C-4), 73.1 (C-2), 78.7 (C-3), 81.3 (C-5), 83.0 (C-1), 165.4 (C=N). HR-ESI-MS: C19H36NO9S3: calcd. 518.1552; found 518.1547.
4. Conclusions
We have developed a general pathway for the synthesis of biologically critical ω-methylsulfanylalkyl GLs involving tailor-made ω-methylsulfanyl nitroalkanes precursors. Synthetic conditions to access those key-intermediates were adjusted according to the length of the alkyl chain that is required. Extension of this work to the synthesis of ω-methylsulfinyl and ω-methylsulfonyl GLs is under current development in our research group.
Acknowledgments
The authors are grateful to Vassilis Dourtoglou (Vioryl S.A., Afidnes, Greece) and the Greek-French Platon PHC. We also wish to thank Agnès Chartier for GC expertise. The multiform support of Gérald Guillaumet (University of Orléans) is sincerely acknowledged.
Author Contributions
P.R. designed the synthetic route and M.M. performed the experimental work, P.R. wrote the manuscript, which was revised by S.C. and S.M.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Blažević, I.; Montaut, S.; Burčul, F.; Rollin, P. Glucosinolates: Novel Sources and Biological Potential. Reference Series in Phytochemistry; Ramawat, K., Mérillon, J.-M., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 3–60. ISBN 978-3-319-25749-5. [Google Scholar]
- Montaut, S.; Rollin, P. Glucosinolates and Their Distribution; Juurlink, B.H.J., Ed.; Nova Science Publishers: New York, NY, USA, 2016; pp. 9–31. ISBN 978-1-63484-314-0. [Google Scholar]
- Hanschen, F.S.; Lamy, E.; Schreiner, M.; Rohn, S. Reactivity and stability of glucosinolates and their breakdown products in foods. Angew. Chem. Int. Ed. 2014, 53, 2–23. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.W.; Zalcmann, A.T.; Talalay, P. The chemical diversity and distribution of glucosinolates and isothiocyanates in plants. Phytochemistry 2001, 56, 5–51. [Google Scholar] [CrossRef]
- Agerbirk, N.; Olsen, C.E. Glucosinolate structures in evolution. Phytochemistry 2012, 77, 16–45. [Google Scholar] [CrossRef] [PubMed]
- Cabello-Hurtado, F.; Gicquel, M.; Esnault, M.-A. Evaluation of the antioxidant potential of cauliflower (Brassica oleracea) from a glucosinolate content perspective. Food Chem. 2012, 132, 1003–1009. [Google Scholar] [CrossRef]
- Montaut, S.; Grandbois, J.; Rossi, L.S.; Kamal, S.; Khouri, J.; Ménard, M.G.; Joly, H.A. Composition of Dithyrea wislizenii fruit extract and the free-radical scavenging activity of its constituents. Can. J. Chem. 2012, 90, 652–659. [Google Scholar] [CrossRef]
- Natella, F.; Maldini, M.; Leoni, G.; Scaccini, C. Glucosinolate redox activities: Can they act as antioxidants? Food Chem. 2014, 149, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Wathelet, J.-P.; Iori, R.; Leoni, O.; Rollin, P.; Quinsac, A.; Palmieri, S. Guidelines for glucosinolate analysis in green tissues used for biofumigation. Agroindustria 2004, 3, 257–266. [Google Scholar]
- Barillari, J.; Cervellati, R.; Paolini, M.; Tatibouët, A.; Rollin, P.; Iori, R. Isolation of 4-methylthio-3-butenyl glucosinolate from Raphanus sativus sprouts (Kaiware daikon) and its redox properties. J. Agric. Food Chem. 2005, 53, 9890–9896. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.N.; Mellon, F.A.; Kroon, P.A. Screening crucifer seeds as sources of specific intact glucosinolates using ion-pair high-performance chromatography negative ion electrospray mass spectrometry. J. Agric. Food Chem. 2004, 52, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Barillari, J.; Gueyrard, D.; Rollin, P.; Iori, R. Barbarea verna as a source of 2-phenylethyl glucosinolate, precursor of cancer chemopreventive phenylethyl isothiocyanate. Fitoterapia 2001, 72, 760–764. [Google Scholar] [CrossRef]
- Barillari, J.; Canistro, D.; Paolini, M.; Ferroni, F.; Pedulli, G.F.; Iori, R.; Valgimigli, L. Direct antioxidant activity of purified glucoerucin, the secondary metabolite contained in rocket (Eruca sativa Mill.) seeds and sprouts. J. Agric. Food Chem. 2005, 53, 2475–2482. [Google Scholar] [CrossRef] [PubMed]
- Gueyrard, D.; Iori, R.; Tatibouët, A.; Rollin, P. Glucosinolate chemistry. Synthesis of O-glycosylated derivatives of glucosinalbin. Eur. J. Org. Chem. 2010, 2010, 3657–3664. [Google Scholar] [CrossRef]
- Rollin, P.; Tatibouët, A. Glucosinolates: The synthetic approach. C. R. Chim. 2011, 14, 194–210. [Google Scholar] [CrossRef]
- Mavratzotis, M.; Dourtoglou, V.; Lorin, C.; Rollin, P. Glucosinolate chemistry. First synthesis of glucosinolates bearing an external thio-function. Tetrahedron Lett. 1996, 37, 5699–5700. [Google Scholar] [CrossRef]
- Morrison, J.J.; Botting, N.P. The synthesis of isotopically labelled glucoraphanin for metabolic studies. Tetrahedron Lett. 2007, 48, 1891–1894. [Google Scholar] [CrossRef]
- Botting, N.P.; Robertson, A.A.B.; Morrison, J.J. The synthesis of isotopically labelled glucosinolates for analysis and metabolic studies. J. Label. Compd. Radiopharm. 2007, 50, 260–263. [Google Scholar] [CrossRef]
- Yamazoe, S.; Hasegawa, K.; Shigemori, H. First total synthesis of 4-methylthio-3-butenyl glucosinolate. Biosci. Biotechnol. Biochem. 2009, 73, 785–787. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lebl, T.; Kulczynska, A.; Botting, N.P. The synthesis of novel hexa-13C-labelled glucosinolates from [13C6]-D-glucose. Tetrahedron 2009, 65, 4871–4876. [Google Scholar] [CrossRef]
- Grundmann, C.; Grünanger, P. The Nitrile Oxides; Springer: Berlin, Germany, 1971; ISBN 3-540-05226-7. [Google Scholar]
- Corey, E.J.; Kim, C.U. New and highly effective method for the oxidation of primary and secondary alcohols to carbonyl compounds. J. Am. Chem. Soc. 1972, 94, 7586–7587. [Google Scholar] [CrossRef]
- Anklam, E. Synthese von α-Halogen-ω-alkylthio-alkanen und α,ω-Bisalkylthio-alkanen. Synthesis 1987, 1987, 841–843. [Google Scholar] [CrossRef]
- Anklam, E.; Aced, G.; Mao, Y. Mechanistic aspects of cyclic sulphonium salt formation and hydrolysis of 1,n-halo(alkylthio) alkanes. Phosphorus Sulfur 1988, 40, 99–104. [Google Scholar] [CrossRef]
- Aced, G.; Darmstädt, E.; Anklam, E. Chromatographic behavior (GC and HPLC) of 1,n-bis(alkylthio)alkanes. Fresenius Z. Anal. Chem. 1988, 331, 740–743. [Google Scholar] [CrossRef]
- Palmer, D.C.; Taylor, E.C. Thietane alkylations. A general synthesis of 3-halopropyl benzyl sulfides. J. Org. Chem. 1986, 51, 846–850. [Google Scholar] [CrossRef]
- Kornblum, N.; Larson, H.O.; Blackwood, R.K.; Mooberry, D.D.; Oliveto, E.P.; Graham, G.E. A new method for the synthesis of aliphatic nitro compounds. J. Am. Chem. Soc. 1956, 78, 1497–1501. [Google Scholar] [CrossRef]
- Kornblum, N. The synthesis of aliphatic and alicyclic nitro compounds. Org. React. 1962, 12, 101–156. [Google Scholar]
- Kornblum, N.; Blackwood, R.K.; Mooberry, D.D. Reaction of aliphatic nitro compounds with nitrite esters. J. Am. Chem. Soc. 1956, 78, 1501–1504. [Google Scholar] [CrossRef]
- Wade, P.A. Synthesis of 3-substituted 2-isoxazolines and 5,6-dihydro-4H-1,2-oxazines. J. Org. Chem. 1978, 43, 2020–2023. [Google Scholar] [CrossRef]
- Bay, P.G. 3-Nitro-2-isoxazoline. U.S. Patent US 3207761, 21 September 1965. [Google Scholar]
- Kloeckner, J.; Schmitz, J.; Holzgrabe, U. Convergent, short synthesis of the muscarinic superagonist iperoxo. Tetrahedron Lett. 2010, 51, 3470–3472. [Google Scholar] [CrossRef]
- Kornblum, N.; Graham, G.E. The regeneration of nitroparaffins from their salts. J. Am. Chem. Soc. 1951, 73, 4041–4043. [Google Scholar] [CrossRef]
- Kornblum, N.; Brown, R.A. The action of acids on nitronic esters and nitroparaffin salts. Concerning the mechanisms of the Nef and the hydroxamic acid forming reactions of nitroparaffins. J. Am. Chem. Soc. 1965, 87, 1742–1747. [Google Scholar] [CrossRef]
- Jensen, S.R.; Kjaer, A. Synthesis of 2-hydroxy-2-butenyl glucosinolates. Acta Chem. Scand. 1971, 25, 3891–3893. [Google Scholar] [CrossRef]
- Viaud, M.-C.; Rollin, P. First synthesis of an indole glucosinolate. Tetrahedron Lett. 1990, 31, 1417–1418. [Google Scholar] [CrossRef]
- Bou-Moreno, R.; Luengo-Arratta, S.; Motherwell, W.B. Observations on the reaction of nitronate anions with oxalyl chloride: A new method for the preparation of germinal chloronitroso compounds. Tetrahedron Lett. 2011, 52, 2097–2099. [Google Scholar] [CrossRef]
- Benn, M.H. Synthesis of thiohydroximates. The addition of thiols to nitrile oxides. Can. J. Chem. 1964, 42, 2393–2397. [Google Scholar] [CrossRef]
- Černy, M.; Vrkoč, J.; Stanĕk, J. Uber die Darstellung von acylierten Derivaten der Glucopyranosylmercaptane. Coll. Czech. Chem. Commun. 1959, 24, 64–69. [Google Scholar] [CrossRef]
- Kjaer, A.; Wagner, S. A convenient synthesis of dl-homomethionine (5-methylthionorvaline). Acta Chem. Scand. 1955, 9, 721–726. [Google Scholar] [CrossRef]
- Kjaer, A.; Christensen, B. Straight-chain ω-methylthioalkyl isothiocyanates and some derivatives. Acta Chem. Scand. 1957, 11, 1298–1307. [Google Scholar] [CrossRef]
- Noshita, T.; Kidachi, Y.; Funayama, H.; Kiyota, H.; Yamaguchi, H.; Ryoyama, K. Anti-nitric oxide production activity of isothiocyanates correlates with their polar surface area rather than their lipophilicity. Eur. J. Med. Chem. 2009, 44, 4931–4936. [Google Scholar] [CrossRef] [PubMed]
- McLeod, A.J.; McLeod, G. Synthesis and natural occurrence of 8-methylthiooctanonitrile and 9-methylthiononanonitrile. Phytochemistry 1977, 16, 907–909. [Google Scholar] [CrossRef]
- Zhao, M.; Lu, W. Visible light-induced oxidative chlorination of alkyl sp3 C-H bonds with NaCl/oxone at room temperature. Org. Lett. 2017, 19, 4560–4563. [Google Scholar] [CrossRef] [PubMed]
- Goti, A.; Brandi, A.; Danza, G.; Guarna, A.; Donati, D.; De Sarlo, F. Rearrangement of isoxazoline-5-spiroderivatives. J. Chem. Soc. Perkin Trans. 1989, 1, 1253–1258. [Google Scholar] [CrossRef]
- Palmieri, A.; Gabrielli, S.; Ballini, R. Easy and direct conversion of tosylates and mesylates into nitroalkanes. Beilstein J. Org. Chem. 2013, 9, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, N.; Allart-Simon, I.; De Nicola, G.R.; Iori, R.; Renault, J.-H.; Rollin, P.; Nuzillard, J.-M. Advanced NMR-based structural investigation of glucosinolates and desulfoglucosinolates. J. Nat. Prod. 2018, 81, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Montaut, S.; Grandbois, J.; Righetti, L.; Barillari, J.; Iori, R.; Rollin, P. Updated glucosinolate profile of Dithyrea wislizenii. J. Nat. Prod. 2009, 72, 889–893. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |
Figure 1.
Glucosinolates (GLs) and their enzymatic degradation.

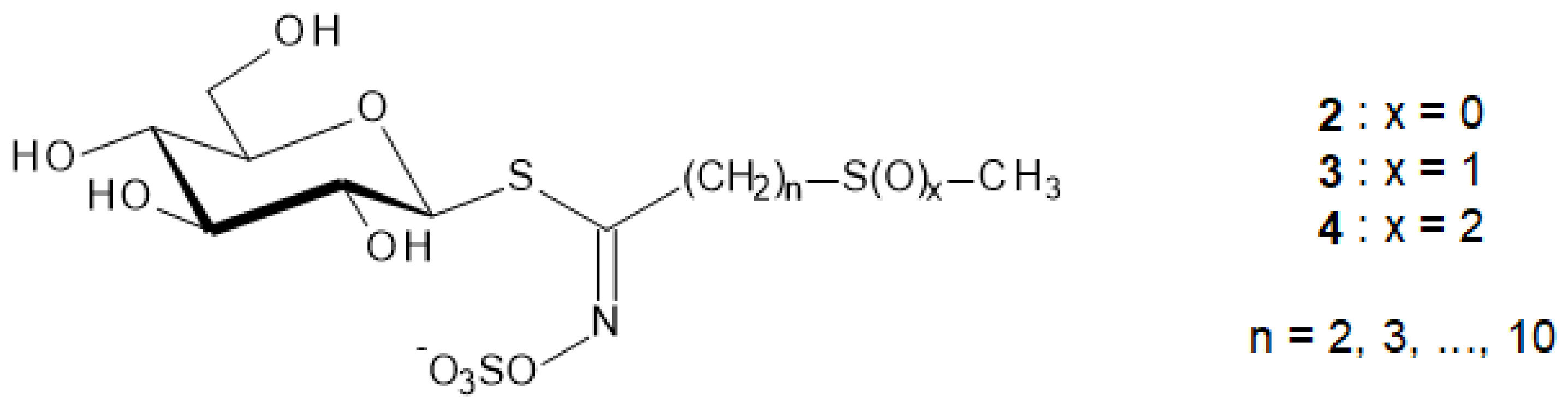
Figure 3.
Methods to generate nitrile oxides [15].
Figure 3.
Methods to generate nitrile oxides [15].
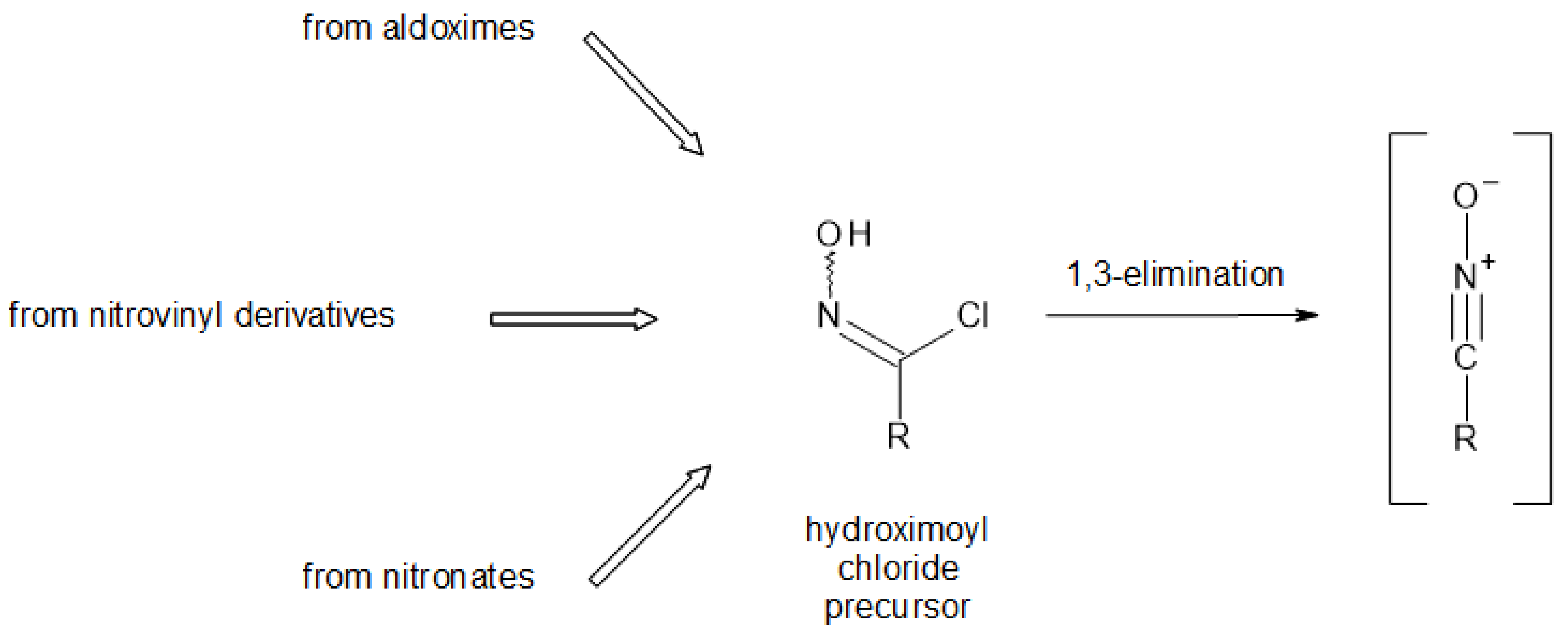
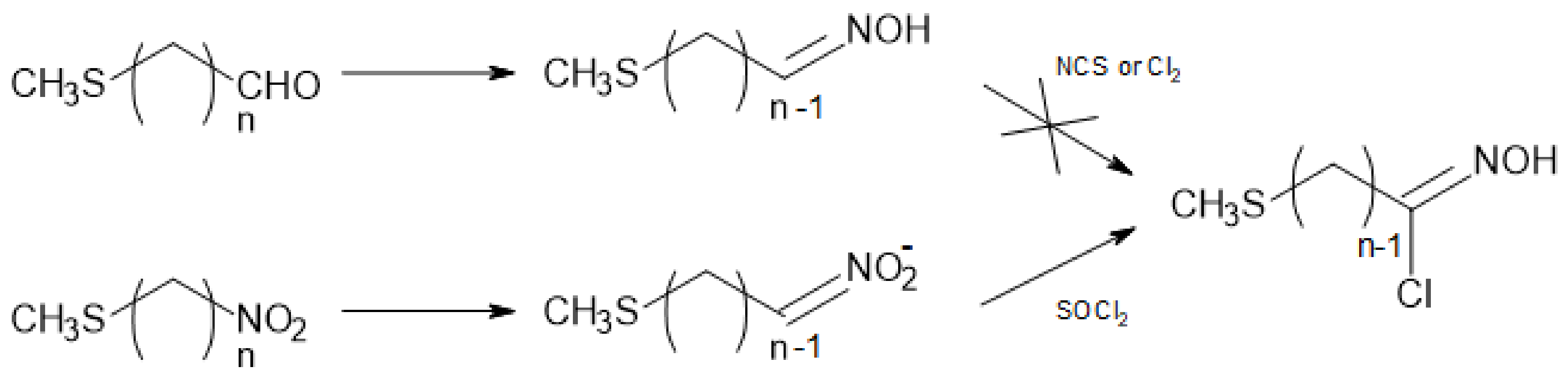
Scheme 1.
Nitronate method vs. oxime method to access hydroximoyl chlorides [20].
Scheme 1.
Nitronate method vs. oxime method to access hydroximoyl chlorides [20].
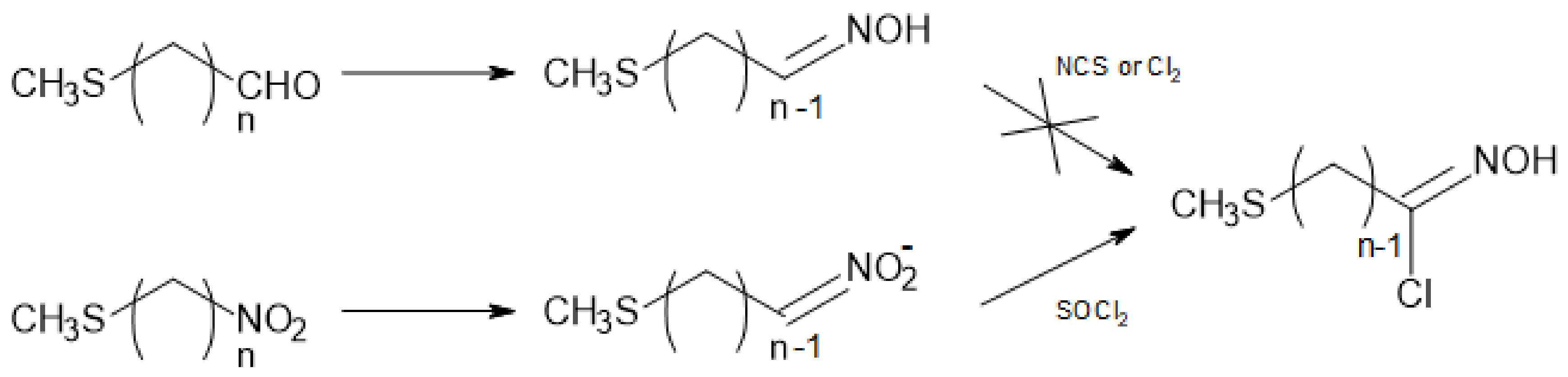
Scheme 2.
Access to small-sized ω-nitroalkyl methyl sulfides 5a–d.
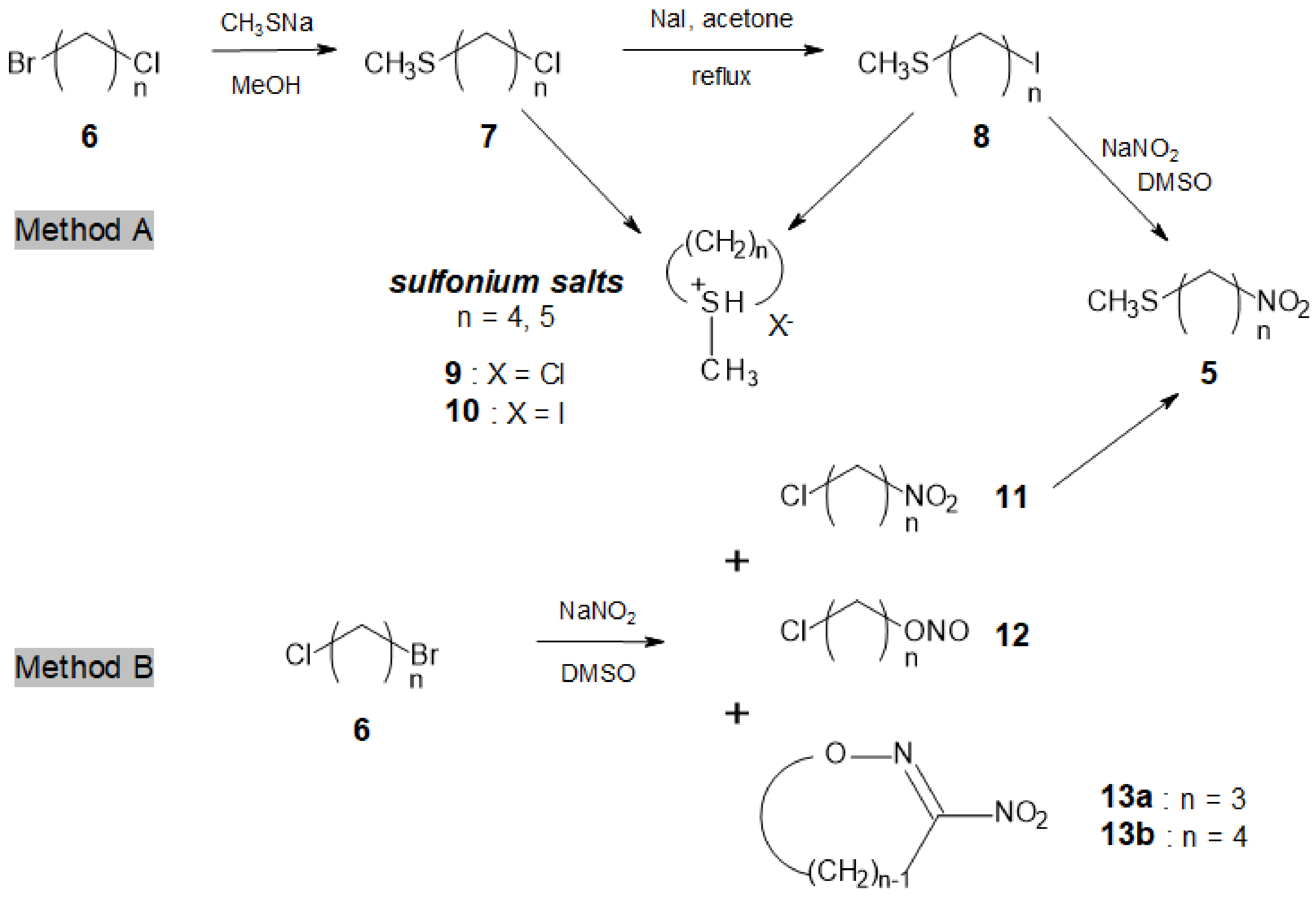
Scheme 3.
Access to longer chain ω-methylsulfanyl nitroalkanes 5e–g.
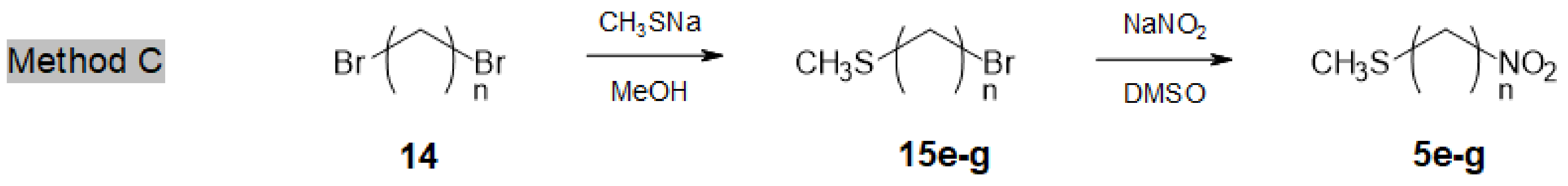
Scheme 4.
Coupling reaction to deliver tetra-O-acetylated ω-methylthioalkyl (Z)-thiohydroximates 18, followed by O-sulfation and deprotection to yield GLs 2.
Scheme 4.
Coupling reaction to deliver tetra-O-acetylated ω-methylthioalkyl (Z)-thiohydroximates 18, followed by O-sulfation and deprotection to yield GLs 2.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
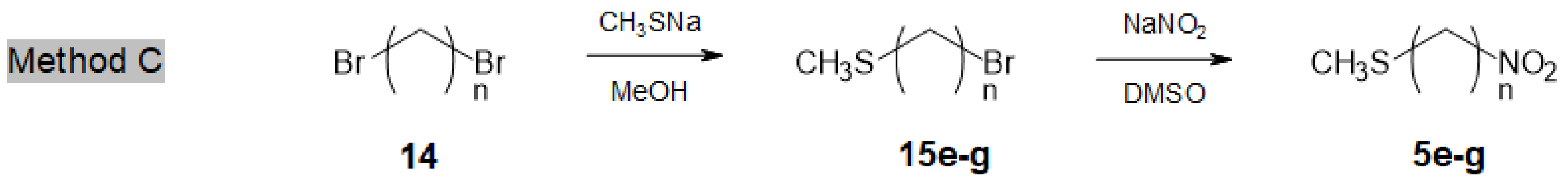{kind=link}
{kind=link}
Table 1.
Preparation of small-sized intermediates.
| Chain Size n | cpd n° | Yield (%) | cpd n° | Yield (%) | cpd n° | Yield (%) |
|---|---|---|---|---|---|---|
| 3 | 7a | 47 | 8a [26] | 68 | 5a | 60 |
| 4 | 7b | 72 | 8b | -- | 5b | -- |
| 5 | 7c | 86 | 8c | -- | 5c | -- |
| 6 | 7d | 94 | 8d | 56 | 5d | 74 |
Table 2.
Preparation of small-sized chloro-nitro derivatives 11a–d.
| Chain Size n | cpd n° | Reaction Time (h) | Nitro/Nitrite Ratio (%) | Isolated Yield (%) |
|---|---|---|---|---|
| 3 | 11a | 2.5 | 46/-- | 39–42 |
| 4 | 11b | 4.0 | 70/15 | 57–60 |
| 5 | 11c | 4.5 | 74/20 | 64–69 |
| 6 | 11d | 4.5 | 76/21 | 73–78 |
Table 3.
Alternative preparation (method B) of small-sized precursors 5.
| Chain Size n | cpd n° | T (°C) | Reaction Time (h) | Yield (%) |
|---|---|---|---|---|
| 3 | 5a | 25 | 40 | 25 |
| 4 | 5b | reflux | 1 | 40 |
| 5 | 5c | reflux | 3 | 87 |
| 6 | 5d | reflux | 4 | 93 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mavratzotis, M.; Cassel, S.; Montaut, S.; Rollin, P. ω-Methylsulfanylalkyl Glucosinolates: A General Synthetic Pathway. Molecules 2018, 23, 786. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23040786
AMA Style
Mavratzotis M, Cassel S, Montaut S, Rollin P. ω-Methylsulfanylalkyl Glucosinolates: A General Synthetic Pathway. Molecules. 2018; 23(4):786. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23040786
Chicago/Turabian StyleMavratzotis, Manolis, Stéphanie Cassel, Sabine Montaut, and Patrick Rollin. 2018. "ω-Methylsulfanylalkyl Glucosinolates: A General Synthetic Pathway" Molecules 23, no. 4: 786. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23040786