
Molecular Docking Evaluation of (E)-5-arylidene-2-thioxothiazolidin-4-one Derivatives as Selective Bacterial Adenylate Kinase Inhibitors

Abstract
:1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Ligand Preparation
4.2. Adenylate Kinases Structure Acquisition and Preparation as Receptors
4.3. Molecular Docking Protocol
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Achenbach-Richter, L.; Stetter, K.O.; Woese, C.R. A possible biochemical missing link among archaebacteria. Nature 1987, 327, 348–349. [Google Scholar] [CrossRef] [PubMed]
- Brügger, K.; Chen, L.; Stark, M.; Zibat, A.; Redder, P.; Ruepp, A.; Awayez, M.; She, Q.; Garrett, R.A.; Klenk, H.-P. The genome of Hyperthermus butylicus: A sulfur-reducing, peptide fermenting, neutrophilic Crenarchaeote growing up to 108 degrees C. Archaea 2007, 2, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, Y.; Ohashi, A.; Parks, D.H.; Yamauchi, T.; Tyson, G.W.; Hugenholtz, P. First genomic insights into members of a candidate bacterial phylum responsible for wastewater bulking. PeerJ 2015, 3, e740. [Google Scholar] [CrossRef] [PubMed]
- Barton, L.L.; Ritz, N.L.; Fauque, G.D.; Lin, H.C. Sulfur Cycling and the Intestinal Microbiome. Dig. Dis. Sci. 2017, 62, 2241–2257. [Google Scholar] [CrossRef] [PubMed]
- Graham, J.S.; Schoneboom, B.A. Historical perspective on effects and treatment of sulfur mustard injuries. Chem. Biol. Interact. 2013, 206, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Geng, J.; Liu, Y.; Yu, S.; Zhao, G. Thiadiazole-a Promising Structure in Medicinal Chemistry. ChemMedChem 2013, 8, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Schippa, S.; Conte, M.P. Dysbiotic events in gut microbiota: Impact on human health. Nutrients 2014, 6, 5786–5805. [Google Scholar] [CrossRef] [PubMed]
- Trotsko, N.; Przekora, A.; Zalewska, J.; Ginalska, G.; Paneth, A.; Wujec, M. Synthesis and in vitro antiproliferative and antibacterial activity of new thiazolidine-2,4-dione derivatives. J. Enzyme Inhib. Med. Chem. 2018, 33, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Garza, I.; Wallace, M.J.; Fernando, D.; Singh, A.; Lee, R.E.; Gerding, J.S.; Franklin, C.; Yendapally, R. Synthesis and Evaluation of Thiazolidine Amide and N-Thiazolyl Amide Fluoroquinolone Derivatives. Arch. Pharm. 2017, 350. [Google Scholar] [CrossRef] [PubMed]
- Mikaili, P.; Maadirad, S.; Moloudizargari, M.; Aghajanshakeri, S.; Sarahroodi, S. Therapeutic uses and pharmacological properties of garlic, shallot, and their biologically active compounds. Iran. J. Basic Med. Sci. 2013, 16, 1031–1048. [Google Scholar] [PubMed]
- Ahmed, S.; Zayed, M.; El-Messery, S.; Al-Agamy, M.; Abdel-Rahman, H. Design, Synthesis, Antimicrobial Evaluation and Molecular Modeling Study of 1,2,4-Triazole-Based 4-Thiazolidinones. Molecules 2016, 21, 568. [Google Scholar] [CrossRef] [PubMed]
- Devi, P.B.; Samala, G.; Sridevi, J.P.; Saxena, S.; Alvala, M.; Salina, E.G.; Sriram, D.; Yogeeswari, P. Structure-guided design of thiazolidine derivatives as Mycobacterium tuberculosis pantothenate synthetase inhibitors. ChemMedChem 2014, 9, 2538–2547. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P.; Evrin, C.; Briozzo, P.; Joly, N.; Bârzu, O.; Gilles, A.-M. Structural and Functional Characterization of Escherichia coli UMP Kinase in Complex with Its Allosteric Regulator GTP. J. Biol. Chem. 2008, 283, 36011–36018. [Google Scholar] [CrossRef] [PubMed]
- Bucurenci, N.; Sakamoto, H.; Briozzo, P.; Palibroda, N.; Serina, L.; Sarfati, R.S.; Labesse, G.; Briand, G.; Danchin, A.; Bărzu, O.; et al. CMP kinase from Escherichia coli is structurally related to other nucleoside monophosphate kinases. J. Biol. Chem. 1996, 271, 2856–2862. [Google Scholar] [CrossRef] [PubMed]
- Wiesmuller, L.; Noegel, A.A.; Barzu, O.; Gerisch, G.; Schleicher, M. cDNA-derived sequence of UMP-CMP kinase from Dictyostelium discoideum and expression of the enzyme in Escherichia coli. J. Biol. Chem. 1990, 265, 6339–6345. [Google Scholar] [PubMed]
- Yamada, M.; Sugahara, M.; Hishitani, Y.; Nobumoto, M.; Nakazawa, A. Isolation and characterization of mutated mitochondrial GTP:AMP phosphotransferase. J. Mol. Biol. 1998, 280, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Panayiotou, C.; Solaroli, N.; Johansson, M.; Karlsson, A. Evidence of an intact N-terminal translocation sequence of human mitochondrial adenylate kinase 4. Int. J. Biochem. Cell Biol. 2010, 42, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Xu, H.; Wei, Z.; Wang, Y.; Lin, Y.; Gong, W. Crystal structure of human adenylate kinase 4 (L171P) suggests the role of hinge region in protein domain motion. Biochem. Biophys. Res. Commun. 2008, 379, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Saint Girons, I.; Gilles, A.M.; Margarita, D.; Michelson, S.; Monnot, M.; Fermandjian, S.; Danchin, A.; Barzu, O. Structural and catalytic characteristics of Escherichia coli adenylate kinase. J. Biol. Chem. 1987, 262, 622–629. [Google Scholar] [PubMed]
- Berry, M.B.; Meador, B.; Bilderback, T.; Liang, P.; Glaser, M.; Phillips, G.N. The closed conformation of a highly flexible protein: The structure of E. coli adenylate kinase with bound AMP and AMPPNP. Proteins 1994, 19, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.G.; Griffith, O.W.; Meister, A. Inhibition of carbamyl phosphate synthetase by P1, P5-di(adenosine 5′)-pentaphosphate: Evidence for two ATP binding sites. J. Biol. Chem. 1977, 252, 3558–3560. [Google Scholar] [PubMed]
- Rose, T.; Glaser, P.; Surewicz, W.K.; Mantsch, H.H.; Reinstein, J.; Le Blay, K.; Gilles, A.M.; Barzu, O. Structural and functional consequences of amino acid substitutions in the second conserved loop of Escherichia coli adenylate kinase. J. Biol. Chem. 1991, 266, 23654–23659. [Google Scholar] [PubMed]
- Müller, C.W.; Schlauderer, G.J.; Reinstein, J.; Schulz, G.E. Adenylate kinase motions during catalysis: An energetic counterweight balancing substrate binding. Structure 1996, 4, 147–156. [Google Scholar] [CrossRef]
- Rundqvist, L.; Aden, J.; Sparrman, T.; Wallgren, M.; Olsson, U.; Wolf-Watz, M. Noncooperative folding of subdomains in adenylate kinase. Biochemistry 2009, 48, 1911–1927. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Makowski, L. Fine structure of conformational ensembles in adenylate kinase. Proteins 2018, 86, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Rogne, P.; Rosselin, M.; Grundstrom, C.; Hedberg, C.; Sauer, U.H.; Wolf-Watz, M. Molecular mechanism of ATP versus GTP selectivity of adenylate kinase. Proc. Natl. Acad. Sci. USA 2018, 115, 3012–3017. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, J.W.; Lee, S.M.; Kim, H.J.; Lee, K.S.; Park, C.; Choe, I.S. Cloning and Expression of Human Adenylate Kinase 2 Isozymes: Differential Expression of Adenylate Kinase 1 and 2 in Human Muscle Tissues. J. Biochem. 1998, 123, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Panayiotou, C.; Solaroli, N.; Karlsson, A. The many isoforms of human adenylate kinases. Int. J. Biochem. Cell Biol. 2014, 49, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Plant, T.; Journal, P. Identification of a Gene Encoding Adenylate Kinase Involved in Antifungal Activity Expression of the Biocontrol Strain Burkholderia pyrrocinia CH-67. Plant Pathol. J. 2012, 28, 373–380. [Google Scholar]
- Russell, P.J.; Conner, J.; Sisson, S. Sulfur specifically inhibits adenylate kinase in assays for creatine kinase. Clin. Chem. 1984, 30, 1555–1557. [Google Scholar] [PubMed]
- Russell, P.J.; Williams, A.; Avtla, D.; Chinn, E.; Taulane, J.P. Characteristics of Rabbit Muscle Adenylate Kinase Inhibition by Sulfur and Recovery by Dithiothreitol. J. Enzyme Inhib. 1995, 9, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Ionescu, M.I.; Radulescu, A.Z.; Banciu, H.L.; Ghiran, D. Inhibition of E.coli adenylate kinase by some 5-aryliden-2-thioxo-thiazolydin-4-one derivatives. Clujul Med. 2005, 3, 638–641. [Google Scholar]
- Ionescu, M.I.; Costache, A.Z.; Oniga, O.; Banciu, H.L.; Lupan, I.; Lupan, I. Inhibition of Streptococcus pneumoniae adenylate kinase by some 5-arylidene-thiazolidin-4-on-2-thione derivates. Rom. Rev. Lab. Med. 2013, 21, 93–99. [Google Scholar] [CrossRef]
- Oniga, O.; Moldovan, C.; Muntean, A.; Verite, P.; Ionescu, M.; Tiperciuc, B. Synthesis and antibacterial activity of some 5-aryliden-2-thioxo-thiazolydin-4-one. Rev. Med. Farm. 2004, 50, 183–186. [Google Scholar]
- Schrank, T.P.; Bolen, D.W.; Hilser, V.J. Rational modulation of conformational fluctuations in adenylate kinase reveals a local unfolding mechanism for allostery and functional adaptation in proteins. Proc. Natl. Acad. Sci. USA 2009, 106, 16984–16989. [Google Scholar] [CrossRef] [PubMed]
- Thach, T.T.; Luong, T.T.; Lee, S.; Rhee, D.K. Adenylate kinase from Streptococcus pneumoniae is essential for growth through its catalytic activity. FEBS Open Bio 2014, 4, 672–682. [Google Scholar] [CrossRef] [PubMed]
- Berry, M.B.; Phillips, G.N.J. Crystal structures of Bacillus stearothermophilus adenylate kinase with bound Ap5A, Mg2+ Ap5A, and Mn2+ Ap5A reveal an intermediate lid position and six coordinate octahedral geometry for bound Mg2+ and Mn2+. Proteins Struct. Funct. Genet. 1998, 32, 276–288. [Google Scholar] [CrossRef]
- Bae, E.; Phillips, G.N. Structures and analysis of highly homologous psychrophilic, mesophilic, and thermophilic adenylate kinases. J. Biol. Chem. 2004, 279, 28202–28208. [Google Scholar] [CrossRef] [PubMed]
- Henzler-Wildman, K.A.; Thai, V.; Lei, M.; Ott, M.; Wolf-Watz, M.; Fenn, T.; Pozharski, E.; Wilson, M.A.; Petsko, G.A.; Karplus, M.; et al. Intrinsic motions along an enzymatic reaction trajectory. Nature 2007, 450, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Criswell, A.R.; Bae, E.; Stec, B.; Konisky, J.; Phillips, G.N., Jr. Structures of Thermophilic and Mesophilic Adenylate Kinases from the Genus Methanococcus. J. Mol. Biol. 2003, 330, 1087–1099. [Google Scholar] [CrossRef]
- Vonrhein, C.; Bönisch, H.; Schäfer, G.; Schulz, G.E. The structure of a trimeric archaeal adenylate kinase. J. Mol. Biol. 1998, 282, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Goodsell, D.S.; Olson, A.J. Automated docking of substrates to proteins by simulated annealing. Proteins Struct. Funct. Genet. 1990, 8, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
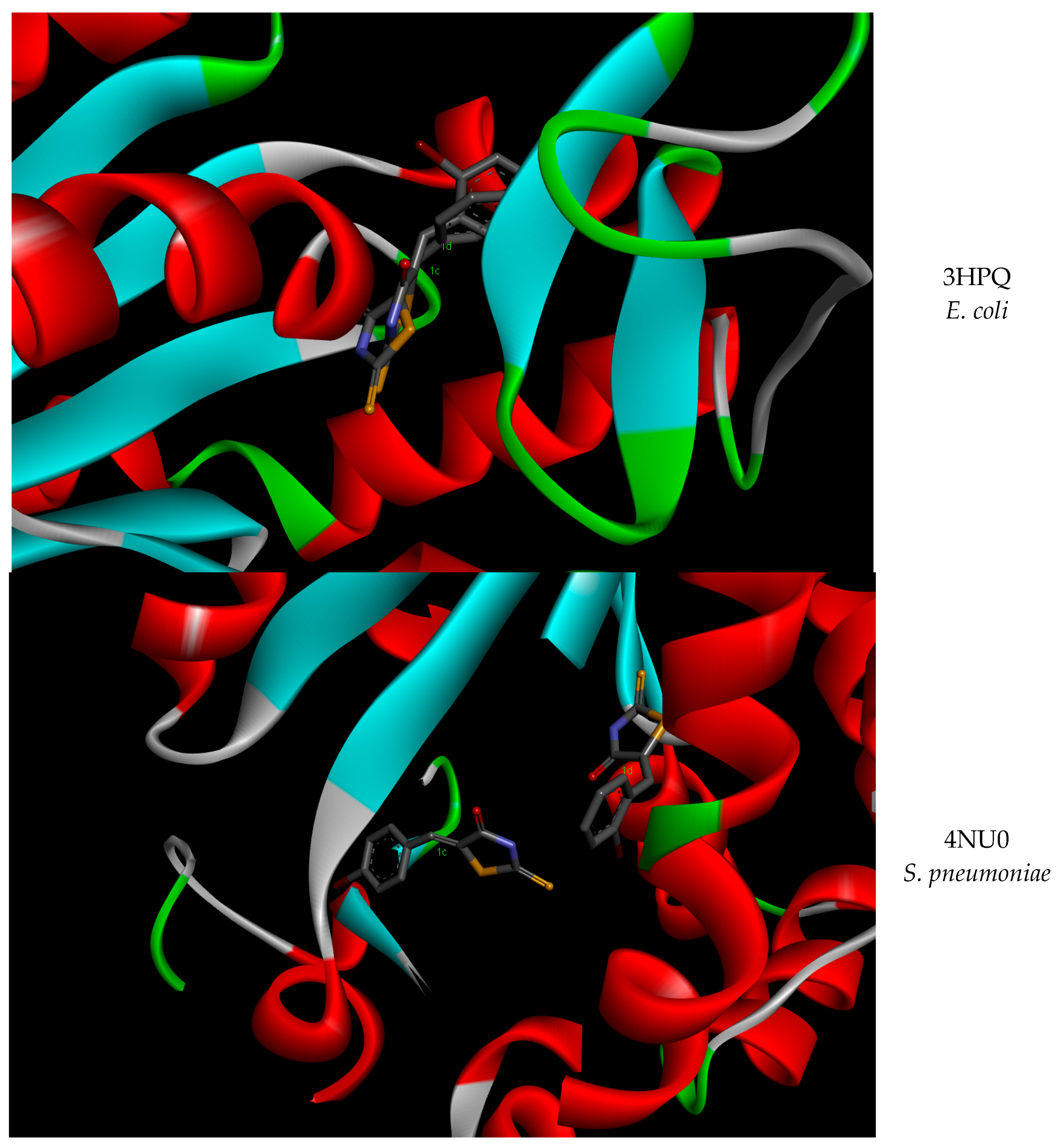

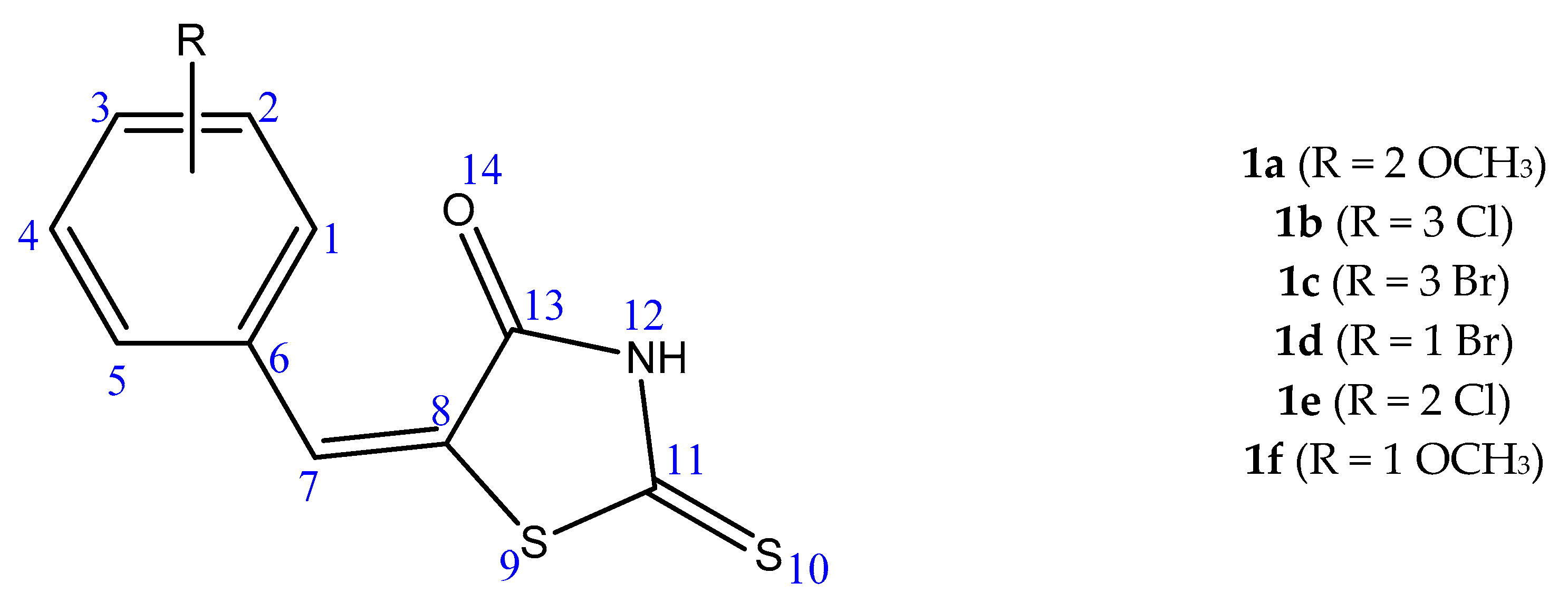

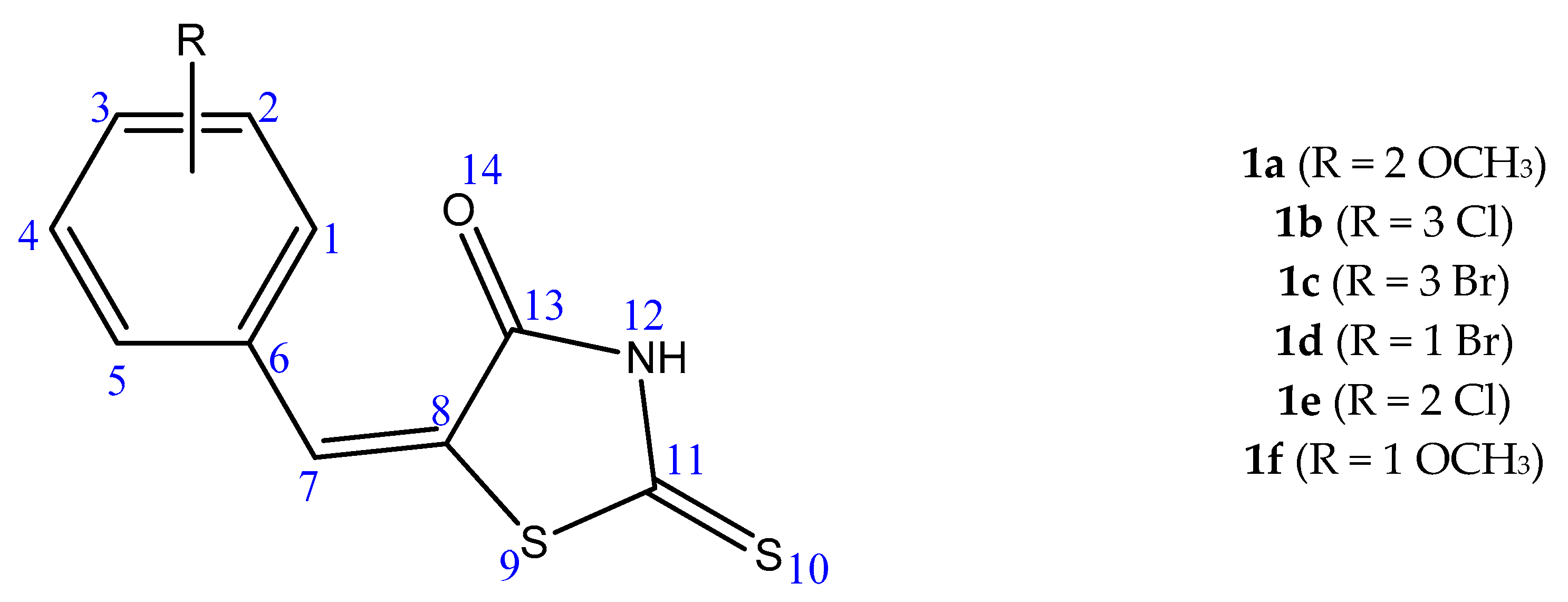
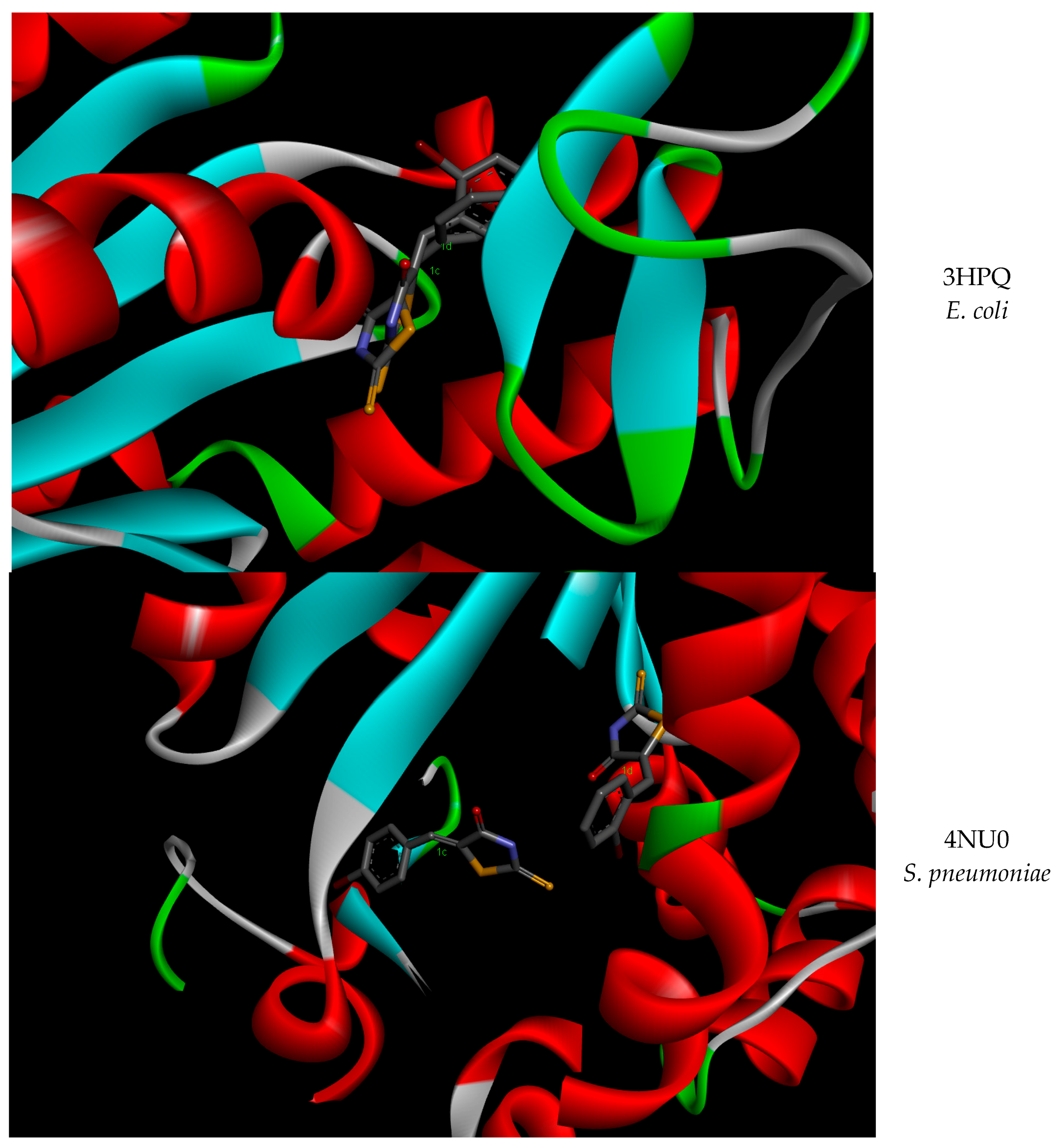{kind=link}
{kind=link}
{kind=link}
| Strain | AK Conformation (PDB ID 1) | ∆G 2 (kcal/mol) Mean (±SD 3) | Ki 4 (µM) Mean (±SD) | p-Value | |
|---|---|---|---|---|---|
| ∆G (kcal/mol) | Ki (µM) | ||||
| E. coli | O 5a (4AKE) | −5.26 (±1.7) | 51.32 (±27.53) | 0.020285 | 0.002378 |
| C 5b (1HPQ) | −7.19 (±0.24) | 5.75 (±2.59) | |||
| S. pneumoniae | O (4NTZ) | −5.59 (±2.48) | 18.12 (±14.01) | 0.2097 | 0.177056 |
| C (4NU0) | −6.97 (±0.42) | 9.23 (±4.55) | |||
| A.aeolicus | O (2RH5) | −5.81 (±0.32) | 59.53 (±22.31) | 0.000898 | 0.000854 |
| C (2RGX) | −6.77 (±0.4) | 13.21 (±9.37) | |||
| S. acidocaldarius6 | O (1NKS/chain A) | −5.87 (±0.09) | 50.61 (±7.75) | 0.000975 | 0.000341 |
| O/C (1NKS/Chain C) | −5.91 (±.44) | 56.04 (±32.03) | |||
| C (1NKS/Chains F) | −6.46 (±0.31) | 34.77 (±14.19) | |||
| M. voltae | O (1KHT/Chain A) | −5.69 (±0.35) | 81.06 (±65.92) | 0.000054 | 0.024538 |
| C (1KHT/Chain B) | −6.88 (±0.26) | 9.73 (±4.31) | |||
| Human–AK4 | O (2AR7) | −6.11 (±0.22) | 34.48 (±13.15) | 0.056913 | 0.034455 |
| C (BBW) | −6.52 (±0.27) | 17.85 (±6.62) | |||
| Human–AK5 | O (2BWJ/Chain A) | −6.41 (±0.27) | 22.19 (±13.11) | 0.010154 | 0.020725 |
| C (2BWJ/Chain B) | −5.77 (±0.41) | 70.03 (±40.66) | |||
| Derivative | 1 ∆G/Ki | t-Value | p-Value | 2 ∆µ | 3 CI 95% |
|---|---|---|---|---|---|
| 1a | ∆G | 0.09867 | 0.923565 | 0.0357 | −0.7821 ≤ ∆µ ≥ 0.8534 |
| Ki | 1.06499 | 0.314617 | 18.4687 | −20.7608 ≤ ∆µ ≥ 57.6981 | |
| 1b | ∆G | 3.84529 | 0.003935 | 0.933 | 0.3841 ≤ ∆µ ≥ 1.4819 |
| Ki | 2.4127 | 0.039076 | 46.6247 | 2.9091 ≤ ∆µ ≥ 90.3402 | |
| 1c | ∆G | 4.49226 | 0.001506 | 0.958 | 0.4756 ≤ ∆µ ≥ 1.4404 |
| Ki | 2.2898 | 0.04779 | 33.761 | 0.4075 ≤ ∆µ ≥ 67.1145 | |
| 1d | ∆G | 1.8825 | 0.09634 | 0.66 | −0.1485 ≤ ∆µ ≥ 1.4685 |
| Ki | 1.87666 | 0.097405 | 21.2967 | −4.8723 ≤ ∆µ ≥ 47.4 | |
| 1e | ∆G | 2.31486 | 0.04587 | 0.8183 | 0.0186 ≤ ∆µ ≥ 1.6180 |
| Ki | 1.53788 | 0.158459 | 58.1810 | −27.4010 ≤ ∆µ ≥ 143.7630 | |
| 1f | ∆G | 1.08505 | 0.309513 | 0.325 | −0.3657 ≤ ∆µ ≥1.0157 |
| Ki | 1.21529 | 0.258901 | 27.4042 | −24.5951 ≤ ∆µ ≥ 79.4034 |
| Strain | PDB ID | 1c | 1d | ||
|---|---|---|---|---|---|
| ∆G 1 | Ki 2 | ∆G | Ki | ||
| E. coli | 4AKE | −5.82 | 54.17 | −6.65 | 13.26 |
| 3HPQ | −7.24 | 4.97 | −7.27 | 4.68 | |
| S. pneumoniae | 4NTZ | −1.91 | 37.12 | −7.33 | 4.22 |
| 4NT0 | −7.13 | 5.92 | −7.74 | 2.13 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ionescu, M.I.; Oniga, O. Molecular Docking Evaluation of (E)-5-arylidene-2-thioxothiazolidin-4-one Derivatives as Selective Bacterial Adenylate Kinase Inhibitors. Molecules 2018, 23, 1076. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23051076
Ionescu MI, Oniga O. Molecular Docking Evaluation of (E)-5-arylidene-2-thioxothiazolidin-4-one Derivatives as Selective Bacterial Adenylate Kinase Inhibitors. Molecules. 2018; 23(5):1076. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23051076
Chicago/Turabian StyleIonescu, Mihaela Ileana, and Ovidiu Oniga. 2018. "Molecular Docking Evaluation of (E)-5-arylidene-2-thioxothiazolidin-4-one Derivatives as Selective Bacterial Adenylate Kinase Inhibitors" Molecules 23, no. 5: 1076. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23051076