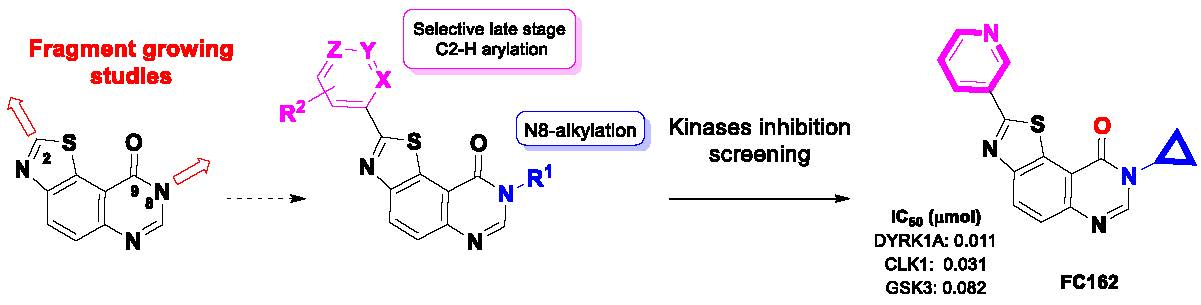
Development of Kinase Inhibitors via Metal-Catalyzed C–H Arylation of 8-Alkyl-thiazolo[5,4-f]-quinazolin-9-ones Designed by Fragment-Growing Studies

 , , and
, , and
Abstract
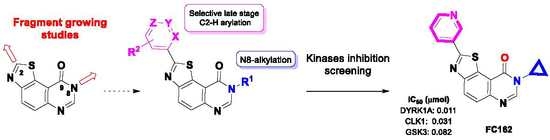
:
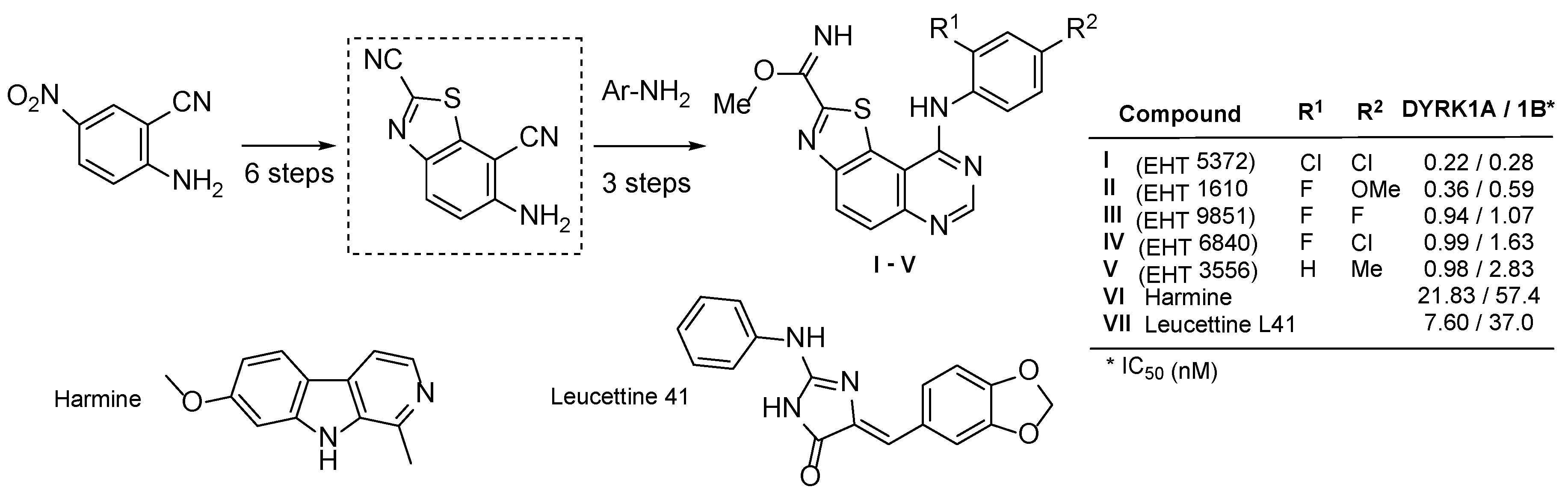
1. Introduction
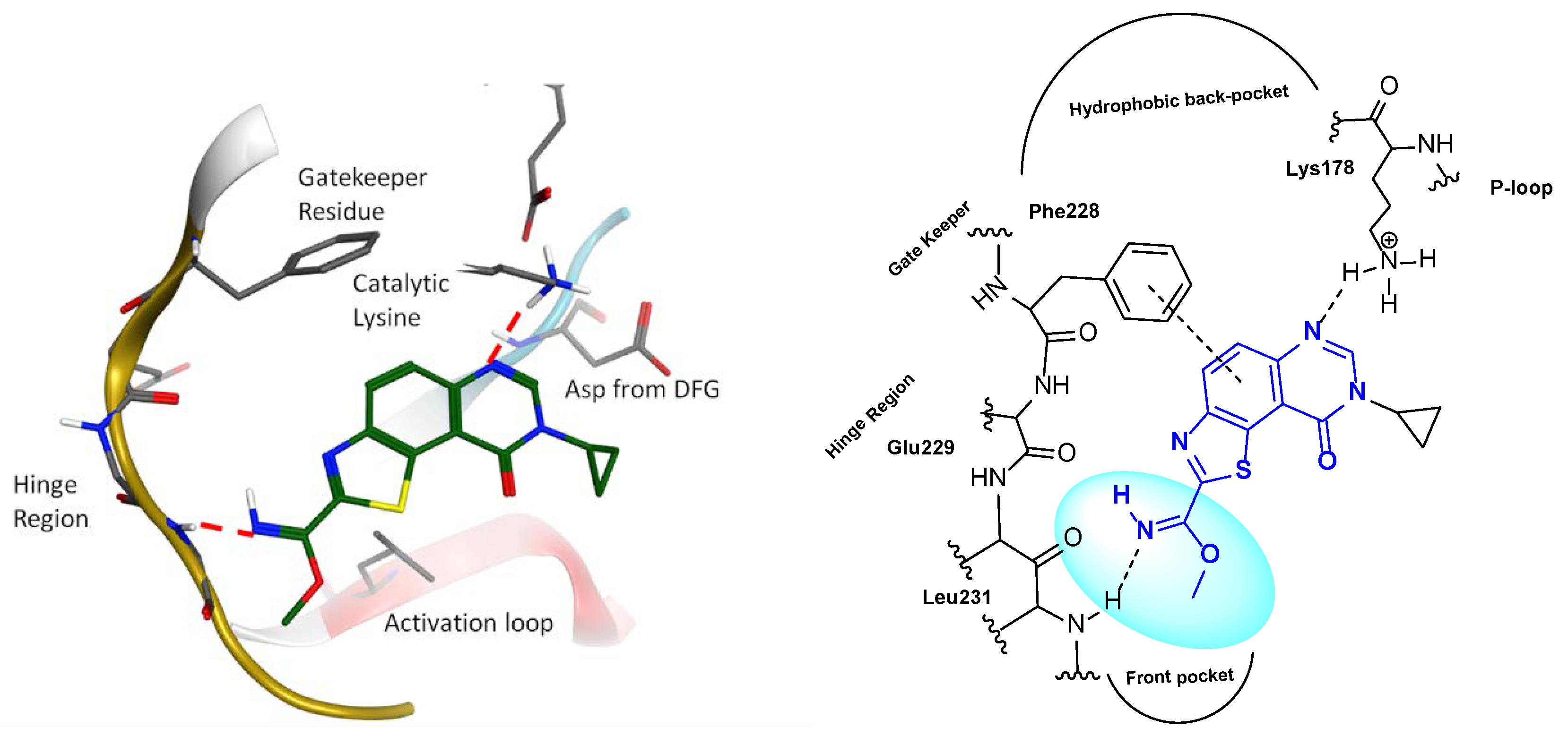
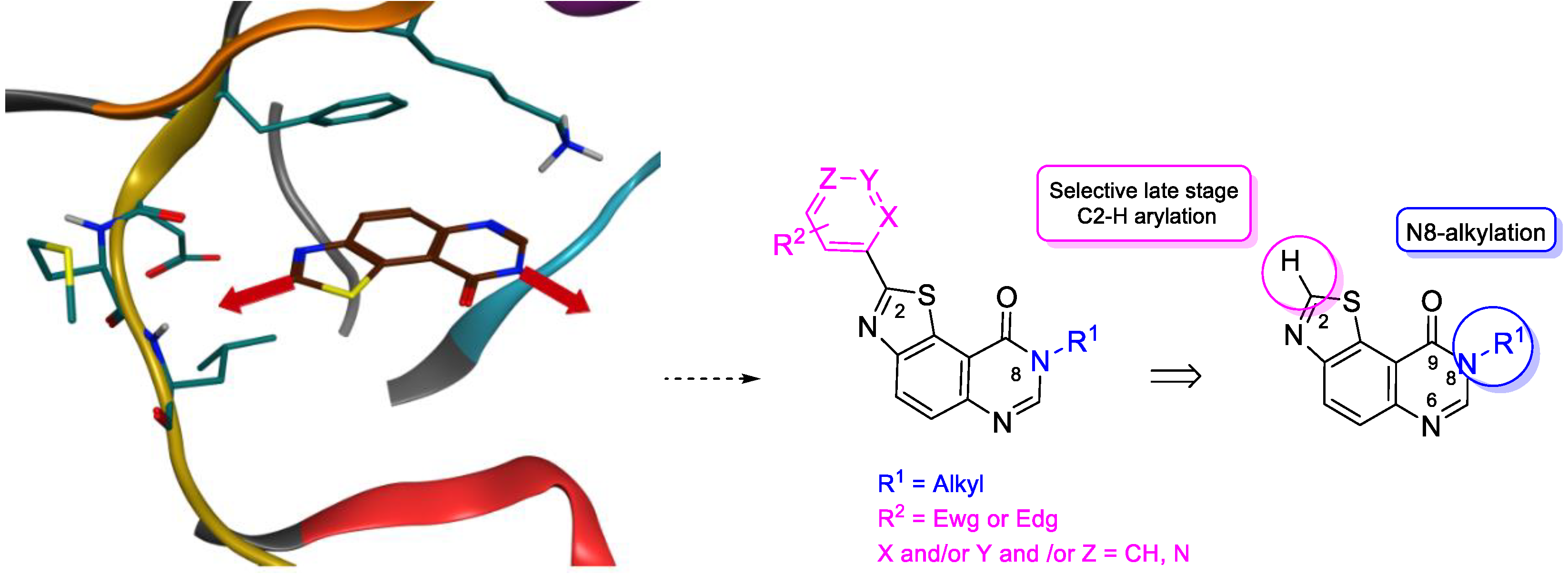
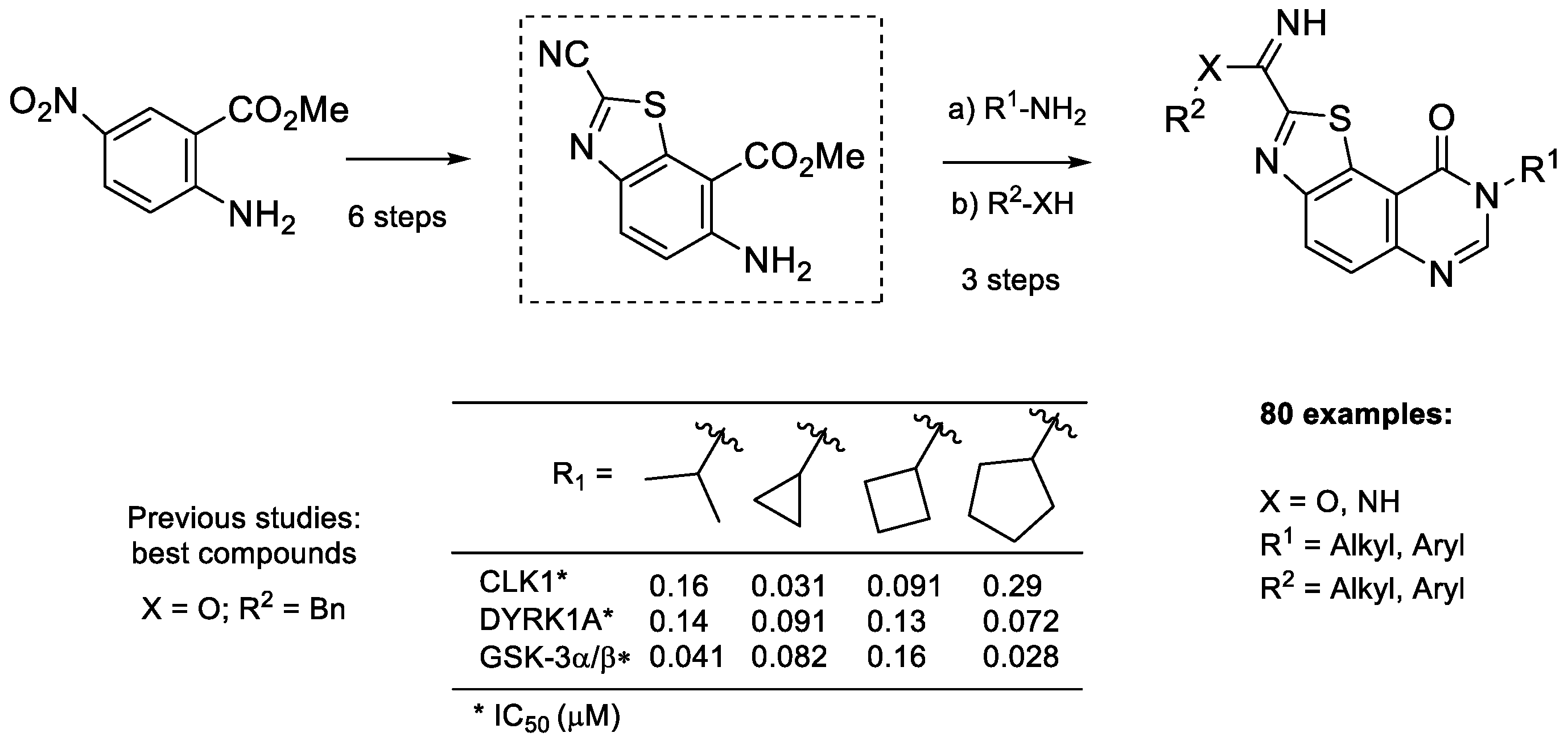
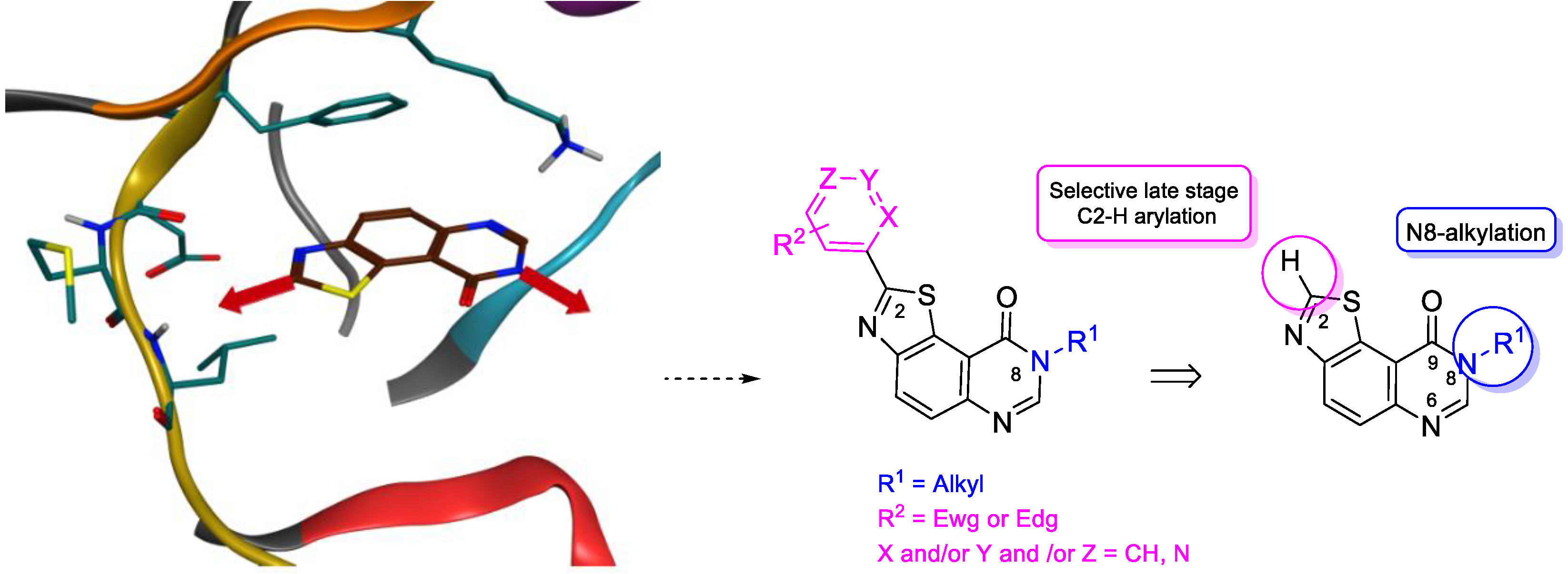
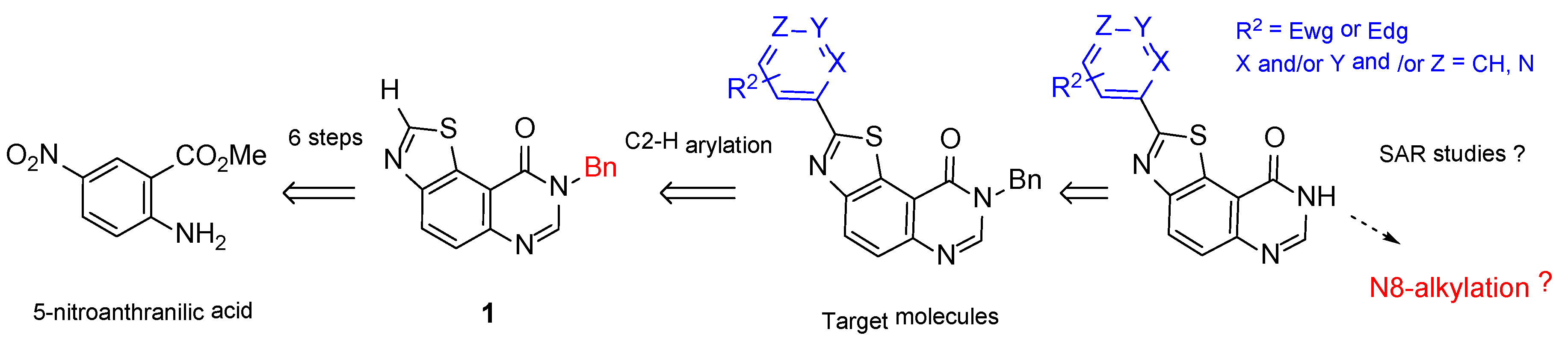
2. Results and Discussion
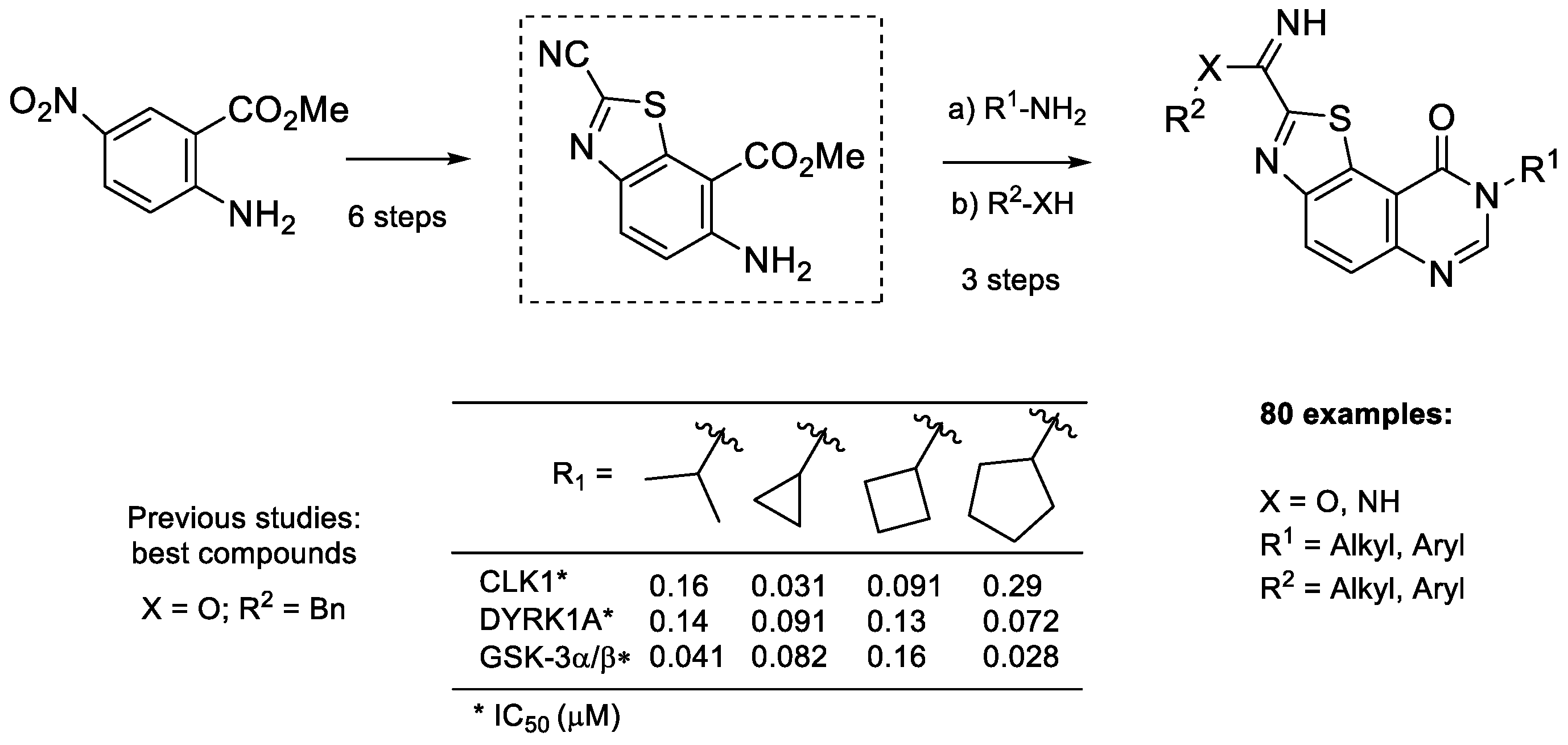
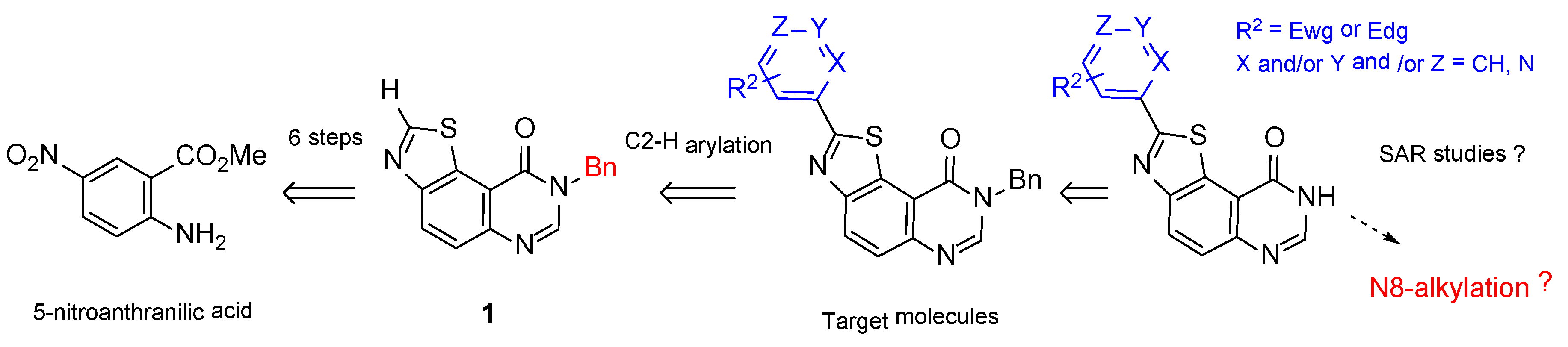
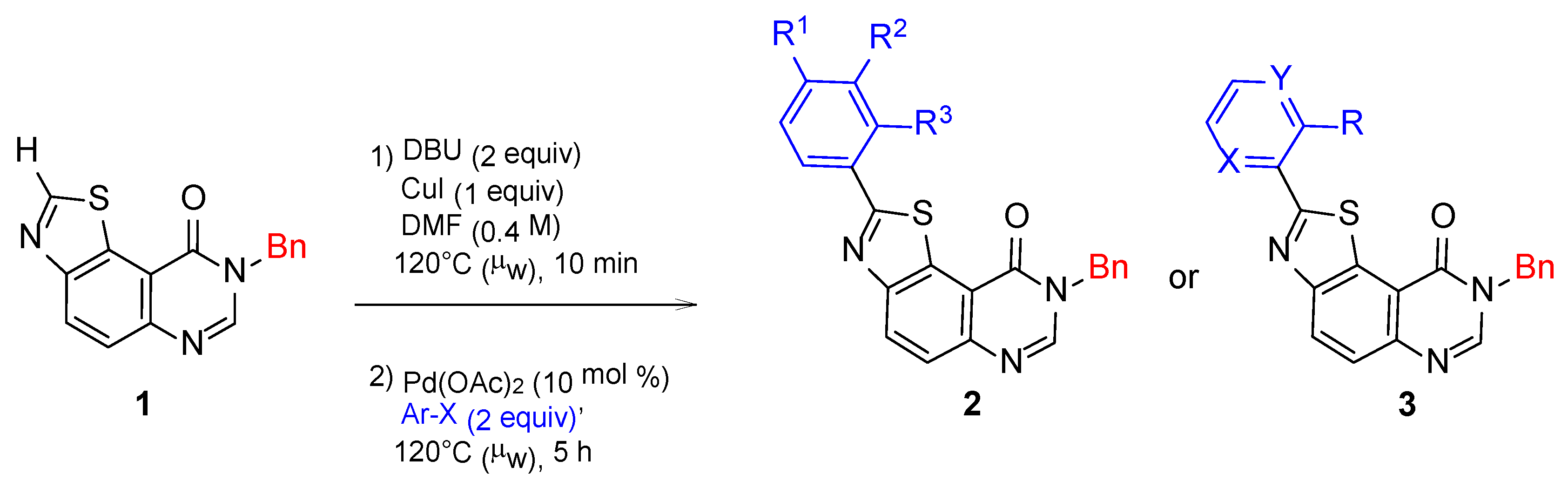
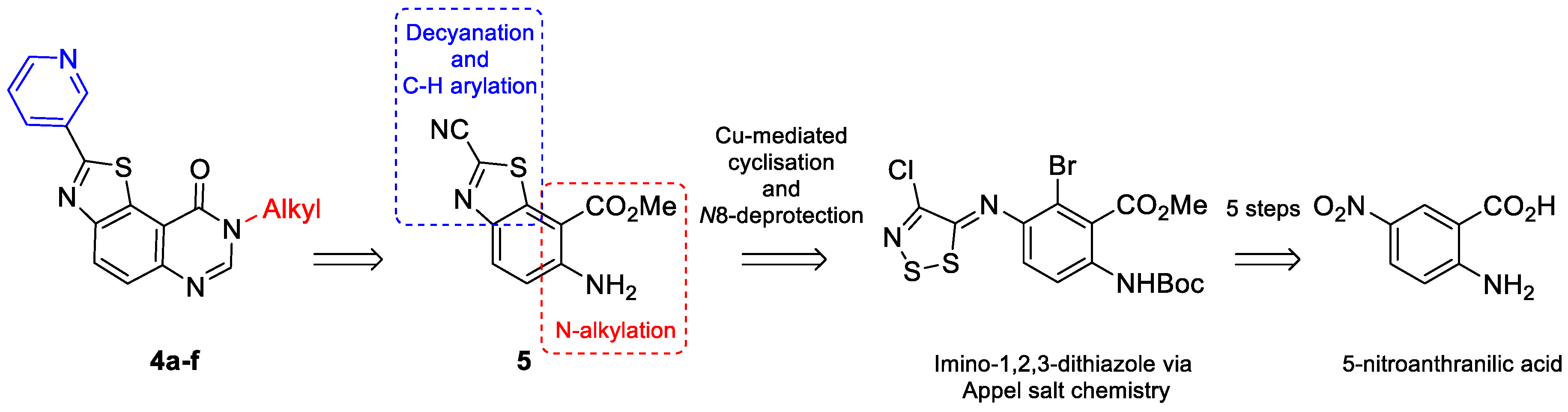
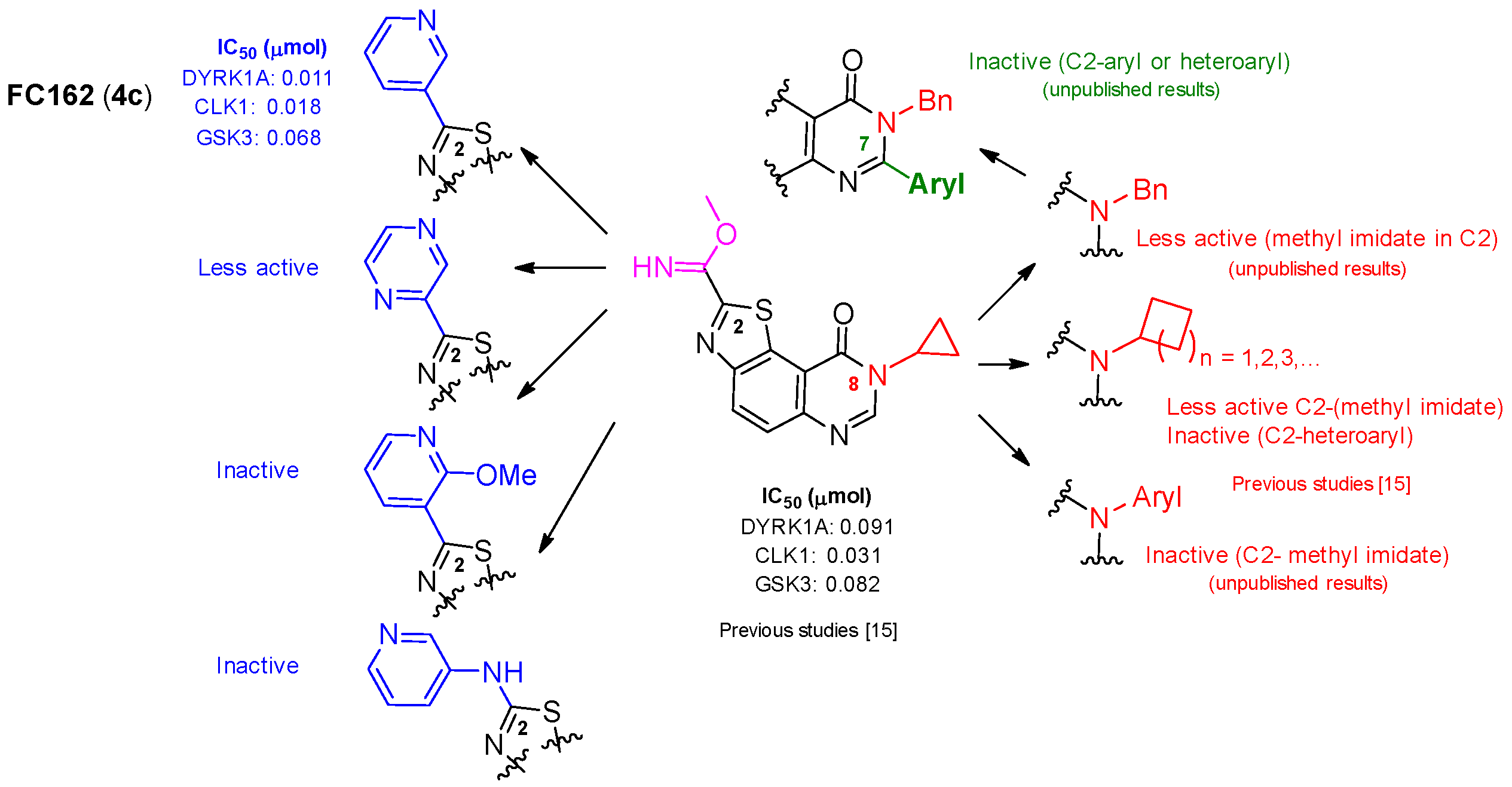
2.1. Chemistry
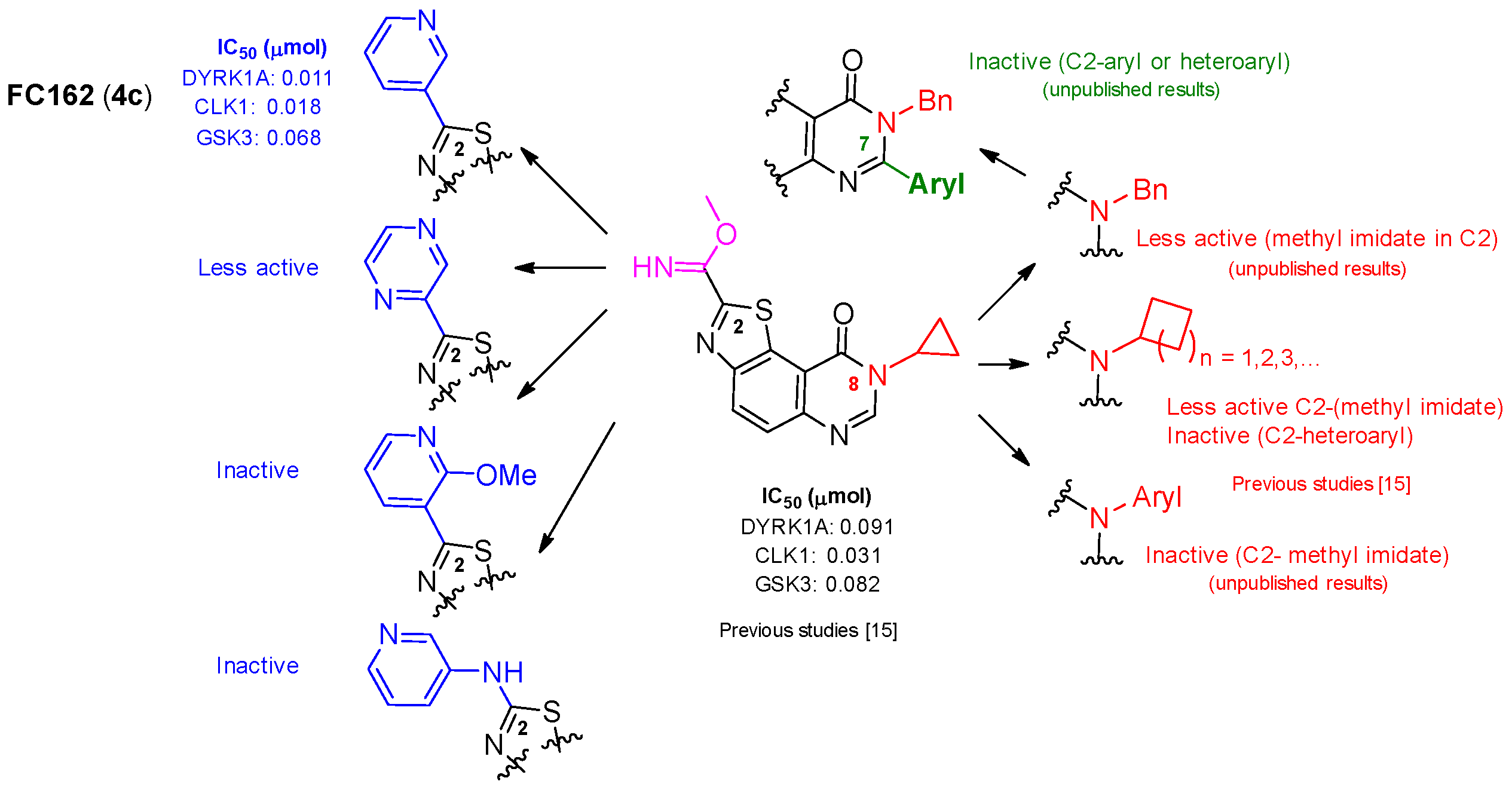
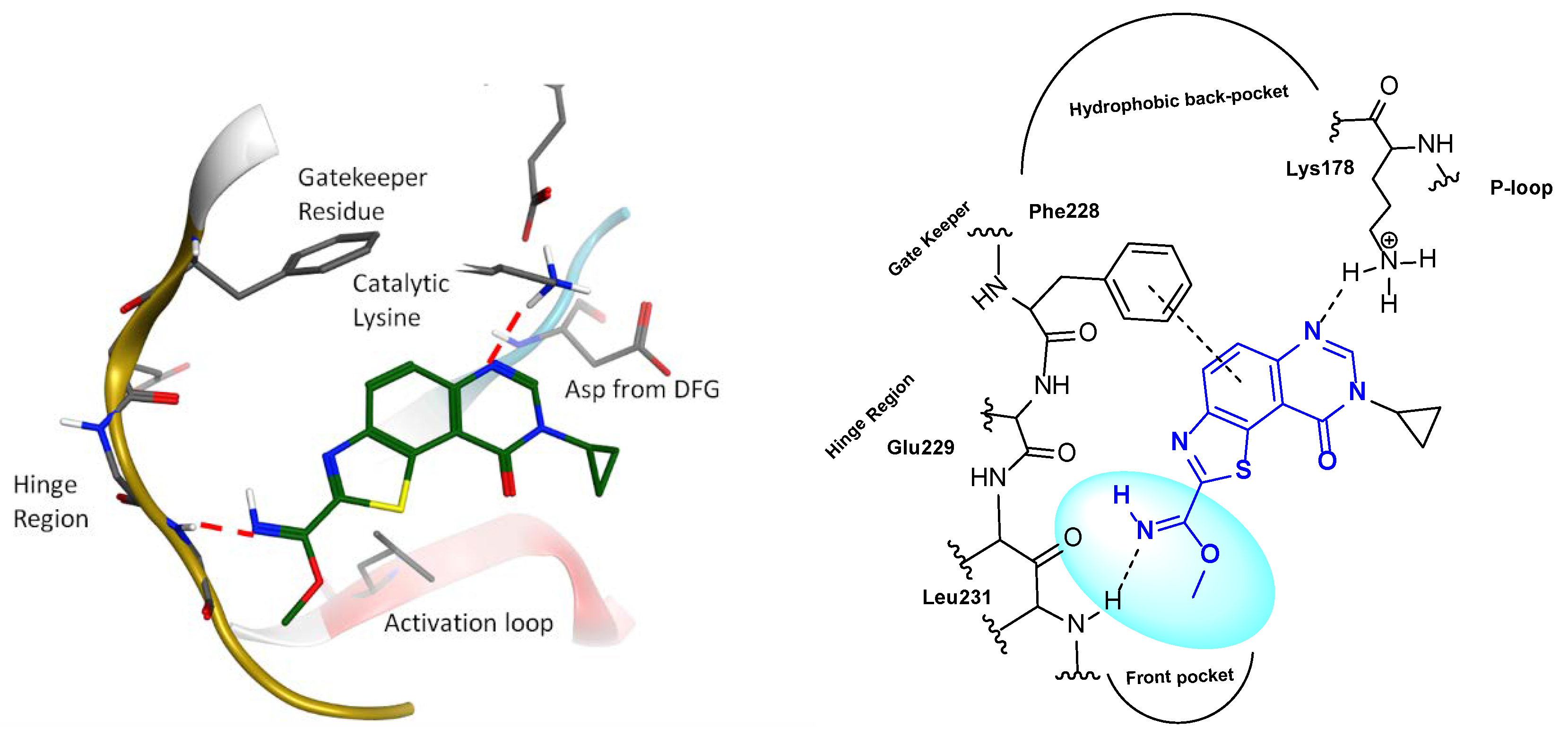
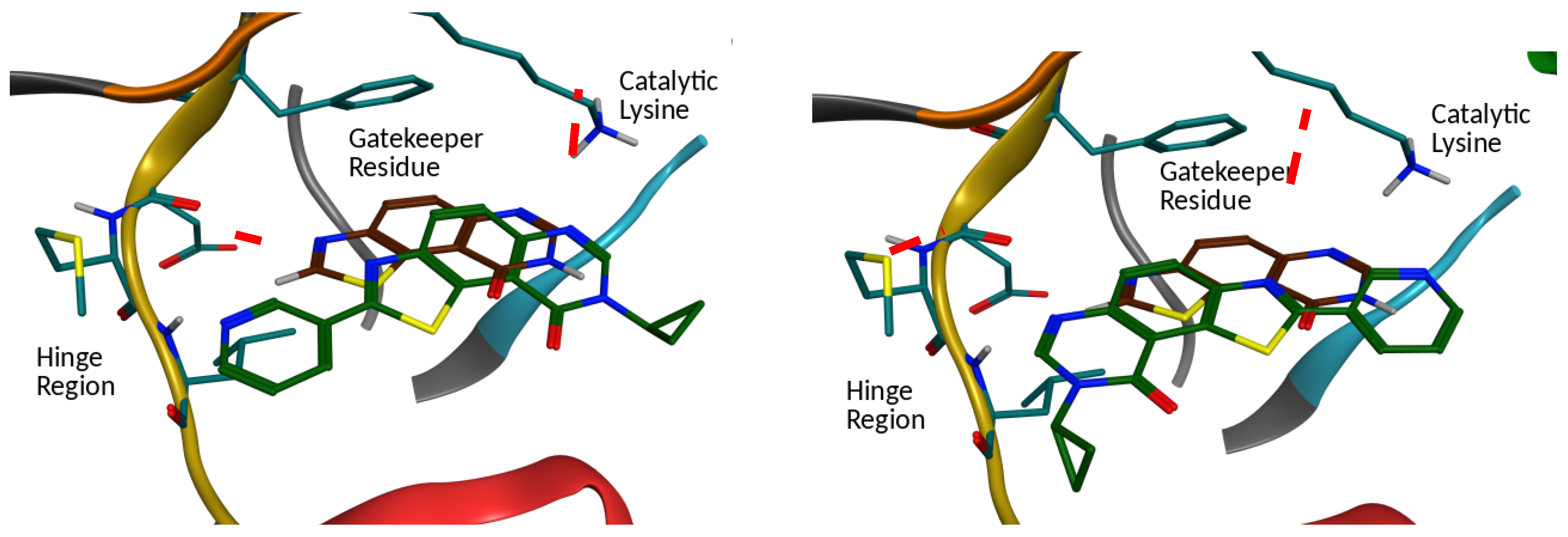
2.2. Kinase Profiling
3. Material and Methods
3.1. General Information
3.2. Chemistry
3.2.1. General Procedure for the Synthesis of 8-Alkyl-thiazolo[5,4-f]quinazolin-9(8H)-one (8a–f) from 8-Alkyl-9-oxo-8,9-dihydrothiazolo[5,4-f]quinazoline-2-carbonitrile (7a–f)
3.2.2. General Procedure for the Synthesis of 8-Alkyl-2-(pyridin-3-yl)thiazolo[5,4-f]quinazolin-9(8H)-one (4a–f) from 8-Alkyl-thiazolo[5,4-f]quinazolin-9(8H)-one (8a–f)
3.3. In Vitro Kinase Preparation and Assays
3.3.1. Buffers
3.3.2. Kinase Preparations and Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATP | adenosine triphosphate |
| CMGC group | group of kinases including cyclin-dependent kinases (CDKs), mitogen-activated protein kinases (MAP kinases), glycogen synthase kinases (GSK) and Cdc2-like kinases (CLKs) |
| DBU | 1,8-diazabicyclo[5.4.0]undec-7-ene |
| DMF | N,N-dimethylformamide |
| DMFDMA | N,N-dimethylformamide dimethyl acetal |
| NBS | N-bromosuccinimide |
| TBD | 1,5,7-triazabicyclo[4.4.0]dec-5-ene |
| SAR | structure–activity relationship |
References
- Bénéteau, V.; Besson, T. Synthesis of novel pentacyclic pyrrolothiazolobenzoquinolinones, analogs of natural marine alkaloids. Tetrahedron Lett. 2001, 42, 2673–2676. [Google Scholar] [CrossRef]
- Lamazzi, C.; Chabane, H.; Thiéry, V.; Pierre, A.; Léonce, S.; Pfeiffer, B.; Renard, P.; Guillaumet, G.; Besson, T. Synthesis and cytotoxic evaluation of novel thiazolocarbazoles. J. Enz. Inhib. Med. Chem. 2002, 17, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Frère, S.; Thiéry, V.; Bailly, C.; Besson, T. Novel 6-substituted benzothiazol-2-yl indolo[1,2-c]quinazolines and benzimidazo[1,2-c]quinazolines. Tetrahedron 2003, 59, 773–779. [Google Scholar] [CrossRef]
- Chabane, H.; Pierre, A.; Léonce, S.; Pfeiffer, B.; Renard, P.; Thiéry, V.; Guillaumet, G.; Besson, T. Synthesis and cytotoxic activity of thiazolofluorenone derivatives. J. Enzym. Inhib. Med. Chem. 2004, 19, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Testard, A.; Picot, L.; Fruitier-Arnaudin, I.; Piot, J.M.; Chabane, H.; Domon, L.; Thiéry, V.; Besson, T. Microwave-assisted synthesis of novel thiazolocarbazoles and evaluation as potential anticancer agents. Part III. J. Enz. Inhib. Med. Chem. 2004, 19, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Logé, C.; Testard, A.; Thiéry, V.; Lozach, O.; Blairvacq, M.; Robert, J.-M.; Meijer, L.; Besson, T. Novel 9-oxo-thiazolo[5,4-f]quinazoline-2-carbonitrile derivatives as dual cyclin-dependent kinase 1 (CDK1)/glycogen synthase kinase-3 (GSK-3) inhibitors: Synthesis, biological evaluation and molecular modeling studies. Eur. J. Med. Chem. 2008, 43, 1469–1477. [Google Scholar] [CrossRef] [PubMed]
- Testard, A.; Logé, C.; Léger, B.; Robert, J.-M.; Lozach, O.; Blairvacq, M.; Meijer, L.; Thiéry, V.; Besson, T. Thiazolo[5,4-f]quinazolin-9-ones, inhibitors of glycogen synthase kinase-3. Bioorg. Med. Chem. Lett. 2006, 16, 3419–3423. [Google Scholar] [CrossRef] [PubMed]
- Loidreau, Y.; Deau, E.; Marchand, P.; Nourrisson, M.-R.; Logé, C.; Coadou, J.M.; Loaëc, N.; Meijer, L.; Besson, T. Synthesis and molecular modelling studies of 8-arylpyrido[3′,2′:4,5]thieno[3,2-d] pyrimidin-4-amines as multitarget Ser/Thr kinases inhibitors. Eur. J. Med. Chem. 2015, 92, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Loidreau, Y.; Marchand, P.; Dubouilh-Benard, C.; Nourrisson, M.-R.; Duflos, M.; Loaëc, N.; Meijer, L.; Besson, T. Synthesis and biological evaluation of N-aryl-7-methoxybenzo[b]furo[3,2-d] pyrimidin-4-amines and their N-arylbenzo[b]thieno[3,2-d]pyrimidin-4-amine analogues as dual inhibitors of CLK1 and DYRK1A kinases. Eur. J. Med. Chem. 2013, 59, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Loidreau, Y.; Marchand, P.; Dubouilh-Benard, C.; Nourrisson, M.-R.; Duflos, M.; Lozach, O.; Loaëc, N.; Meijer, L.; Besson, T. Synthesis and biological evaluation of N-arylbenzo[b]thieno[3,2-d]pyrimidin-4-amines and their pyrido and pyrazino analogues as Ser/Thr kinase inhibitors. Eur. J. Med. Chem. 2012, 58, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Foucourt, A.; Dubouilh-Benard, C.; Chosson, E.; Corbière, C.; Buquet, C.; Iannelli, M.; Leblond, B.; Marsais, F.; Besson, T. Microwave-accelerated Dimroth rearrangement for the synthesis of 4-anilino-6-nitroquinazolines. Application to an efficient synthesis of a microtubule destabilizing agent. Tetrahedron 2010, 66, 4495–4502. [Google Scholar] [CrossRef]
- Foucourt, A.; Hédou, D.; Dubouilh-Benard, C.; Désiré, L.; Casagrande, A.-S.; Leblond, B.; Loaëc, N.; Meijer, L.; Besson, T. Design and synthesis of thiazolo[5,4-f]quinazolines as DYRK1A inhibitors, Part I. Molecules 2014, 19, 15546–15571. [Google Scholar] [CrossRef] [PubMed]
- Foucourt, A.; Hédou, D.; Dubouilh-Benard, C.; Désiré, L.; Casagrande, A.-S.; Leblond, B.; Loaëc, N.; Meijer, L.; Besson, T. Design and synthesis of thiazolo[5,4-f]quinazolines as DYRK1A inhibitors, Part II. Molecules 2014, 19, 15411–15439. [Google Scholar] [CrossRef] [PubMed]
- Leblond, B.; Casagrande, A.-S.; Désiré, L.; Foucourt, A.; Besson, T. DYRK1 inhibitors and uses thereof WO 2013026806. Chem. Abstr. 2013, 158, 390018. [Google Scholar]
- Hédou, D.; Godeau, J.; Loaëc, N.; Meijer, L.; Fruit, C.; Besson, T. Synthesis of Thiazolo[5,4-f]quinazolin-9(8H)-ones as Multi-Target Directed Ligands of Ser/Thr Kinases. Molecules 2016, 21, 578. [Google Scholar] [CrossRef] [PubMed]
- Hédou, D.; Dubouilh-Benard, C.; Loaëc, N.; Meijer, L.; Fruit, C.; Besson, T. Synthesis of Bioactive 2-(Arylamino)thiazolo[5,4-f]-quinazolin-9-ones via the Hügershoff Reaction or Cu-Catalyzed Intramolecular C-S Bond Formation. Molecules 2016, 21, 794. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.; Latypova, X.; Wilson, C.M.; Magnaudeix, A.; Perrin, M.-L.; Terro, F. Tau protein kinases: Involvement in Alzheimer’s disease. Ageing Res. Rev. 2013, 12, 289–309. [Google Scholar] [CrossRef] [PubMed]
- Flajolet, M.; He, G.; Heiman, M.; Lin, A.; Nairn, A.C.; Greengard, P. Regulation of Alzheimer’s disease amyloid-β formation by casein kinase I. Proc. Nat. Acad. Sci. USA 2007, 104, 4159–4164. [Google Scholar] [CrossRef] [PubMed]
- Weinmann, H.; Metternich, R. Drug discovery process for kinase Inhibitors. Chembiochem 2005, 6, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. Small-molecule kinase inhibitors: An analysis of FDA-approved drugs. Drug Discovery Today 2016, 21, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Carles, F.; Bourg, S.; Meyer, C.; Bonnet, P. PKIDB: A Curated, Annotated and updated database of protein kinase inhibitors in clinical trials. Molecules 2018, 23, 908. [Google Scholar] [CrossRef] [PubMed]
- Jarhed, D.B.; Mashelkar, K.K.; Kim, H.-R.; Noh, M.; Jeong, L.S. Dual-dspecificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) inhibitors as potential therapeutics. J. Med. Chem. 2018, 61. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Fruit, C.; Herault, Y.; Meijer, L.; Besson, T. Dual-specificity tyrosine phosphorylation-regulated kinase 1a (dyrk1a) inhibitors: A survey of recent patent literature. Expert Opin. Ther. Pat. 2017, 27, 1183–1199. [Google Scholar] [CrossRef] [PubMed]
- Abbassi, R.; Johns, T.G.; Kassiou, M.; Munoz, L. DYRK1A in neurodegeneration and cancer: Molecular basis and clinical implications. Pharmacol. Ther. 2015, 151, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Medda, F.; Smith, B.; Gokhale, V.; Shaw, A.Y.; Dunckley, T.; Hulme, C. Beyond secretases: Kinase inhibitors for the treatment of Alzheimer’s disease. Annu. Rep. Med. Chem. 2013, 48, 57–71. [Google Scholar] [CrossRef]
- Smith, B.; Medda, F.; Gokhale, V.; Dunckley, T.; Hulme, C. Recent advances in the design, synthesis, and biological evaluation of selective DYRK1A inhibitors: A new avenue for a disease modifying treatment of Alzheimer’s? ACS Chem. Neurosci. 2012, 3, 857–872. [Google Scholar] [CrossRef] [PubMed]
- Varjosalo, M.; Keskitalo, S.; Van Drogen, A.; Nurkkala, H.; Vichalkovski, A.; Aebersold, R.; Gstaiger, M. The protein interaction landscape of the human CMGC kinase group. Cell Rep. 2013, 3, 1306–1320. [Google Scholar] [CrossRef] [PubMed]
- Coutadeur, S.; Benyamine, H.; Delalonde, L.; de Oliveira, C.; Leblond, B.; Foucourt, A.; Besson, T.; Casagrande, A.-S.; Taverne, T.; Girard, A.; et al. A Novel DYRK1A (Dual specificity tyrosine phosphorylation-regulated kinase 1A) inhibitor for the treatment of Alzheimer’s disease: Effect on Tau and amyloid pathologies in vitro. J. Neurochem. 2015, 133, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Chaikuad, A.; Diharce, J.; Schröder, M.; Foucourt, A.; Leblond, B.; Casagrande, A.-S.; Désiré, L.; Bonnet, P.; Knapp, S.; Besson, T. An unusual binding mode of the methyl 9-anilinothiazolo[5,4-f]quinazoline-2-carbimidates (EHT 1610 and EHT 5372) confers high selectivity for DYRK kinases. J. Med. Chem. 2016, 59, 10315–10321. [Google Scholar] [CrossRef] [PubMed]
- Bournez, C.; Gally, J.-M.; Krezel, P.; Aci-Sèche, S.; Bonnet, P. Frags2Drugs: An in silico tool to discover new molecules based on 3D fragment network. J. Chem. Inf. Model. 2018, in press. [Google Scholar]
- Harari, M.; Couly, F.; Fruit, C.; Besson, T. Late-stage C–H Pd-catalyzed and copper assisted regioselective sequential C2 and C7 arylation of thiazolo[5,4-f]quinazolin-9(8H)-one with aryl halides. Org. Lett. 2016, 18, 3282–3285. [Google Scholar] [CrossRef] [PubMed]
- Couly, F.; Dubouilh-Benard, C.; Besson, T.; Fruit, C. Arylation of thiazolo[5,4-f]quinazolin-9(8H)-one backbone: Synthesis of an array of potential kinase inhibitors. Synthesis 2017, 49, 4615–4622. [Google Scholar] [CrossRef]
- Besson, T.; Fruit, C. Recent developments in microwave-assisted metal-catalyzed C-H functionalization of heteroarenes for medicinal chemistry and material applications. Synthesis 2016, 48, 3879–3889. [Google Scholar] [CrossRef]
- Appel, R.; Janssen, H.; Siray, M.; Knoch, F. Synthese und reaktionen des 4,5-dichlor-1,2,3-dithiazolium-chlorids. Eur. J. Inorg. Chem. 1985, 118, 1632–1643. [Google Scholar] [CrossRef]
- Topliss, J.G. Utilization of operational schemes for analog synthesis in drug design. J. Med. Chem. 1972, 15, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Turočkin, A. 1,5,7-Triazabicyclo[4.4.0]dec-5-ene (TBD) as a Lewis Base. Synlett 2014, 25, 894–895. [Google Scholar] [CrossRef] [Green Version]
- Tahtouh, T.; Elkins, J.M.; Filippakopoulos, P.; Soundararajan, M.; Burgy, G.; Durieu, E.; Cochet, C.; Schmid, R.S.; Lo, D.C.; Delhommel, F.; et al. Selectivity, cocrystal structures and neuroprotective properties of leucettines, a family of protein kinase inhibitors derived from the marine sponge alkaloid leucettamine B. J. Med. Chem. 2012, 55, 9312–9330. [Google Scholar] [CrossRef] [PubMed]
- Primot, A.; Baratte, B.; Gompel, M.; Borgne, A.; Liabeuf, S.; Romette, J.L.; Jho, E.H.; Costantini, F.; Meijer, L. Purification of GSK-3 by affinity chromatography on immobilized axin. Protein Expr. Purif. 2000, 20, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, J.; Ferandin, Y.; Meijer, L. Purification of CK1 by affinity chromatography on immobilised axin. Protein Expr. Purif. 2007, 54, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Gadewar, M.; Tripathi, R.; Prasad, S.K.; Patel, D.K. A review on medicinal importance, pharmacological activity and bioanalytical aspects of beta-carboline alkaloid “Harmine”. Asian Pac. J. Trop. Biomed. 2012, 2, 660–664. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of compound FC162 (4c) are available from the authors for academic studies with Material Transfer Agreement (MTA). |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
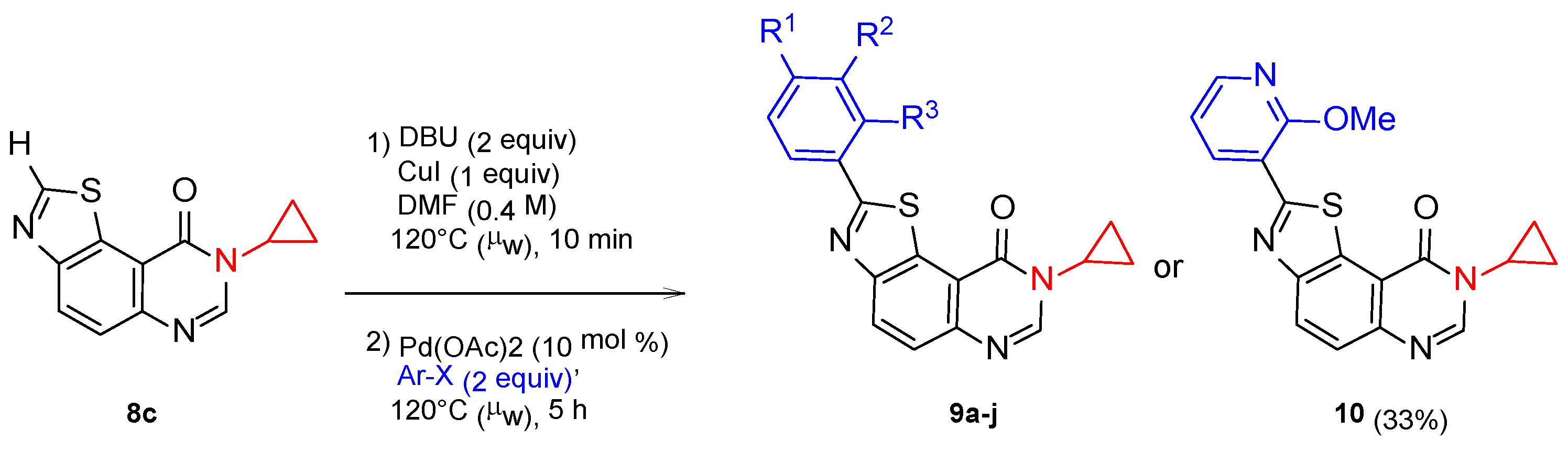{kind=link}
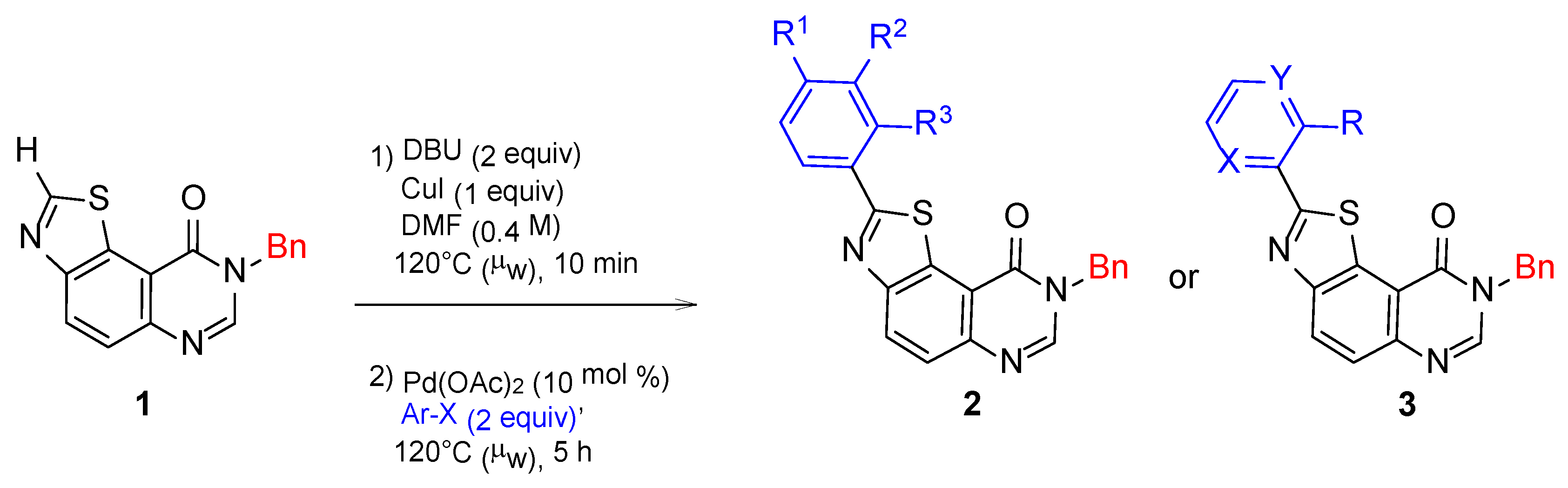
| Product | R1 | R2 | R3 | X(Ar) | Yield c (%) |
| 2a | H | H | H | Br | 92 |
| 2b | Me | H | H | Br | 86 |
| 2c | MeO | H | H | I | 63 |
| 2d | Cl | H | H | I | 87 |
| 2e | F | H | H | I | 71 |
| 2f | CN | H | H | I | 59 |
| 2h | NMe2 | H | H | Br | 87 |
| 2i | Cl | H | Cl | I | 62 |
| 2j | Cl | Cl | H | I | 64 |
| Product | X | Y | R | X(Ar) | Yield c (%) |
| 3a | CH | N | H | I | 69 |
| 3b | N | N | H | I | 47 |
| 3c | CH | N | OMe | I | 29 |
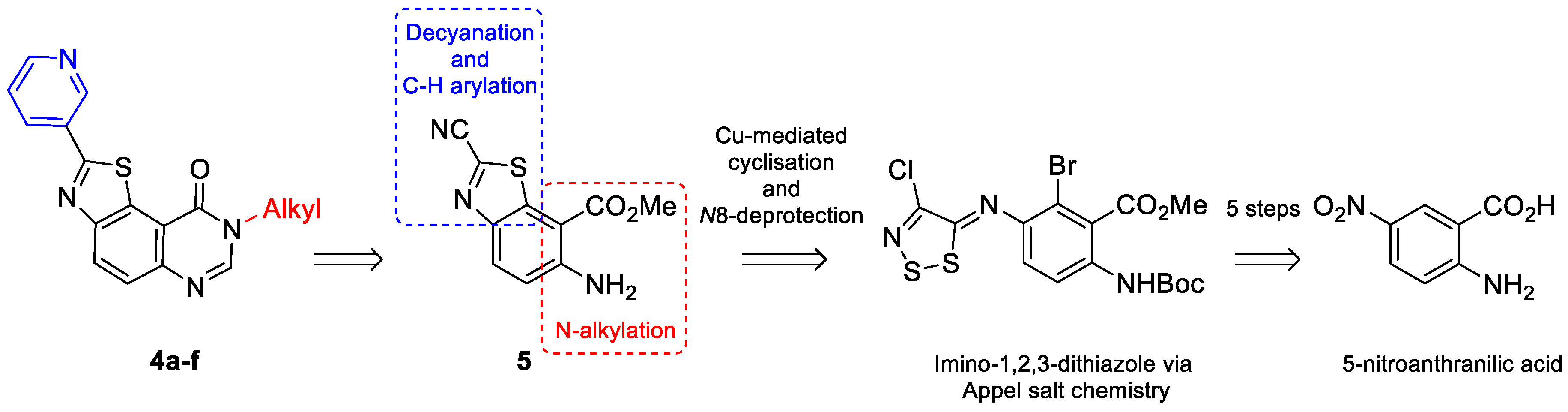
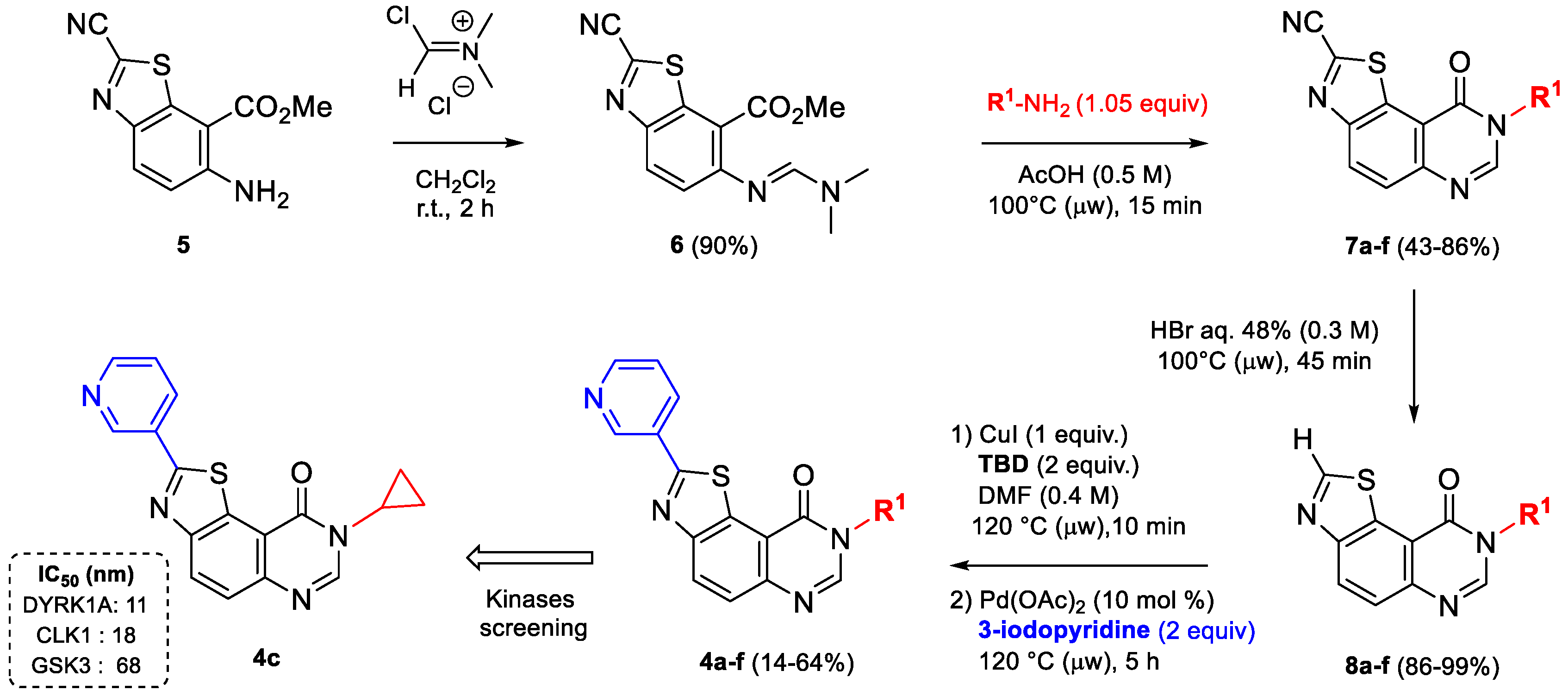
| –R1 | Compound | Yield a (%) | Compound | Yield a (%) | Compound | Yield a (%) |
|---|---|---|---|---|---|---|
 | 7a | 70 | 8a | 98 | 4a | 14 |
 | 7b | 43 | 8b | 99 | 4b | 64 |
 | 7c | 86 | 8c | 86 | 4c | 43 |
 | 7d | 65 | 8d | 97 | 4d | 57 |
 | 7e | 60 | 8e | 98 | 4e | 55 |
 | 7f | 50 | 8f | 98 | 4f | 54 |
| Product | R1 | R2 | R3 | X(Ar) | Yield b (%) |
|---|---|---|---|---|---|
| 9a | H | H | H | Br | 58 |
| 9b | Me | H | H | Br | 64 |
| 9c | MeO | H | H | I | 76 |
| 9d | Cl | H | H | I | 67 |
| 9e | F | H | H | I | 52 |
| 9f | CN | H | H | I | - c |
| 9h | NMe2 | H | H | Br | 31 |
| 9i | Cl | H | Cl | I | 65 |
| 9j | Cl | Cl | H | I | 69 |
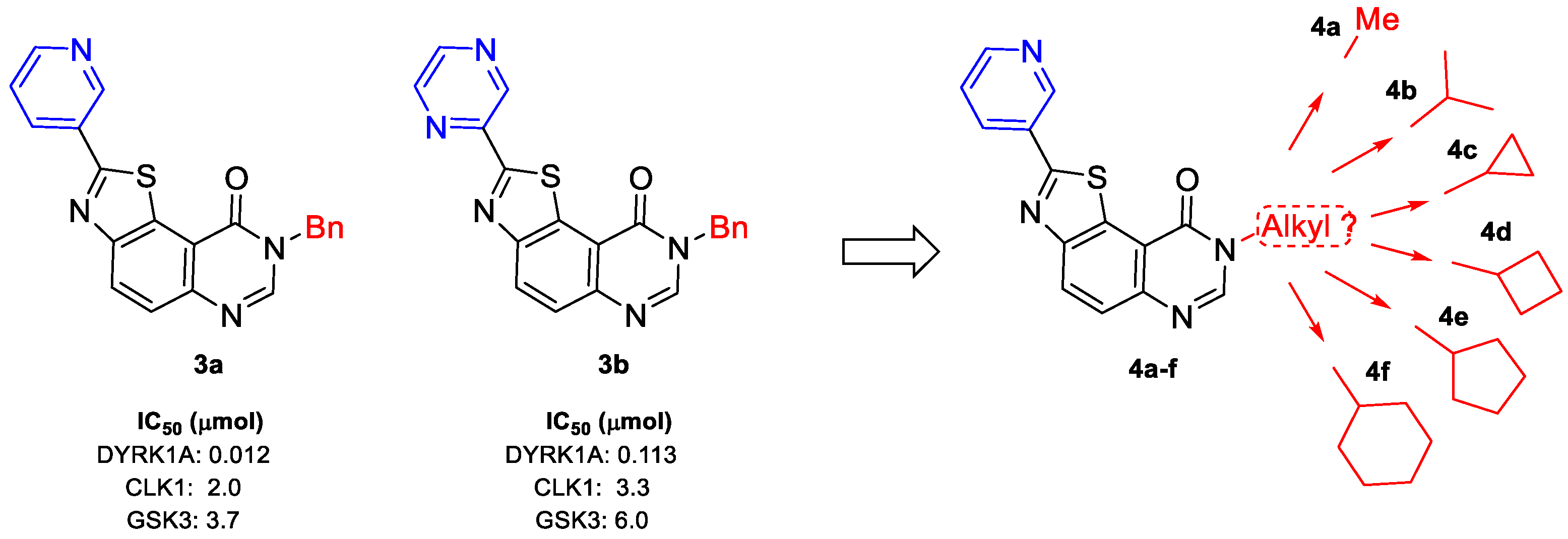
| Compounds | CDK5/p25 | CK1δ/ε | CLK1 | DYRK1A | GSK-3α/β |
|---|---|---|---|---|---|
| 2a–j | >10 | >10 | >10 | >10 | >10 |
| 3a | >10 | >10 | 2.0 | 0.012 | 3.7 |
| 3b | >10 | >10 | 3.33 | 0.133 | 6.0 |
| 3c | >10 | >10 | >10 | >10 | >10 |
| 4a | n.t. d | >10 | >10 | >10 | >10 |
| 4b | n.t. | >10 | >10 | >10 | >10 |
| 4c (FC162) | n.t. | 6.0 | 0.018 | 0.011 | 0.068 |
| 4d | n.t. | >10 | >10 | >10 | >10 |
| 4e | n.t. | >10 | >10 | >10 | >10 |
| 4f | n.t. | >10 | >10 | >10 | >10 |
| 9a–i | >10 | >10 | >10 | >10 | ≥10 |
| 10 | >10 | >10 | >10 | >10 | ≥10 |
| Harmine | >10 | 1.5 | 0.026 | 0.029 | >10 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Couly, F.; Harari, M.; Dubouilh-Benard, C.; Bailly, L.; Petit, E.; Diharce, J.; Bonnet, P.; Meijer, L.; Fruit, C.; Besson, T. Development of Kinase Inhibitors via Metal-Catalyzed C–H Arylation of 8-Alkyl-thiazolo[5,4-f]-quinazolin-9-ones Designed by Fragment-Growing Studies. Molecules 2018, 23, 2181. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23092181
Couly F, Harari M, Dubouilh-Benard C, Bailly L, Petit E, Diharce J, Bonnet P, Meijer L, Fruit C, Besson T. Development of Kinase Inhibitors via Metal-Catalyzed C–H Arylation of 8-Alkyl-thiazolo[5,4-f]-quinazolin-9-ones Designed by Fragment-Growing Studies. Molecules. 2018; 23(9):2181. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23092181
Chicago/Turabian StyleCouly, Florence, Marine Harari, Carole Dubouilh-Benard, Laetitia Bailly, Emilie Petit, Julien Diharce, Pascal Bonnet, Laurent Meijer, Corinne Fruit, and Thierry Besson. 2018. "Development of Kinase Inhibitors via Metal-Catalyzed C–H Arylation of 8-Alkyl-thiazolo[5,4-f]-quinazolin-9-ones Designed by Fragment-Growing Studies" Molecules 23, no. 9: 2181. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23092181