
Microwave (MW), Ultrasound (US) and Combined Synergic MW-US Strategies for Rapid Functionalization of Pharmaceutical Use Phenols


Abstract
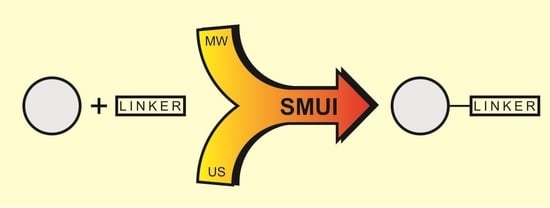
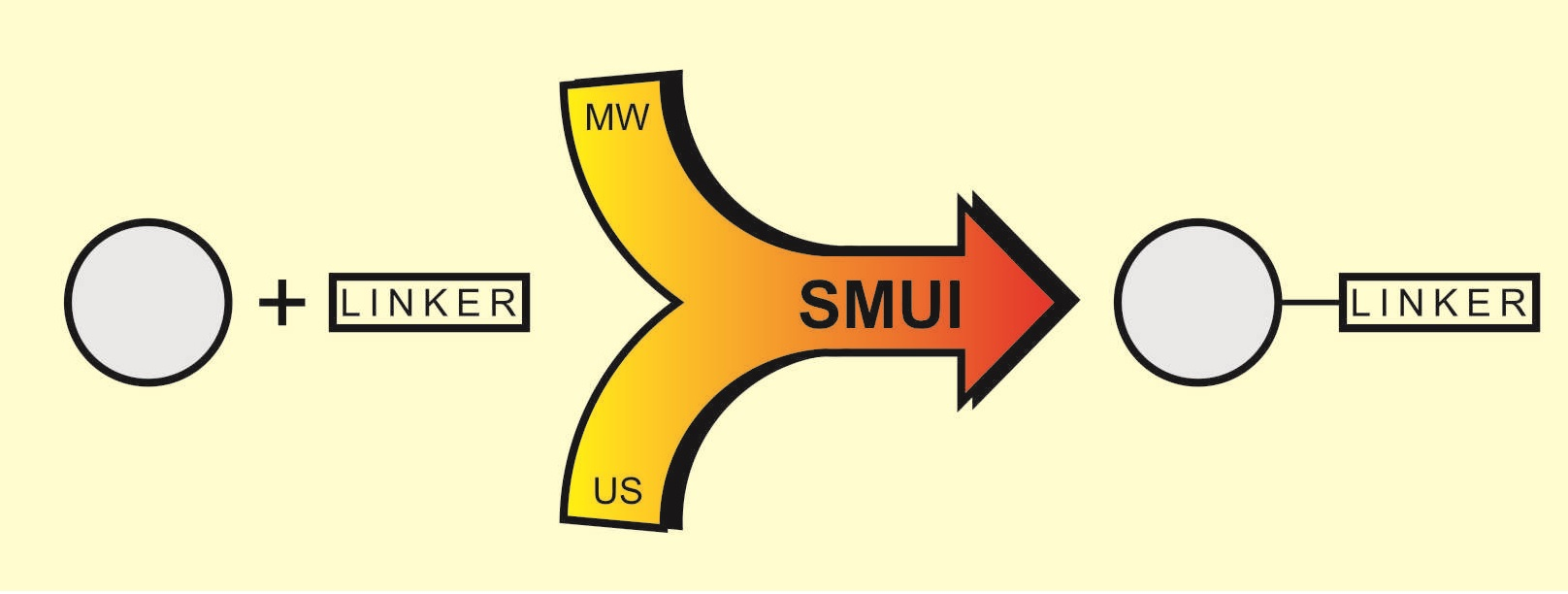
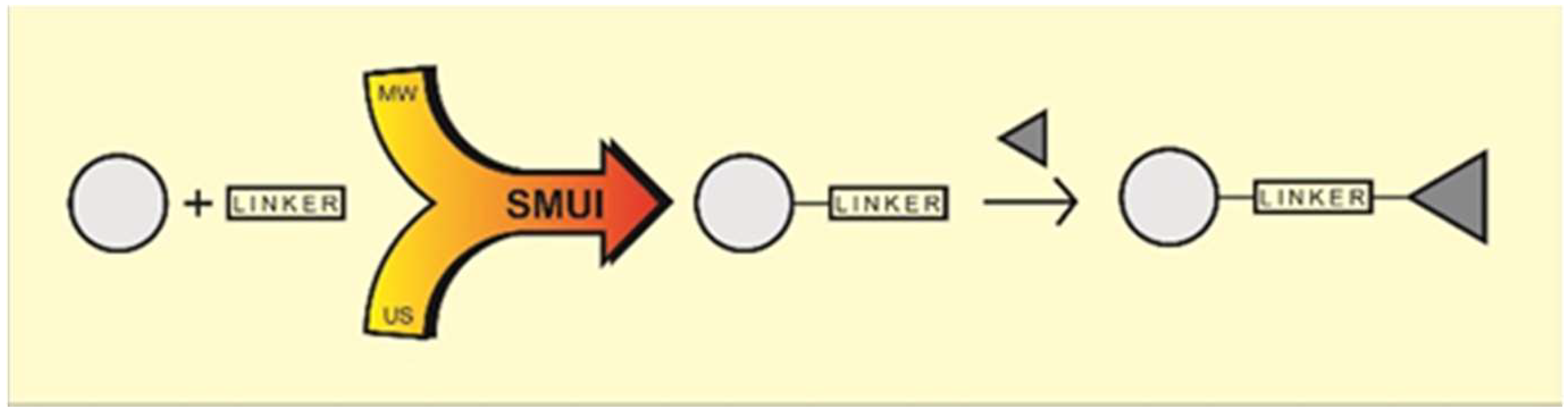
:
1. Introduction
- Synergism by synthesis process intensification by means of combining two non-conventional factors: microwaves and ultrasound. These two effects of process intensification have been used to great effect in various chemical processes and engineering applications. Microwaves and ultrasounds are significantly different in terms of their nature; however, simultaneously used, they form the green SMUI technology (Simultaneous Microwave and Ultrasound Irradiation), allowing significant optimization of the chemical reaction parameters. The combination of microwaves and ultrasound has become a real chance for effective, economical and, above all, green synthetic procedures [11,12,13,14,15]. The favorable results are due to the combination of the phenomena of both factors—unique microwave heating and the phenomenon of cavitation. The microwave medium is usually combined with effective heating, and the sonification factor with efficient temperature stimulation of the chemical process. Thanks to the use of effective microwave heating, there is no loss of energy associated with conventional heating and thermal pollution of the environment is avoided. Acoustic cavitation allows thorough mixing of substrates, including two-phase mixtures, limiting the use of surfactants that have a negative impact on the environment. Thanks to cavitation, it is possible to increase the contact between the liquid substrate and the low solubility due to its fragmentation, which has a positive effect on the reaction results [11,12,13,14,15].
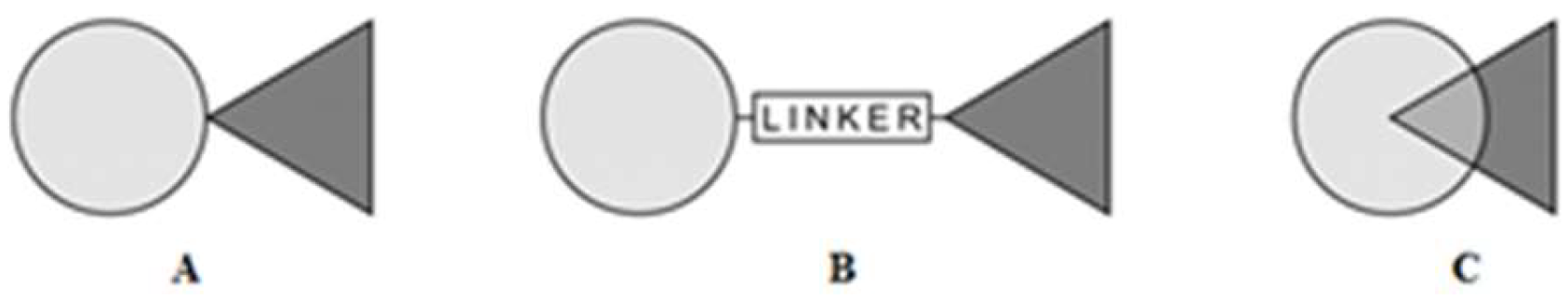
- Synergism by combining various active chemistry building blocks—pharmacomodulation of two biologically active structures by chemical hybridization methods leads to a new combined structure with interesting biological activity. According to the recent literature, the compounds derived from different bioactive molecules are often characterized by a synergy of their individual component activities. Many trends have been proposed for the design of new drugs containing different structures (dimers, heterodimers, heteromers, adducts, associates, complexes, biooligomers, dendrimers, dual-, bivalent-, multifunction drugs and codrugs, identical or non-identical twin drugs, mixed or combo drugs, supramolecular particles and various nanoindividuals). Chemical association of two (or more) structures into one new molecule depends on their functionality, cross-reactivity, and can be implemented by way of various methods [23,24]. Generally, the most popular molecules integration modes are: direct no-linker mode (fused hybrids), intermediate linker mode and overlap mode (merged hybrids) (Figure 1).
2. Results and Discussion
- monophenols: 5-methyl-2-(propan-2-yl)phenol (thymol) (1), 2-methoxy-4-(prop-2-enyl)phenol (eugenol) (2), 2-hydroxybenzoic acid (salicylic acid) (3), 4-hydroxybenzoic acid (4), its methyl ester-nipagin M (5) and propyl ester-nipagin P (6), 4-acetaminophenol (paracetamol) (7), 1-naphtol (8) and 2-naphtol (9);
- diphenols: 1,2-dihydroxybenzene (hydroquinone) (10), 1,3-dihydroxybenzene (resorcinol) (11), 1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-dien-3,5-dione (curcumin) (12);
- triphenols: 1,3,5-trihydroxybenzene (phloroglucinol) (13), 1,2,3-trihydroxybenzene (pyrogallol) (14), genistein (5,7-dihydroxy-3-(4-hydroxyphenyl)chromen-4-one) (15).
- monophenol: linker: NaOH (1:1:2)
- diphenol: linker: NaOH (1:2:3)
- triphenol: linker: NaOH (1:3:4)
3. Materials and Methods
3.1. Equipment and General Procedures
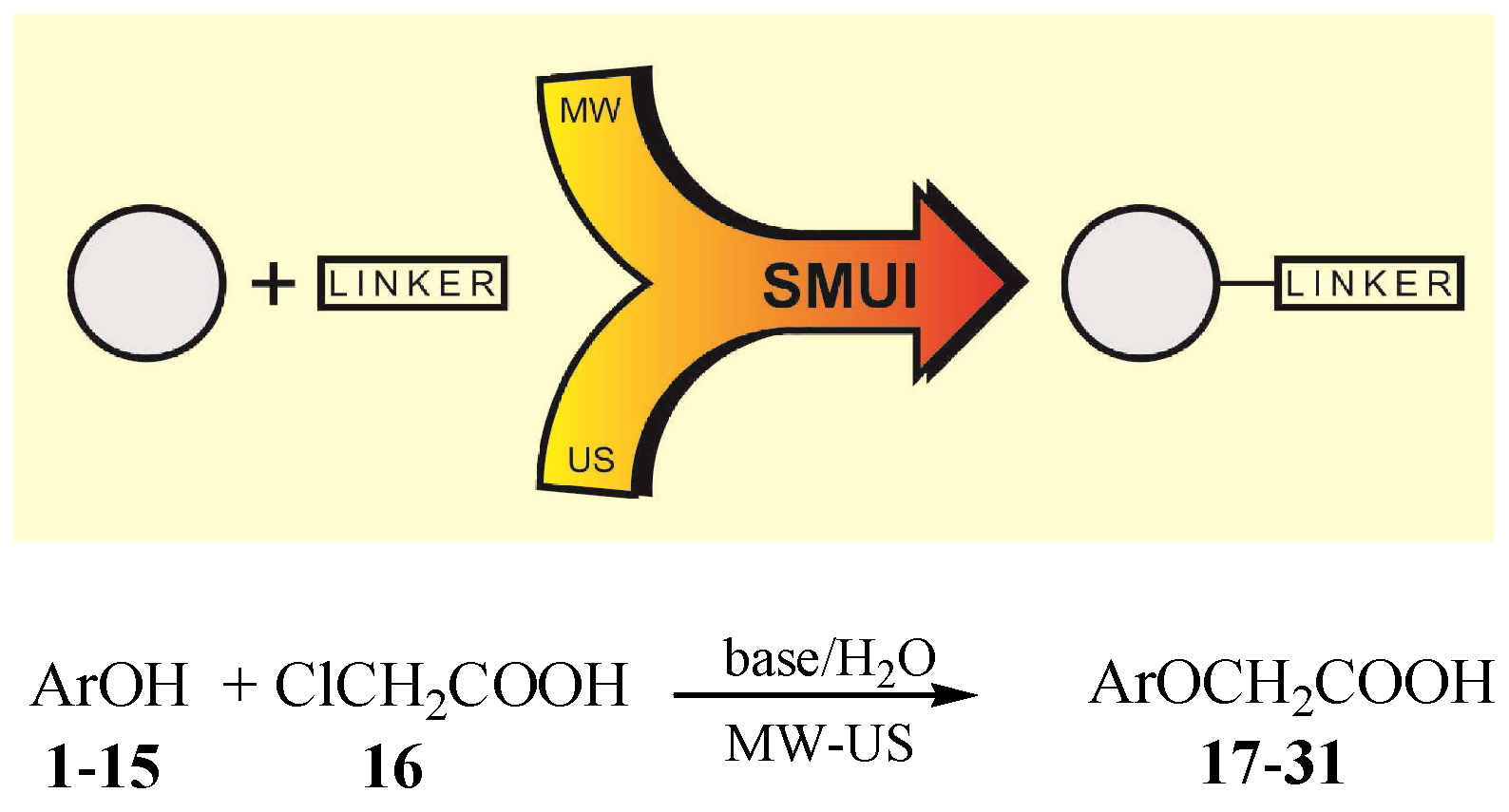
3.2. Synthetic Procedures—General Procedures for Synthesis of Functionalized Phenols (17–31)
3.2.1. US Method
- 1.2 mmol (0.11 g) of 16 and 2.3 mmol of base for monophenols (1–9)
- 2.2 mmol (0.21 g) of 16 and 3.3 mmol of base for diphenols (10–12)
- 3.2 mmol (0.30 g) of 16 and 4.3 mmol of base for triphenols (13–15)
3.2.2. MW Method
- 1.2 mmol (0.11 g) of 16 and 2.3 mmol of base for monophenols (1–9)
- 2.2 mmol (0.21 g) of 16 and 3.3 mmol of base for diphenols (10–12)
- 3.2 mmol (0.30 g) of 16 and 4.3 mmol of base for triphenols (13–15)
3.2.3. MW-US Method
- 1.2 mmol (0.11 g) of 16 and 2.3 mmol of base for monophenols (1–9)
- 2.2 mmol (0.21 g) of 16 and 3.3 mmol of base for diphenols (10–12)
- 3.2 mmol (0.30 g) of 16 and 4.3 mmol of base for triphenols (13–15)
3.2.4. Classical Method
- 1.2 mmol (0.11 g) of 16 and 2.3 mmol of base for monophenols (1–9)
- 2.2 mmol (0.21 g) of 16 and 3.3 mmol of base for diphenols (10–12)
- 3.2 mmol (0.30 g) of 16 and 4.3 mmol of base for triphenols (13–15)
3.3. The Data of Compounds (17–31) Obtained by MS-US Method
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cathcart, C. Green Chemistry in the Emerald Isle. Chem. Ind. 1990, 5, 684–687. [Google Scholar]
- Linthorst, J.A. An Overview: Origins and Development of Green Chemistry. Found. Chem. 2010, 12, 55–68. [Google Scholar] [CrossRef]
- Anastas, P.T.; Warner, J.C. Green Chemistry: Theory and Practice; Oxford University Press: New York, NY, USA; Oxford, UK, 1998; ISBN 9780198506980. [Google Scholar]
- Gawande, M.B.; Bonifácio, V.D.; Luque, R.; Branco, P.S.; Varma, R.S. Solvent-free and catalysts-free chemistry: A benign pathway to sustainability. ChemSusChem 2014, 7, 24–44. [Google Scholar] [CrossRef] [PubMed]
- Calcio Gaudino, E.; Manzoli, M.; Carnaroglio, D.; Wu, Z.; Grillo, G.; Rotolo, L.; Medlock, J.; Bonrath, W.; Cravotto, G. Sonochemical preparation of alumina-spheres loaded with Pd nanoparticles for 2-butyne-1,4-diol semi-hydrogenation in a continuous flow microwave reactor. RSC Adv. 2018, 8, 7029–7039. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Toda, F. Solvent-Free Organic Synthesis. Chem. Rev. 2000, 100, 1025–1074. [Google Scholar] [CrossRef] [PubMed]
- Maiuolo, L.; De Nino, A.; Merino, P.; Russo, B.; Stabile, G.; Nardi, M.; D’Agostino, N.; Bernardi, T. Rapid, efficient and solvent free microwave mediated synthesis of aldo- and ketonitrones. Arab. J. Chem. 2016, 9, 25–31. [Google Scholar] [CrossRef]
- Maiuolo, L.; Merino, P.; Algieri, V.; Nardi, M.; Di Gioia, M.L.; Russo, B.; Delso, I.; Tallarida, M.A.; De Nino, A. Nitrones and nucleobase-containing spiroisoxazolidines derived from isatin and indanone: Solvent-free microwave-assisted stereoselective synthesis and theoretical calculations. RSC Adv. 2017, 7, 48980–48988. [Google Scholar] [CrossRef]
- Kappe, C.O. Controlled Microwave Heating in Modern Organic Synthesis. Angew. Chem. Int. Ed. 2004, 43, 6250–6284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, H.M.; Lodeiro, C.; Capelo-Martinez, J. The Power of Ultrasounds. In Ultrasounds in Chemistry: Analytical Applications; WILEY-VCH Verlag GmbH & Co KgaA: Weinheim, Germany, 2009; pp. 1–16, ISBN 10 3527319344. [Google Scholar]
- Peng, Y.; Song, G. Simultaneous Microwave and Ultrasound Irradiation: A Rapid Synthesis of Hydrazides. Green Chem. 2001, 3, 302–304. [Google Scholar] [CrossRef]
- Cravotto, G.; Cintas, P. The Combined Use of Microwaves and Ultrasound: Improved Tools in Process Chemistry and Organic Synthesis. Chem. Eur. J. 2007, 13, 1902–1909. [Google Scholar] [CrossRef] [PubMed]
- Martina, K.; Tagliapietra, S.; Bargre, A.; Cravotto, G. Combined Microwaves/Ultrasound, a Hybrid Technology. Top. Curr. Chem. 2016, 374–379. [Google Scholar] [CrossRef]
- Leonellia, C.; Mason, T.J. Microwave and Ultrasonic Processing: Now a Realistic Option for Industry. Chem. Eng. Process. 2010, 49, 885–900. [Google Scholar] [CrossRef]
- Zbancioc, G.; Zbancioc, A.M.; Mangalagiu, I.I. Ultrasound and Microwave Assisted Synthesis of Dihydroxyacetophenone Derivatives with or without 1,2-Diazine Skeleton. Ultrason. Sonochem. 2014, 21, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Nardi, M.; Costanzo, P.; De Nino, A.; Di Gioia, M.L.; Olivito, F.; Sindona, G.; Procopioc, A. Water excellent solvent for the synthesis of bifunctionalized cyclopentenones from furfural. Green Chem. 2017, 19, 5403–5411. [Google Scholar] [CrossRef]
- Nardi, M.; Herrera Cano, N.; Costanzo, P.; Oliverio, M.; Sindona, G.; Procopio, A. Aqueous MW eco-friendly protocol for amino group protection. RSC Adv. 2015, 5, 18751–18760. [Google Scholar] [CrossRef]
- Oliverio, M.; Nardi, M.; Cariati, L.; Vitale, E.; Bonacci, S.; Procopio, A. “On Water” MW-Assisted Synthesis of Hydroxytyrosol Fatty Esters. ACS Sustain. Chem. Eng. 2016, 4, 661–665. [Google Scholar] [CrossRef]
- Oliverio, M.; Costanzo, P.; Nardi, M.; Calandruccio, C.; Salerno, R.; Procopio, A. Tunable microwave-assisted method for the solvent-free and catalyst-free peracetylation of natural products. Beilstein J. Org. Chem. 2016, 12, 2222–2233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nardi, M.; Bonacci, S.; Cariati, L.; Costanzo, P.; Oliverio, M.; Sindona, G.; Procopio, A. Synthesis and antioxidant evaluation of lipophilic oleuropein aglycone derivatives. Food Funct. 2017, 8, 4684–4692. [Google Scholar] [CrossRef] [PubMed]
- Cravotto, G.; Boffa, L.; Mantegna, S.; Perego, P.; Avogadro, M.; Cintas, P. Improved extraction of vegetable oils under high-intensity ultrasound and/or microwaves. Ultrason. Sonochem. 2008, 15, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Woerly, E.M.; Roy, J.; Burke, M.D. Synthesis of Most Polyene Natural Product Motifs Using Just 12 Building Blocks and One Coupling Reaction. Nat. Chem. 2014, 6, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Pawełczyk, A.; Sowa-Kasprzak, K.; Olender, D.; Zaprutko, L. Molecular Consortia-Various Structural Concepts and Powerful Approach in More Effective Therapeutics Synthesis. Int. J. Mol. Sci. 2018, 19, 1104. [Google Scholar] [CrossRef] [PubMed]
- Decker, M. Design of Hybrid Molecules for Drug Development, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2017; ISBN 535 9780081010112. [Google Scholar]
- Decker, M. Towards Gaseous Mediator Hybrid Drugs. In Design of Hybrid Molecules for Drug Development, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2017; p. 73, ISBN 535 9780081010112. [Google Scholar]
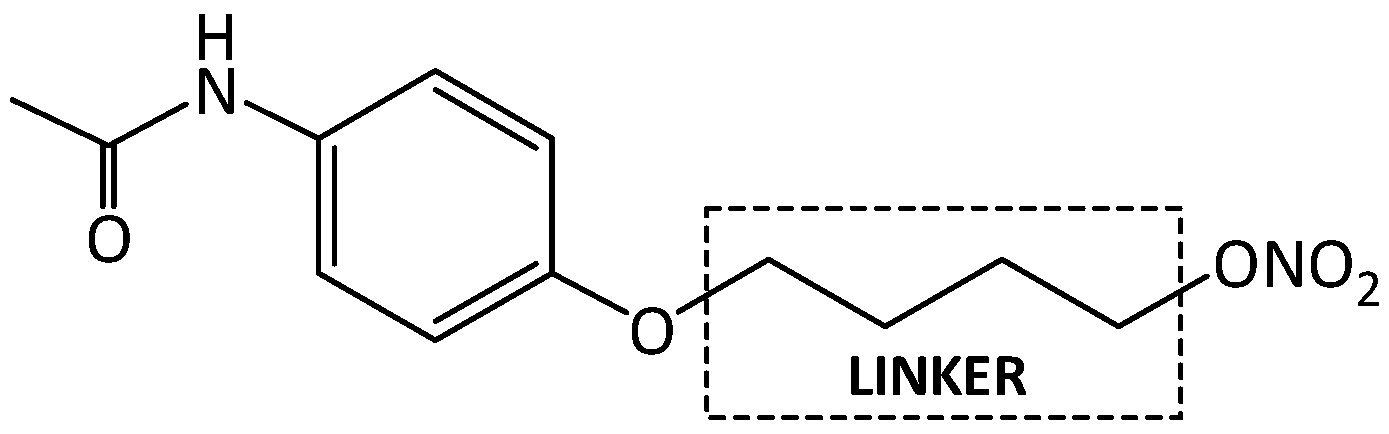
- Nitric Oxide Releasing Derivatives of Paracetamol. Available online: https://patents.google.com/patent/US8207222B2/en (accessed on 9 August 2018).
- Williamson, A.W. On Etherification. J. Chem. Soc. 1852, 4, 229–239. [Google Scholar] [CrossRef]
- Peng, Y.; Song, G. Combined Microwave and Ultrasound Assisted Williamson Ether Synthesis in the Absence of Phase-Transfer Catalysts. Green Chem. 2002, 4, 349–351. [Google Scholar] [CrossRef]
- Sandler, S.R.; Karo, W. Organic Functional Group Preparations, I; Academic Press: New York, NY, USA, 1983; p. 132. ISBN 9780126186031. [Google Scholar]
- Ragaini, V.; Pirola, C.; Borrelli, S.; Ferrari, C.; Longo, I. Simultaneous Ultrasounds and Microwave New Reactor: Detailed Description and Energetic Considerations. Ultrasonics Sonochem. 2012, 19, 872–876. [Google Scholar] [CrossRef] [PubMed]
- UWave-1000. Available online: www.sineomicrowave.com/Upload/%E4%BA%A7%E5%93%81%E5%9B%BE%E7%89%87/hechengyi/UWave1000-20315522903.pdf (accessed on 8 August 2018).
- Nelson, K.M.; Dahlin, J.L.; Bisson, J.; Graham, J.; Pauli, G.F.; Walters, M.A. The Essential Medicinal Chemistry of Curcumin. J. Med. Chem. 2017, 60, 1620–1637. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Qadir, M.A.; Hameed, A.; Arshad, M.N.; Asiri, A.M.; Muddassar, M. Sulfonamides Containing Curcumin Scaffold: Synthesis, Characterization, Carbonic Anhydrase Inhibition and Molecular Docking Studies. Bioorg. Chem. 2018, 76, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Wang, J.; Zhang, X.; Hong, C.; Wang, C.J. Design, Synthesis and Evaluation of Genistein-Polyamine Conjugates as Multi-Functional Anti-Alzheimer Agents. Med. Chem. 2014, 4, 617–622. [Google Scholar] [CrossRef]
- Pawełczyk, A.; Sowa-Kasprzak, K.; Zaprutko, L. Synthesis of Selected Azoles Derivatives Using the Cross-Combination of Microwave and Ultrasound Factors. In Proceedings of the 21st International Electronic Conference on Synthetic Organic Chemistry, Poznan, Poland, 1–30 November 2017. Sciforum Electronic Conference Series, 21. [Google Scholar] [CrossRef]
- Niederl, J.B.; Natelson, S. The Synthesis of Thymol, Chlorothymol and Homologs of Thymol by the Intramolecular Rearrangement of meta-Cresyl Ethers. J. Am. Chem. Soc. 1932, 54, 1063–1070. [Google Scholar] [CrossRef]
- Abraham, D.J.; Kennedy, P.E.; Mehanna, A.S.; Patwa, D.C.; Williams, F.L. Design, Synthesis, and Testing of Potential Antisickling Agents. 4. Structure-Aactivity Relationships of Benzyloxy and Phenoxy Acids. J. Med. Chem. 1984, 27, 967–978. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.E.; Smith, G.; Freney, D.; Byriel, K.A.; Kennard, C.H.L. Molecular Cocrystals of Carboxylic Acids. XV. Preparation and Characterization of Heterocyclic Base Adducts with a Series of Carboxylic Acids, and the Crystal Structures of the Adducts of 2-Aminopyrimidine With 2,6-Dihydroxybenzoic Acid, 4-Aminobenzoic Acid, Phenoxyacetic Acid, (2,4-Dichlorophenoxy)acetic Acid, (3,4-Dichlorophenoxy)-acetic Acid and Salicylic Acid, and 2-Aminopyridine With 2,6-Dihydroxybenzoic Acid. Aust. J. Chem. 1994, 47, 1097–1115. [Google Scholar] [CrossRef]
- Wang, X.; Li, Z.; Da, Y.; Chen, J.; Wei, T. Phase Transfer Catalyzed Syntheses of 4-Carboxylphenoxyacetic Acid Derivatives. Synth. Commun. 1999, 29, 4153–4161. [Google Scholar] [CrossRef]
- Howard, C.C. Ueber p-Amidophenoxylessigsäure und Derivate Derselben. Chem. Ber. 1897, 30, 548. [Google Scholar] [CrossRef]
- Villemin, D.; Hammadi, M. Environmentally Desirable Synthesis without Use of Organic Solvent. Synthesis of Aryloxyacetic Acids. Synth. Commun. 1996, 26, 4337–4341. [Google Scholar] [CrossRef]
- Wei, T.; Chen, J.; Wang, X.; Zhang, Y.; Wang, L. Phase Transfer Catalyzed Synthesis of Diaryl 1,4-Phenylene Dioxydiacetate. Synth. Commun. 1996, 26, 1447–1454. [Google Scholar] [CrossRef]
- Sougoule, A.S.; Mei, Z.; Xiao, X.; Balde, C.A.; Samoura, S.; Dolo, A.; Zhu, D. A Novel Macrocyclic Organotin Carboxylate Containing a Penta-Nuclear Long Ladder. J. Organomet. Chem. 2014, 758, 19–24. [Google Scholar] [CrossRef]
- Kovacs-Kulyassa, A.; Herczegh, P.; Sztaricskai, F.J.; Szabo, P. Cephalosporin Podand Derivatives. J. Antibiot. 2000, 53, 1207–1211. [Google Scholar] [CrossRef] [PubMed]
- Giacosa, P. Vortheilhafte Darstellung der Phenolglycolsäure und über die Pyrogallotriglycolsäure. J. Prakt. Chem. 1879, 2, 396. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 17–31 are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | SUBSTRAT | No | PRODUCT | |
|---|---|---|---|---|
| MONOPHENOLS | 1 |  | 17 |  |
| 2 |  | 18 |  | |
| 3 |  | 19 |  | |
| 4 |  | 20 |  | |
| 5 |  | 21 |  | |
| 6 |  | 22 |  | |
| 7 |  | 23 |  | |
| 8 |  | 24 |  | |
| 9 |  | 25 |  | |
| DIPHENOLS | 10 |  | 26 |  |
| 11 |  | 27 |  | |
| 12 |  | 28 |  | |
| TRIPHENOLS | 13 |  | 29 |  |
| 14 |  | 30 |  | |
| 15 |  | 31 |  |
| Substrat Product | Name | US 1 | MW 2 | MW-US 3 | Classic 4 | ||||
|---|---|---|---|---|---|---|---|---|---|
| Time min | % | Time min | % | Time min | % | Time h | % | ||
| 1 | 5-methyl-2-(propan-2-yl)phenol (thymol) | 30 | 40 | 10 | 95 | 5 | 89 | 8 | 19 |
| 17 | 2-[5-methyl-2-(propan-2-yl)phenoxy]acetic acid | ||||||||
| 2 | 2-methoxy-4-(prop-2-enyl)phenol (eugenol) | 30 | 45 | 10 | 92 | 5 | 86 | 8 | 21 |
| 18 | 2-[2-methoxy-4-(prop-2-enyl)phenoxy]acetic acid | ||||||||
| 3 | 2-Hydroxybenzoic acid (salicylic acid) | 30 | 22 | 10 | 75 | 7 | 81 | - | - |
| 19 | 2-(Carboxymethoxy)benzoic acid | ||||||||
| 4 | 4-Hydroxybenzoic acid | 30 | 37 | 10 | 89 | 5 | 96 | - | - |
| 20 | 4-(Carboxymethoxy)benzoic acid | ||||||||
| 5 | 4-Hydroxybenzoic acid methyl ester (nipagin M) | 30 | 20 * | 10 | 58 * | 5 | 75 * | 8 | 25 |
| 21 | 4-(Carboxymethoxy)benzoic acid methyl ester | ||||||||
| 6 | 4-Hydroxybenzoic acid propyl ester (nipagin P) | 30 | 31 * | 10 | 67 * | 7 | 68 * | 8 | 35 |
| 22 | 4-(Carboxymethoxy)benzoic acid propyl ester | ||||||||
| 7 | N-(4-hydroxyphenyl)acetamide (paracetamol) | 30 | 27 | 10 | 68 | 5 | 86 | 7 | 23 |
| 23 | 2-(4-acetamidophenoxy)acetic acid | ||||||||
| 8 | 1-Naphtol | 30 | 35 | 10 | 72 | 10 | 88 | 8 | 33 |
| 24 | 2-Naphtalen-1-yloxyacetic acid | ||||||||
| 9 | 2-Naphtol | 30 | 38 | 10 | 75 | 10 | 91 | - | - |
| 25 | 2-Naphtalen-2-yloxyacetic acid | ||||||||
| 10 | 1,4-Dihybroxybenzen (hydrochinon) | 30 | 40 | 10 | 54 | 10 | 84 | 8 | 36 |
| 26 | 2-[4-(Carboxymethoxy)phenoxy]acetic acid | ||||||||
| 11 | 1,3-Dihydroxybenzen (resorcinol) | 30 | 32 | 10 | 64 | 10 | 87 | - | - |
| 27 | 2-[3-(Carboxymetoxy)phenoxy]acetic acid | ||||||||
| 12 | 1,7-Bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-dien-3,5-dione (curcumin) | 30 | - | 10 | 15 | 10 | 36 | 8 | - |
| 28 | 2-{4-[7-(4-hydroxy-3-metoxyphenyl)-3,5-dioxohepta-1,6-dienyl]-2-metoxyphenoxy}acetic acid | ||||||||
| 13 | 1,3,5-Trihydroksybenzen (phloroglucinol) | 30 | 39 | 10 | 66 | 10 | 79 | 8 | 38 |
| 29 | 2-[3,5-(Bis-carboxymetoxy)phenoxy]acetic acid | ||||||||
| 14 | 1,2,3-Trihydroxybezen (pyrogallol) | 30 | 29 | 10 | 71 | 10 | 83 | - | - |
| 30 | 2-[2,3-Bis(carboxymetoxy)phenoxy]acetic acid | ||||||||
| 15 | 5,7-Dihydroxy-3-(4-hydroxy phenyl)chromen-4-one (genistein) | 30 | - | 10 | - | 30 | 41 | 8 | - |
| 31 | 2-[5-Hydroxy-3-(4-hydroxyphenyl)-4-oxo-4H-chromen-7-yloxy] acetic acid | ||||||||
| Method | Condition | Time | [%] |
|---|---|---|---|
| Classic | NaOH/H2O/reflux | 7 h | 23 |
| Classic | DMF/K2CO3/rt | 24 h | - |
| Classic | Aceton/K2CO3/rt | 24 h | - |
| US | US-800W/60–70 °C | 30 min | 27 |
| MW/solvent free | MW/200W/NaOH | 10 min | 68 |
| MW | MW/200W/NaOH/H2O/~100 °C | 10 min | 52 |
| MW-US | MW-200W/US-800W/~100 °C | 5–10 min | 86 |
| US | MW | MW-US | |
|---|---|---|---|
| Power | 800 W | 200 W | ∑MW+US |
| Frequency | 28 KHz | 2450 MHz | ∑MW+US |
| Temperature | 60–70 °C | 95–100 °C | 95–100 °C |
| Time | 30 min | 10 min | 5–10 min |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pawełczyk, A.; Sowa-Kasprzak, K.; Olender, D.; Zaprutko, L. Microwave (MW), Ultrasound (US) and Combined Synergic MW-US Strategies for Rapid Functionalization of Pharmaceutical Use Phenols. Molecules 2018, 23, 2360. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23092360
Pawełczyk A, Sowa-Kasprzak K, Olender D, Zaprutko L. Microwave (MW), Ultrasound (US) and Combined Synergic MW-US Strategies for Rapid Functionalization of Pharmaceutical Use Phenols. Molecules. 2018; 23(9):2360. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23092360
Chicago/Turabian StylePawełczyk, Anna, Katarzyna Sowa-Kasprzak, Dorota Olender, and Lucjusz Zaprutko. 2018. "Microwave (MW), Ultrasound (US) and Combined Synergic MW-US Strategies for Rapid Functionalization of Pharmaceutical Use Phenols" Molecules 23, no. 9: 2360. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23092360