
Molecules that Inhibit Bacterial Resistance Enzymes

1
Institute of Comparative Medicine, College of Veterinary Medicine, Yangzhou University, Yangzhou 225009, China
2
Jiangsu Co-innovation Center for Prevention and Control of Important Animal Infectious Diseases and Zoonoses, Yangzhou 225009, China
*
Author to whom correspondence should be addressed.
Molecules 2019, 24(1), 43; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24010043
Submission received: 20 November 2018
/
Revised: 18 December 2018
/
Accepted: 19 December 2018
/
Published: 22 December 2018
(This article belongs to the Collection Antibiotics & Superbugs: New Strategies to Combat Antimicrobial Resistance)
Abstract
:Antibiotic resistance mediated by bacterial enzymes constitutes an unmet clinical challenge for public health, particularly for those currently used antibiotics that are recognized as “last-resort” defense against multidrug-resistant (MDR) bacteria. Inhibitors of resistance enzymes offer an alternative strategy to counter this threat. The combination of inhibitors and antibiotics could effectively prolong the lifespan of clinically relevant antibiotics and minimize the impact and emergence of resistance. In this review, we first provide a brief overview of antibiotic resistance mechanism by bacterial secreted enzymes. Furthermore, we summarize the potential inhibitors that sabotage these resistance pathways and restore the bactericidal activity of inactive antibiotics. Finally, the faced challenges and an outlook for the development of more effective and safer resistance enzyme inhibitors are discussed.
1. Introduction
The emergence, prevalence, and rapid spread of antibiotic resistance in pathogens represent an urgent threat to our medical system for the treatment of bacterial infectious diseases [1,2,3]. According to the Centers for Disease Control and Prevention (CDC) reports, at least 2,000,000 illnesses are caused by resistant bacteria annually in the USA that result in approximately 23,000 deaths [4]. There is no doubt that we have entered the post-antibiotic age [5], in which multidrug resistant (MDR) bacteria are increasingly disseminating in the community and cannot be effectively treated by commonly used antibiotics. Worryingly, the sluggish progress in the development of new antibiotics with unique modes of action further exacerbates this problem [6,7]. Although billions of dollars and longer than a decade have been invested, only a few antibiotics were approved by the Food and Drug Administration (FDA) in the past decades [8,9]. Despite these ongoing efforts [10,11,12,13,14,15,16,17,18,19,20], the rigorous challenges faced in novel antibiotics discovery require additional strategies to tackle multidrug-resistant bacteria. Antibiotic adjuvants [21,22], an alternative to antibiotics (i.e., antibodies, probiotics, vaccines, and anti-virulence drugs) [23,24,25], offer two feasible approaches [26]. Given that these alternatives to antibiotics are mainly administered as adjunctive or preventive therapies in a clinical setting [27], antibiotic adjuvants such as resistance enzyme inhibitors are currently the most promising in the fight against antibiotic-resistant bacteria from both scientific and commercial sides.
These compounds that directly inhibit antibiotic resistance could be explored as antibiotic adjuvants. Thus, elucidation of antibiotic resistance mechanisms would contribute to novel antibiotic adjuvants’ discovery. To date, the molecular mechanisms of antibiotic resistance involve reduced cell membrane permeability, chemical structure changes of targets, active efflux of the intracellular antibiotics, and inactivation of antibiotics by resistance enzymes [5,28,29]. Taking into account these resistance mechanisms, the inhibitors of inactivating enzymes and efflux pumps have great promise as antibiotics’ adjuvants. Theretofore, small molecule inhibitors that target the bacterial efflux pumps have been previously reported [30,31,32,33,34] and systematically reviewed [35,36,37]. Notably, the antibiotic resistance mediated by bacterial enzymes poses an unmet clinical challenge, particularly for those antibiotics that are recognized as “last-resort” defense. For example, metallo-β-lactamases (MBLs) such as New Delhi metallo-β-lactamase 1 (NDM-1) [38] could hydrolyze the carbapenems, and plasmid-mediated colistin resistance (mediated by MCR-1) [39] could decrease the affinity between colistin and lipopolysaccharide (LPS) of the outer membrane in Gram-negative bacteria [40]. Therefore, inhibition of resistance enzyme activity provides a potential therapeutic approach for fighting antibiotic-resistant bacteria.
Different from conventional antibiotics that directly target the components or progress that are essential for bacterial growth, inhibitors of resistance enzyme always have no or little direct antibacterial activity. However, these inhibitors could decrease the activity of resistance enzymes by specifically binding to the active site or inhibiting the transcription and translation of resistance genes. For example, clavulanic acid, a β-lactam-containing natural product, is a valuable inhibitor of β-lactamase [41]. Most notably, enzyme inhibitors may have the significant advantage that they do not exert as much selective pressure as conventional antibacterial agents. They do not directly target growth or viability, thereby conceivably minimizing the evolution of resistance [21]. This review offers a brief overview of antibiotic resistance mechanisms promoted by bacterial secreted enzymes and the development of potent resistance enzyme inhibitors in the battle against antibiotic-resistant bacteria. Besides, challenges and outlook for future efforts to discover therapeutic resistance inhibitors are also discussed.
2. Antibiotic Resistance Mediated by Bacterial Enzymes
Most antibiotics act by specifically binding to their targets, thereby preventing the physiological function of these targets. However, in the long-term competition with antibiotics, some bacteria have evolved multi-detoxifying enzymes to inactivate clinically relevant antibiotics such as β-lactams, carbapenems, and aminoglycosides [42]. Based on their inactivation mechanisms, these resistance enzymes are majorly divided into hydrolytic enzymes and modifying enzymes (Figure 1).
2.1. Inactivation of Antibiotics by Hydrolytic Enzymes
β-Lactamases are one of the most common and important hydrolytic enzymes that inactivate penicillins, cephalosporins, carbapenems, and monobactams [43]. In particular, the evolution of β-lactam antibiotic classes with improved properties is associated with the emergence of novel β-lactamases with altered spectra of activity. For example, the early β-lactamases (penicillinase) are only active against first-generation β-lactams [44], whereas the subsequent extended-spectrum β-lactamases (ESBLs) [45] have hydrolytic activity against [46]. Alarmingly, the spread of diverse ESBLs and carbapenemases, including the imipenemase (IMP), Verona integron-encoded metallo-β-lactamase (VIM), Klebsiella pneumoniae carbapenemase (KPC) [47], oxacillinase (OXA) [48], and New Delhi metallo-β-lactamase (NDM) [49] enzymes in Gram-negative bacteria such as K. pneumoniae, Escherichia coli, Pseudomonas aeruginosa, and Acinetobacter baumannii, has facilitated the emergence of isolates that are resistant to all β-lactam antibiotics [50].
β-Lactamases hydrolytically cleave β-lactam antibiotics by means of two main molecular strategies: Serine residue covalently capture and destroy antibiotic activity, whereas Zn2+ atoms activate a water molecule to hydrolyze the antibiotics (Figure 2).
Based on a classification that was first proposed by Ambler, β-lactamases are classified into four classes, namely, types A to D [51,52]. Among them, type A (penicillinase), C (cephalosporins enzyme), and D (oxacillinase) belong to Ser-β-lactamases, whereas type B refers to Zn2+-dependent metallo β-lactamases [53,54]. As a result of the increasing number of bacteria carrying ESBL genes, the clinical use of carbapenem antibiotics has significantly increased over the past decade. In turn, this has been accompanied by increasing clinical isolates carrying Zn2+-dependent metallo (type B)-β-lactamases [55,56], particularly for NDM. Since its first description in India in 2009 [57], NDM carbapenemase has become one of the most widespread carbapenemases [58,59] and it is found worldwide in numerous Gram-negative pathogens, including A. baumannii, E. coli, and K. pneumoniae [60,61]. Worryingly, NDM-1 and its numerous variants [62,63,64,65,66,67,68,69] confer resistance to all β-lactams except for monocyclic aztreonam [70]. Such hydrolysis also occurs for other classes of antibiotics such as macrolides and fosfomycin. Macrolide antibiotics such as erythromycin kill bacteria by binding to the large subunit of ribosomes and interfering with protein synthesis [71]. Macrolides are cyclized via an ester bond, which is the critical step for their activity. However, macrolide esterases could destroy the antibiotic’s cyclic structure by opening the ring, which [72] is then followed by an internal cyclization and dehydration through intramolecular condensation (Figure 3A) [73,74]. As a result, the ring-opened macrolides lose their antibacterial activity. Fosfomycin [75], an epoxide antibiotic, targets the enzyme UDP-N-acetylglucosamine enolpyruvyl transferase (MurA) that is essential for the biosynthesis of peptidoglycan [76]. Enzymatic hydrolysis to fosfomycin at position 1 occurs through the destruction of the reactive epoxide via either a thiol-containing co-substrate or a water-mediated ring opening, which depend on three similar but mechanistically distinct fosfomycin-resistance proteins, i.e., FosA, FosB, and FosX (Figure 3B) [77]. FosA, purified from Gram-negative bacteria, is a Mn2+- and K+-dependent glutathione transferase. FosB is a Mg2+-dependent l-cysteine thioltransferase, whereas FosX is a Mn2+-dependent fosfomycin-specific epoxide hydrolase [78].
2.2. Inactivation of Antibiotic by Modifying Enzymes
2.2.1. Modification on Antibiotics
Antibiotic-modifying enzymes (also termed group transferases) are the most diverse and largest family of bacterial resistance enzymes, which inactivate antibiotics by preventing binding to their respective targets. This inactivation of antibiotics depends on the modifying enzymes’ function, i.e., the addition of chemical groups to vulnerable sites of the antibiotic molecule or antibiotic targets. Various different chemical groups can be transferred, including acyl [79,80], phosphate [81], nucleotidyl [82,83], ribitoyl [42], glycosyl [84], and thiol groups [75,85,86] (Figure 4).
Aminoglycoside antibiotics are particularly vulnerable to modification as they tend to be large molecules with many exposed hydroxyl and amide groups [87]. Aminoglycoside-modifying enzymes confer high levels of resistance to antibiotics via group modification [88]. There are three principal classes of aminoglycoside-modifying enzymes: nucleotidyltransferases (ANTs), phosphotransferases (APHs), and acetyltransferases (AACs) [89,90,91]. These transferases are evolutionarily diverse and vary in the aminoglycosides that they can modify and in the part of the molecule that is modified. In addition, several other antibiotic classes are susceptible to modification, including lincomycin [92], chlormamphenicol [93] (Figure 5), and macrolides [29] (Figure 6). Except for these groups, hydroxylation mediated by redox enzymes could also confer antibiotics resistance [94]. For example, the hydroxylation of tetracycline antibiotics at position 11, which is required for antibacterial activity, disrupts the Mg2+-binding site of the antibiotic [95,96] (Figure 7A). Another example of redox-mediated resistance regards the antibiotic streptogramin, in which the key ketone group at position 16 is turned to hydroxymethyl, thereby making the drug inactive [97] (Figure 7B).
2.2.2. Modification of Antibiotic Targets
The vast majority of modifying enzymes inactivate antibiotics by transferring chemical groups on antibiotics’ molecules. However, there is a growing concern that bacteria can also modify antibiotics’ targets to inactivate the antibiotics. A recent example is the plasmid-borne colistin resistance mediated by MCR-1 [39]. Colistin was discovered in the 1950s [98,99] and widely used for bacterial infection control from the late 1950s to the late 1970s. This cationic peptide antibiotic was then gradually withdrawn from clinical practice owing to its potential neurotoxicity [100,101] and nephrotoxicity [102]. However, the widespread emergence of carbapenem-resistant Enterobacteriaceae (CRE) led to the re-emergence of colistin [103,104], which is currently recognized as a last-resort antibiotic against multidrug resistant Gram-negative bacteria. Colistin acts by interfering with anionic lipopolysaccharide (LPS) present in Gram-negative bacterial outer membrane [105]. However, the overuse and misuse of colistin in the clinical setting and food-producing animals have led to the emergence of colistin resistance [106]. In 2015, the first plasmid-mediated colistin resistant gene mcr-1 in Enterobacteriaceae was reported in China [39]. The phosphoethanolamine (pEtN) transferase coded by mcr-1 catalyzes the addition of the pEtN moiety to the phosphate groups in the lipid A component of LPS, resulting in decreased affinity between colistin and LPS (Figure 8) [39]. Most notably, the high transferability of mcr-1 led to its global distribution in Gram-negative bacteria [107]. Meanwhile, a series of mcr-1 variants (mcr-2/3/4/5/6/7/8) has been reported indicative of genetic adaptability [108,109,110,111,112,113,114]. Another example is the wide dissemination of plasmid-encoded chloramphenicol-florfenicol resistance (cfr) methyltransferase, which specifically methylates the adenine of position 2503 in the 23S rRNA [115,116]. Such an enzyme thereby confers resistance to a wide range of ribosome-targeted antibiotics, including phenicols, pleuromutilins, streptogramins, lincosamides, selected 16-membered macrolides, and also oxazolidinones (such as linezolid) [117,118]. Notably, cfr methyltransferase has been reported in Gram-positive and Gram-negative pathogens of both human and animal origins [119,120].
3. Bacterial Resistance Enzymes Inhibitors
An alternative strategy to counter the antibiotic resistance mediated by resistance enzyme is the use of inhibitors that specifically bind to the active site of enzymes and restore the activity of antibiotics. During the past decades, many enzyme inhibitors have been identified and introduced into the clinical setting, particularly for β-lactamases. In the following, we will briefly summarize the development of hydrolytic enzyme inhibitors and modifying enzyme inhibitors occurred in the past decades and highlight the novel, recently identified enzyme inhibitors.
3.1. Hydrolytic Enzymes Inhibitors
3.1.1. Ser-β-lactamases Inhibitors
Clavulanic acid (Figure 9-(1)), a β-lactam-containing natural product, was the first identified β-lactamases inhibitor in the 1970s, isolated from a β-lactam antibiotic-producing strain of Streptomyces clavuligerus [41,122]. Clavulanic acid has poor antibiotic activity but it does show potent and irreversible inactivation of Ser-β-lactamases [123]. This unprecedented discovery gave rise to the first antibiotic/adjuvant combination that could be successfully approved in clinical use. For example, Augmentin (amoxicillin/clavulanic acid) has been applied in the clinic for over 30 years [124]. The wide use of clavulanic acid facilitated the development of other β-lactamase inhibitors, including the β-lactam sulfones tazobactam (Figure 9-(2)) and sulbactam (Figure 9-(3)) that are paired with piperacillin and ampicillin, respectively [125,126]. However, these β-lactam sulfones inhibitors are predominantly restricted to type A Ser-β-lactamases. The increase in type C and type D β-lactamases such as plasmid-borne AmpC [127] and oxacillinases [128] have progressively eroded the efficacy of these three inhibitors. To counter this threat, a new class of non-β-lactam β-lactamase inhibitor, diazabicyclooctanes (DABCOs), was introduced into clinical practice [129]. For example, avibactam (Figure 9-(4)) maintains the capacity to covalently acylate its β-lactamase targets [130], including CTX-M [131,132,133], AmpCs, and K. pneumoniae carbapenemase (KPC) [134,135]. Avycaz (a combination of ceftazidime and acibactam) was approved for clinical use by the FDA in 2015 [136]. Besides, DABCO relebactam (Figure 9-(5)) paired with imipenem or cilastatin are also being investigated [137,138]. Notably, neither avibactam nor relebactam are effective against A. baumannii infections, owing to a wide variety of class D β-lactamases expressed by this micoorganism [139]. By modifying DABCO scaffold to extend its spectrum of activity to a broad range of class D enzymes, ETX2514 (Figure 9-(6)) was optimized as the best candidate with high inhibition activity towards clinically relevant class A, C, and D β-lactamases and penicillin-binding proteins (PBPs). Importantly, the combination of sulbactam and ETX2514 shows high efficacy in MDR A. baumannii mouse infection models [140]. In addition, a novel structural β-lactamases inhibitor, Vaborbactam (Figure 9-(7)) based on a cyclic boronic acid pharmacophore was discovered, which is used paired with meropenem against Gram-negative bacteria, particularly for KPC-producing carbapenem-resistant Enterobacteriaceae (CRE) [141] (Figure 9).
3.1.2. Metallo-β-lactamases Inhibitors
Except for Ser-β-lactamase inhibitors, many efforts are being made to identify potential metallo-β-lactamases inhibitors because of the increasing concern for MDR Gram-negative bacterial infections. There are two different strategies to identify clinically useful metallo-β-lactamases inhibitors, namely, Zn2+-dependent and Zn2+-independent strategies. Because Zn2+ ions are essential for enzymes catalysis, most studies have focused on zinc ion chelation. For example, the well-known blood pressure-modulating drug l-captopril and its d-stereoisomer have proven to be useful inhibitors of several metallo-β-lactamases enzymes, including NDM-1, VIM-1, and IMP-7 in vitro [142,143]. Besides, some previously approved thiol-containing drugs and their derivatives, including thiorphan, tiopronin, and dimercaprol, inhibit the activity of NDM-1, VIM-1, and IMP-7 [144]. By means of an NMR counter-screening of selected compounds, Schofield’s group found that isoquinoline showed affinity for VIM-2 but not for other enzymes. Crystallographic analysis revealed that the exocyclic carboxylate displays stronger hydrogen-bonding interactions with the conserved Arg228 and Asn233 of the enzymes [145]. Notably, this is the first report about non-metal ligand inhibitors of metallo-β-lactamases, providing a new avenue for inhibitors screening. Nevertheless, a few effective inhibitors both in vitro and in vivo were identified [146]. Aspergillomarasmine A (Figure 9-(8)), a fungus-derived natural product [147,148] discovered by a cell-based screen for inhibitors of NDM-1, fully restored the activity of meropenem in mice infection models expressing NDM-1 K. pneumoniae [149]. Besides, several inhibitors were also discovered that target other hydrolytic enzymes. For instance, several simple phosphonates, including the antiviral compound phosphonoformate (Ki = 0.4 ± 0.1 µM, Kd ≈ 0.2 µM), phosphonoacetate, and acetylphosphonate (Figure 10-(9/10/11)), were shown to be potent inhibitors of the fosfomycin-resistance protein FosA [150].
3.2. Modifying Enzymes Inhibitors
3.2.1. Aminoglycoside Modifying Enzymes Inhibitors
Aminoglycoside antibiotics are the foremost bactericidal drugs that specifically target the ribosome [151], which show potent antibacterial activity against both Gram-negative and Gram-positive pathogens. For instance, gentamicin, tobramycin, and amikacin are still widely used in clinical settings [152]. The prevalent resistance mechanisms of aminoglycoside antibiotics have been briefly described in Section 2.2, especially the aminoglycoside-modifying enzymes (AMEs) including nucleotidyltransferases (ANTs), phosphotransferases (APHs), and acetyltransferases (AACs). Some AMEs inhibitors have been reported to inhibit ANTs or multiple enzymes and restore antibiotics’ activity against resistant bacteria (Figure 11). The oxidopyrylium cycloaddition/ring-opening strategy was applied to determine the inhibition activity of a library of synthetic a-hydroxytropolones against ANT(2″)-Ia. As a consequence, two of these synthetic compounds, including α-hydroxytropolones 3e and 3o (Figure 11-(12/13)), restored gentamicin activity against ANT-(2″)-Ia-expressing bacteria [153]. Besides, some compounds showed evidence that they simultaneously inhibit multiple AMEs. A library of 45 non-carbohydrate AME inhibitors (Figure 11-(14)) was designed based on the 1,3-diamine pharmacophore found in aminoglycoside structures and were shown to be competitive inhibitors of ANT(2″)-Ia and APH(3′)-IIIa [154]. A detailed structure–activity relationship study of non-carbohydrate inhibitors revealed that the 3-(dimethylamino) propylamine moiety was critical for their inhibitory activity [155].
Similarly, several potent inhibitors of APHs were identified (Figure 12). Because of the high similarity of APHs to eukaryotic protein kinases in both protein structure and mechanism, protein kinase inhibitors were investigated as potential inhibitors of APHs, including CKI-7 [156], the aforementioned natural product kinase inhibitor quercetin [157], and damnacanthal (Figure 12-(15/16/17)) [158]. Besides, the lipid PI-3 kinase inhibitor wortmannin (Figure 12-(18)) also displays inhibitory activity towards APH(2´´)-Ia [159], owing to the analogous protein conformation between lipid kinases and APHs. However, these inhibitors may nonspecifically bind to eukaryotic protein kinases or lipid kinases in vivo, resulting in undesirable side effects. Therefore, compounds with a highly selective binding mechanism to AMEs would have a beneficial outcome, particularly for their safety in vivo. Meaningfully, pyrazolopyrimidines such as 2a (Figure 12-(19)) binds to and inhibits APH(3´)-I (a dominant resistance mechanism in Gram-negative pathogens) by a pathway that is distinct from the binding mode of human kinases [160], offering a potential route for derivatives that selectively inhibit APHs over human targets.
The development of computer-assisted technology undoubtedly facilitates a versatile screening approach for novel resistance enzyme inhibitors, particularly for high-throughput screening (HTS). For instance, in silico molecular docking simulation provides a viable strategy to identify enzyme inhibitors [161,162,163]. One classic example is Glide, which offers a rapid, accurate docking and scoring approach for the screening of potential inhibitors [164]. Unlike other methods for docking ligands to the rigid 3D structure of a known protein receptor, Glide approximates a complete systematic search of the orientation, conformation, and positional space of the docked ligand [165,166]. These characters confirm its higher accuracy with respect to other methods such as GOLD [167,168], FlexX [169,170], and Surflex [171,172]. By employing this strategy to screen a collection of 280,000 compounds, 1-[3-(2-aminoethyl)benzyl]-3-(piperidin-1-ylmethyl)pyrrolidin-3-ol (Figure 13-(20)) was found. It inhibits the acetylation of aminoglycosides in vitro with IC50 values of 39.7 and 34.9 μM using kanamycin A or amikacin as substrates [173]. To screen the inhibitors of AACs from Mycobacterium tuberculosis, Keith et al. tested 23,000 molecules, of which 300 (1.3%) showed a reasonable degree of inhibition against AACs, and one compound (Figure 13-(21)) showed IC50 values of 0.364 μM [174]. Aranorosin (Figure 13-(22)), a natural product isolated from Gymnascella aurantiaca, circumvents aminoglycoside antibiotics resistance by inhibiting AME AAC(6′)-Ie/APH(2′′)-Ia [175] (Figure 13).
3.2.2. MCR-1 Inhibitors
Two successful strategies were applied for screening MCR-1 inhibitors, including directly looking for molecules interacting with MCR-1 protein and targeting its mRNA (Figure 14). Pterostilene (Figure 14-(23), trans-3,5-dimethoxy-4′-hydroxystilbene), isolated from fresh leaves or fruit, has been previously reported owing to its potent anticancer [176,177], anti-inflammatory [178], and antioxidant activities [179]. Recently, pterostilene was found to be a novel MCR-1 inhibitor [180], which rescues the efficacy of polymyxin B both in vitro and in vivo. A significant synergistic effect between pterostilbene and polymyxin B (fractional inhibitory concentration index 0.156 or 0.188) against MCR-producing E. coli of both human and animal origins was observed. However, more mechanistic studies are required to elucidate the potentiation activity of pterostilene. Transcription inhibition of enzyme-coding genes also provides a potential approach to rescue antibiotic activity, such as specifically designed antisense oligonucleotides [181,182,183], which suppress bacterial genes expression on the basis of complementary interaction. The obstacle that mostly limits the adjuvant potential of antisense oligonucleotides is the highly impermeable bacterial membrane [184]. Besides, bacteria do not initiatively uptake oligonucleotides from their environment [185]. Therefore, conjugation with vectors such as lipids [186], cell-penetrating peptides [187], polymers [188], and nanoparticles [189] would facilitate the delivery of antisense oligonucleotides to various bacterial cells. For example, an optimized cationic peptide-conjugated phosphorodiamidate morpholino oligomer (PPMO) Mcr1-0545 (Figure 14-(24)) was designed to target mcr-1 mRNA, preventing translation and modulating colistin resistance [190]. Excitingly, the combination of Mcr1-0545 with colistin offered a significant survival advantage in the murine sepsis infection model. Meaningfully, such antisense RNA strategies could be expanded to other applications for combating antibiotic bacteria by targeting different function genes [191,192,193], including bacterial essential gene (thus killing bacteria, like antibiotics) [194,195,196,197], resistance genes (thus rescuing antibiotic activity, like adjuvants) [190,198], and virulence genes (thus preventing bacterial pathogenesis, like anti-virulence agents) [199,200,201,202].
4. Outlook and Conclusions
Although numerous compounds that inhibit different bacterial resistance enzymes have been found, instances of their clinical use are limited to β-lactamases inhibitors. There are two major concerns, that is, low efficacy and unspecific toxicity of these compounds in vivo. It is noteworthy that some candidates failed to demonstrate significant efficacy in vivo due to their liability to degradation by proteases. Thus, activity screening in vitro alone is not enough to show evidence of their therapeutic potential. Alternatively, some animal infection models could be straightforwardly utilized to evaluate the therapeutic effectiveness of identified leads, such as the silkworm infection model, which has conserved immune response and comparable pharmacokinetics with mammals [203]. In addition, the toxicity of some enzymes inhibitors is also a key factor that leads to failure during safety evaluation. This problem could be addressed to identify more specific resistance enzyme inhibitors, which specifically bind to prokaryotic (but not eukaryotic) enzymes. Such a solution requires a better understanding of the conformation of resistance enzymes and of the binding sites of the inhibitors.
Antibiotics resistance, caused by bacterial resistance enzymes that have hydrolytic and modifying effects, poses a foremost and serious threat to public health worldwide. Despite the challenges faced in the screening of novel resistance enzyme inhibitors and in bringing them into the market, the discovery of enzyme inhibitors provides a complementary strategy to new antibiotic discovery. Importantly, these inhibitors can potentiate or restore the antibacterial activity of some promising antibiotics that are vulnerable and can be hydrolyzed or modified. The wide application of β-lactamase inhibitors in the clinic over the past decades is the most robust proof of concept. In addition, the pairing of inhibitors with antibiotics also contributes to minimizing the burden and emergence of antibiotic resistance. Overall, resistance enzyme inhibitors show great promise. Nevertheless, other novel strategies should be continuously explored to preserve the efficacy of antibiotics in this resistance era.
Funding
This work was supported by the National Key Research and Development Program of China (2016YFD0501310) and A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Conflicts of Interest
The authors declare no competing financial interest.
References
- Kupferschmidt, K. Resistance fighter. Science 2016, 352, 758–761. [Google Scholar] [CrossRef] [PubMed]
- Beatson, S.A.; Walker, M.J. Microbiology. Tracking antibiotic resistance. Science 2014, 345, 1454–1455. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Modarai, M.; Naylor, N.R.; Boyd, S.E.; Atun, R.; Barlow, J.; Holmes, A.H.; Johnson, A.; Robotham, J.V. Quantifying drivers of antibiotic resistance in humans: A. systematic review. Lancet. Infect. Dis. 2018, 18, e368–e378. [Google Scholar] [CrossRef]
- CDC Threat Report: ‘We Will Soon Be in a Post-Antibiotic Era’. Available online: https://www.wired.com/2013/09/cdc-amr-rpt1/ (accessed on 22 December 2018).
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ding, S.; Shen, J.; Zhu, K. Nonribosomal antibacterial peptides that target multidrug-resistant bacteria. Nat. Prod. Rep. 2018. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.D.; Wright, G.D. Antibacterial drug discovery in the resistance era. Nature 2016, 529, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug. Discov. 2007, 6, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, M.A.; Walsh, C.T. Antibiotics for emerging pathogens. Science 2009, 325, 1089–1093. [Google Scholar] [CrossRef]
- Zipperer, A.; Konnerth, M.C.; Laux, C.; Berscheid, A.; Janek, D.; Weidenmaier, C.; Burian, M.; Schilling, N.A.; Slavetinsky, C.; Marschal, M.; et al. Human commensals producing a novel antibiotic impair pathogen colonization. Nature 2016, 535, 511–516. [Google Scholar] [CrossRef]
- Johnston, C.W.; Skinnider, M.A.; Dejong, C.A.; Rees, P.N.; Chen, G.M.; Walker, C.G.; French, S.; Brown, E.D.; Berdy, J.; Liu, D.Y.; et al. Assembly and clustering of natural antibiotics guides target identification. Nat. Chem. Biol. 2016, 12, 233–239. [Google Scholar] [CrossRef]
- Chu, J.; Vila-Farres, X.; Inoyama, D.; Ternei, M.; Cohen, L.J.; Gordon, E.A.; Reddy, B.V.; Charlop-Powers, Z.; Zebroski, H.A.; Gallardo-Macias, R.; et al. Discovery of MRSA active antibiotics using primary sequence from the human microbiome. Nat. Chem. Biol. 2016, 12, 1004–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schaberle, T.F.; Hughes, D.E.; Epstein, S.; et al. A new antibiotic kills pathogens without detectable resistance. Nature 2015, 517, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, H.; Urai, M.; Ishii, K.; Yasukawa, J.; Paudel, A.; Murai, M.; Kaji, T.; Kuranaga, T.; Hamase, K.; Katsu, T.; et al. Lysocin E is a new antibiotic that targets menaquinone in the bacterial membrane. Nat. Chem. Biol. 2015, 11, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Cociancich, S.; Pesic, A.; Petras, D.; Uhlmann, S.; Kretz, J.; Schubert, V.; Vieweg, L.; Duplan, S.; Marguerettaz, M.; Noell, J.; et al. The gyrase inhibitor albicidin consists of p-aminobenzoic acids and cyanoalanine. Nat. Chem. Biol. 2015, 11, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Reynolds, K.A.; Kersten, R.D.; Ryan, K.S.; Gonzalez, D.J.; Nizet, V.; Dorrestein, P.C.; Moore, B.S. Direct cloning and refactoring of a silent lipopeptide biosynthetic gene cluster yields the antibiotic taromycin A. Proc. Natl. Acad. Sci. USA 2014, 111, 1957–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, P.A.; Koehler, M.F.T.; Girgis, H.S.; Yan, D.; Chen, Y.; Chen, Y.; Crawford, J.J.; Durk, M.R.; Higuchi, R.I.; Kang, J.; et al. Optimized arylomycins are a new class of Gram-negative antibiotics. Nature 2018, 561, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-X.; Zhong, Z.; Zhang, W.-P.; Qian, P.-Y. Discovery of cationic nonribosomal peptides as Gram-negative antibiotics through global genome mining. Nat. Commun. 2018, 9, 3273. [Google Scholar] [CrossRef]
- Hover, B.; Kim, S.; Katz, M.; Charlop-Powers, Z.; Owen, J.; Ternei, M.; Maniko, J.; Estrela, A.; Molina, H.; Park, S.; et al. Culture-independent discovery of the malacidins as calcium-dependent antibiotics with activity against multidrug-resistant Gram-positive pathogens. Nat. Microbiol. 2018, 3, 415–422. [Google Scholar] [CrossRef]
- Liu, Y.; Song, M.; Ding, S.; Zhu, K. Discovery of linear low-cationic peptides to target methicillin-resistant Staphylococcus aureus in vivo. ACS Infect. Dis. 2018. [Google Scholar] [CrossRef]
- Wright, G.D. Antibiotic adjuvants: Rescuing antibiotics from resistance. Trends Microbiol. 2016, 24, 862–871. [Google Scholar] [CrossRef]
- Farha, M.A.; Brown, E.D. Discovery of antibiotic adjuvants. Nat. Biotechnol. 2013, 31, 120. [Google Scholar] [CrossRef] [PubMed]
- Czaplewski, L.; Bax, R.; Clokie, M.; Dawson, M.; Fairhead, H.; Fischetti, V.A.; Foster, S.; Gilmore, B.F.; Hancock, R.E.; Harper, D. Alternatives to antibiotics—A pipeline portfolio review. Lancet Infect. Dis. 2016, 16, 239–251. [Google Scholar] [CrossRef]
- Johnson, B.; Abramovitch, R. Small molecules that sabotage bacterial virulence. Trends Pharmacol. Sci. 2017, 38, 339–362. [Google Scholar] [CrossRef] [PubMed]
- Garland, M.; Loscher, S.; Bogyo, M. Chemical strategies to target bacterial virulence. Chem. Rev. 2017, 117, 4422–4461. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.D.; Sutherland, A.D. New strategies for combating multidrug-resistant bacteria. Trends Mol. Med. 2007, 13, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Gill, E.E.; Franco, O.L.; Hancock, R.E. Antibiotic adjuvants: Diverse strategies for controlling drug-resistant pathogens. Chem. Biol. Drug Des. 2015, 85, 56–78. [Google Scholar] [CrossRef] [PubMed]
- Blair, J.M.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol 2015, 13, 42–51. [Google Scholar] [CrossRef]
- Alekshun, M.N.; Levy, S.B. Molecular mechanisms of antibacterial multidrug resistance. Cell 2007, 128, 1037–1050. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Goswami, S.; Gorityala, B.K.; Domalaon, R.; Lyu, Y.; Kumar, A.; Zhanel, G.G.; Schweizer, F.M. A tobramycin vector enhances synergy and efficacy of efflux pump inhibitors against multidrug-resistant Gram-negative bacteria. J. Med. Chem. 2017, 60, 3913–3932. [Google Scholar] [CrossRef]
- Lamers, R.; Cavallari, J.; Burrows, L. The efflux inhibitor phenylalanine-arginine beta-naphthylamide (PAβN) permeabilizes the outer membrane of Gram-negative bacteria. PLoS ONE 2013, 8, e60666. [Google Scholar] [CrossRef]
- Abdali, N.; Parks, J.M.; Haynes, K.M.; Chaney, J.L.; Green, A.T.; Wolloscheck, D.; Walker, J.K.; Rybenkov, V.V.; Baudry, J.; Smith, J.C. Reviving antibiotics: Efflux pump inhibitors that interact with AcrA, a membrane fusion protein of the AcrAB-TolC multidrug efflux pump. ACS Infect. Dis. 2016, 3, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, S.T.; Kwasny, S.M.; Ding, X.; Cardinale, S.C.; McCarthy, C.T.; Kim, H.-S.; Nikaido, H.; Peet, N.P.; Williams, J.D.; Bowlin, T.L. Structure–activity relationships of a novel pyranopyridine series of Gram-negative bacterial efflux pump inhibitors. Bioorg. Med. Chem. 2015, 23, 2024–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hequet, A.; Burchak, O.N.; Jeanty, M.; Guinchard, X.; Le Pihive, E.; Maigre, L.; Bouhours, P.; Schneider, D.; Maurin, M.; Paris, J.M. 1-(1H-Indol-3-yl) ethanamine derivatives as potent Staphylococcus aureus NorA efflux pump inhibitors. ChemMedChem 2014, 9, 1534–1545. [Google Scholar] [CrossRef] [PubMed]
- Opperman, T.J.; Nguyen, S.T. Recent advances toward a molecular mechanism of efflux pump inhibition. Front. Microbiol. 2015, 6, 421. [Google Scholar] [CrossRef] [PubMed]
- Pagès, J.-M.; Masi, M.; Barbe, J. Inhibitors of efflux pumps in Gram-negative bacteria. Trends Mol. Med. 2005, 11, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Venter, H.; Ma, S. Efflux pump inhibitors: A novel approach to combat efflux-mediated drug resistance in bacteria. Curr. Drug Targets 2016, 17, 702–719. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.R.; Weeks, J.; Livermore, D.M.; Toleman, M.A. Dissemination of NDM-1 positive bacteria in the New Delhi environment and its implications for human health: An environmental point prevalence study. Lancet Infect. Dis. 2011, 11, 355–362. [Google Scholar] [CrossRef]
- Liu, Y.-Y.; Wang, Y.; Walsh, T.R.; Yi, L.-X.; Zhang, R.; Spencer, J.; Doi, Y.; Tian, G.; Dong, B.; Huang, X.; et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: A microbiological and molecular biological study. Lancet Infect. Dis. 2016, 16, 161–168. [Google Scholar] [CrossRef]
- Gao, R.; Hu, Y.; Li, Z.; Sun, J.; Wang, Q.; Lin, J.; Ye, H.; Liu, F.; Srinivas, S.; Li, D. Dissemination and mechanism for the MCR-1 colistin resistance. PLoS Pathog. 2016, 12, e1005957. [Google Scholar] [CrossRef]
- Reading, C.; Cole, M. Clavulanic acid: A beta-lactamase-inhibiting beta-lactam from Streptomyces clavuligerus. Antimicrob. Agents Chemother. 1977, 11, 852–857. [Google Scholar] [CrossRef]
- Wright, G.D. Bacterial resistance to antibiotics: Enzymatic degradation and modification. Adv. Drug Deliv. Rev. 2005, 57, 1451–1470. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, G.A.; Munoz-Price, L.S. The new β-lactamases. N. Engl. J. Med. 2005, 352, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.P.; Chain, E. An enzyme from bacteria able to destroy penicillin. 1940. Rev. Infect. Dis. 1940, 10, 677. [Google Scholar]
- Paterson, D.L.; Bonomo, R.A. Extended-spectrum β-lactamases: A. clinical update. Clin. Microbiol. Rev. 2005, 18, 657–686. [Google Scholar] [CrossRef] [PubMed]
- Rd, L.J.; Clark, N.M.; Zhanel, G.G. Evolution of antimicrobial resistance among Enterobacteriaceae (focus on extended spectrum β-lactamases and carbapenemases). Expert Opin. Pharmacother. 2013, 14, 199–210. [Google Scholar]
- Fishbain, J.T.; Sinyavskiy, O.; Riederer, K.; Hujer, A.M.; Bonomo, R.A. Detection of extended spectrum β-lactamase (ESBL) and Klebsiella pneumoniae Carbapenemase (KPC) genes directly from blood cultures using a nucleic acid microarray. J. Clin. Microbiol. 2012. [Google Scholar] [CrossRef]
- Liu, X.; Thungrat, K.; Boothe, D.M. Occurrence of OXA-48 carbapenemase and other β-lactamase genes in ESBL-producing multidrug resistant Escherichia coli from dogs and cats in the United States, 2009–2013. Front. Microbiol. 2016, 7, 1057. [Google Scholar] [CrossRef] [PubMed]
- Chereau, F.; Herindrainy, P.; Garin, B.; Huynh, B.-T.; Randrianirina, F.; Padget, M.; Piola, P.; Guillemot, D.; Delarocque-Astagneau, E. ESBL–and NDM-1-producing Enterobacteriaceae colonization among pregnant women in the community in a low income country: A potential reservoir for transmission of multi-resistant Enterobacteriaceae to neonates. Antimicrob. Agents Chemother. 2015, 59, 3652–3655. [Google Scholar]
- Woodford, N.; Turton, J.F.; Livermore, D.M. Multiresistant Gram-negative bacteria: The role of high-risk clones in the dissemination of antibiotic resistance. FEMS Microbiol. Rev. 2011, 35, 736–755. [Google Scholar] [CrossRef]
- Livermore, D.M. beta-lactamases in laboratory and clinical resistance. Clin. Microbiol. Rev. 1995, 8, 557–584. [Google Scholar] [CrossRef]
- Ambler, R.P. The structure of β-lactamases. Phil. Trans. R. Soc. Lond. B 1980, 289, 321–331. [Google Scholar] [CrossRef]
- Bush, K.; Jacoby, G.A.; Medeiros, A.A. A functional classification scheme for beta-lactamases and its correlation with molecular structure. Antimicrob. Agents Chemother. 1995, 39, 1211. [Google Scholar] [CrossRef] [PubMed]
- Ambler, R.; Coulson, A.; Frere, J.-M.; Ghuysen, J.-M.; Joris, B.; Forsman, M.; Levesque, R.; Tiraby, G.; Waley, S. A standard numbering scheme for the class A beta-lactamases. Biochem. J. 1991, 276, 269. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.R.; Toleman, M.A.; Poirel, L.; Nordmann, P. Metallo-β-lactamases: The quiet before the storm? Clin. Microbiol. Rev. 2005, 18, 306–325. [Google Scholar] [CrossRef] [PubMed]
- Lisa, M.-N.; Palacios, A.R.; Aitha, M.; González, M.M.; Moreno, D.M.; Crowder, M.W.; Bonomo, R.A.; Spencer, J.; Tierney, D.L.; Llarrull, L.I. A general reaction mechanism for carbapenem hydrolysis by mononuclear and binuclear metallo-β-lactamases. Nat. Commun. 2017, 8, 538. [Google Scholar] [CrossRef] [PubMed]
- Kumarasamy, K.K.; Toleman, M.A.; Walsh, T.R.; Bagaria, J.; Butt, F.; Balakrishnan, R.; Chaudhary, U.; Doumith, M.; Giske, C.G.; Irfan, S. Emergence of a new antibiotic resistance mechanism in India, Pakistan, and the UK: A molecular, biological, and epidemiological study. Lancet Infect. Dis. 2010, 10, 597–602. [Google Scholar] [CrossRef]
- Patrice, N.; Laurent, P.; Walsh, T.R.; Livermore, D.M. The emerging NDM carbapenemases. Trends Microbiol. 2011, 19, 588–595. [Google Scholar]
- Johnson, A.P.; Woodford, N. Global spread of antibiotic resistance: The example of New Delhi metallo-β-lactamase (NDM)-mediated carbapenem resistance. J. Med. Microbiol. 2013, 62, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Dortet, L.; Poirel, L.; Nordmann, P. Worldwide dissemination of the NDM-type carbapenemases in Gram-negative bacteria. Biomed Res. Int. 2014, 2014, 249856. [Google Scholar] [CrossRef]
- Nordmann, P.; Naas, T.; Poirel, L. Global spread of carbapenemase-producing Enterobacteriaceae. Emerg. Infect. Dis. 2011, 17, 1791. [Google Scholar] [CrossRef]
- Mohibur, R.; Sanket Kumar, S.; Kashi Nath, P.; Ovejero, C.M.; Binod Kumar, P.; Aparna, T.; Avinash, S.; Srivastava, A.K.; Bruno, G.Z. Prevalence and molecular characterisation of New Delhi metallo-β-lactamases NDM-1, NDM-5, NDM-6 and NDM-7 in multidrug-resistant Enterobacteriaceae from India. Int. J. Antimicrob. Agents 2014, 44, 30–37. [Google Scholar]
- Laurent, D.; Laurent, P.; Nadia, A.; Patrice, N. New Delhi Metallo-β-Lactamase 4–producingEscherichia coliin Cameroon. Emerg. Infect. Dis. 2012, 18, 1540–1542. [Google Scholar]
- Tada, T.; Miyoshi-Akiyama, T.; Dahal, R.K.; Sah, M.K.; Ohara, H.; Kirikae, T.; Pokhrel, B.M. NDM-8 metallo-β-lactamase in a multidrug-resistant Escherichia coli strain isolated in Nepal. Antimicrob. Agents Chemother. 2013, 57, 2394–2396. [Google Scholar] [CrossRef]
- Tada, T.; Shrestha, B.; Miyoshiakiyama, T.; Shimada, K.; Ohara, H.; Kirikae, T.; Pokhrel, B.M. NDM-12, a novel New Delhi metallo-β-Lactamase variant from a carbapenem-resistant Escherichia coli clinical isolate in Nepal. Antimicrob. Agents Chemother. 2014, 58, 6302–6305. [Google Scholar] [CrossRef]
- Shrestha, B.; Tada, T.; Miyoshiakiyama, T.; Shimada, K.; Ohara, H.; Kirikae, T.; Pokhrel, B.M. Identification of a Novel NDM Variant, NDM-13, from a multidrug-resistant Escherichia coli clinical isolate in Nepal. Antimicrob. Agents Chemother. 2015, 59, 5847. [Google Scholar] [CrossRef] [PubMed]
- Zou, D.; Huang, Y.; Zhao, X.; Liu, W.; Dong, D.; Li, H.; Wang, X.; Huang, S.; Wei, X.; Yan, X. A novel New Delhi metallo-β-lactamase variant, NDM-14, isolated in a Chinese hospital possesses increased enzymatic activity against carbapenems. Antimicrob. Agents Chemother. 2015, 59, 2450–2453. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, J.; Wang, X.; Liu, D.; Ke, Y.; Wang, Y.; Shen, J. Novel variant of New Delhi metallo-β-lactamase, NDM-20, in Escherichia coli. Front. Microbiol. 2018, 9, 248. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, Y.; Walsh, T.R.; Liu, D.; Shen, Z.; Zhang, R.; Yin, W.; Yao, H.; Li, J.; Shen, J. Plasmid-mediated novel blaNDM-17 gene encoding a carbapenemase with enhanced activity in a ST48 Escherichia coli strain. Antimicrob. Agents Chemother. 2018, 61, e02233-16. [Google Scholar]
- Nordmann, P.; Poirel, L.; Carrër, A.; Toleman, M.A.; Walsh, T.R. How to detect NDM-1 producers. J. Clin. Microbiol. 2011, 49, 718–721. [Google Scholar] [CrossRef]
- Seiple, I.B.; Zhang, Z.; Jakubec, P.; Langlois-Mercier, A.; Wright, P.M.; Hog, D.T.; Yabu, K.; Allu, S.R.; Fukuzaki, T.; Carlsen, P.N. A platform for the discovery of new macrolide antibiotics. Nature 2016, 533, 338. [Google Scholar] [CrossRef]
- Golkar, T.; Zieliński, M.; Berghuis, A.M. Look and outlook on enzyme-mediated macrolide resistance. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef]
- Morar, M.; Pengelly, K.; Koteva, K.; Wright, G.D. Mechanism and diversity of the erythromycin esterase family of enzymes. Biochemistry 2012, 51, 1740–1751. [Google Scholar] [CrossRef] [PubMed]
- Dinos, G.P. The macrolide antibiotic renaissance. Br. J. Pharmacol. 2017, 174, 2967–2983. [Google Scholar] [CrossRef] [PubMed]
- Falagas, M.E.; Vouloumanou, E.K.; Samonis, G.; Vardakas, K.Z. Fosfomycin. Clin. Microbiol. Rev. 2016, 29, 321–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahan, F.M.; Kahan, J.S.; Cassidy, P.J.; Kropp, H. The mechanism of action of fosfomycin (phosphonomycin). Ann. N. Y. Acad. Sci. 1974, 235, 364–386. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.K.; Keithly, M.E.; Sulikowski, G.A.; Armstrong, R.N. Diversity in fosfomycin resistance proteins. Perspect. Sci. 2015, 4, 17–23. [Google Scholar] [CrossRef]
- Rigsby, R.E.; Fillgrove, K.L.; Beihoffer, L.A.; Armstrong, R.N. Fosfomycin resistance proteins: A nexus of glutathione transferases and epoxide hydrolases in a metalloenzyme superfamily. Methods Enzymol. 2005, 401, 367–379. [Google Scholar] [PubMed]
- Green, K.D.; Chen, W.; Houghton, J.L.; Fridman, M.; Garneau-Tsodikova, S. Exploring the substrate promiscuity of drug-modifying enzymes for the chemoenzymatic generation of N-acylated aminoglycosides. Chembiochem 2010, 11, 119–126. [Google Scholar] [CrossRef]
- Galimand, M.; Fishovitz, J.; Lambert, T.; Barbe, V.; Zajicek, J.; Mobashery, S.; Courvalin, P. AAC(3)-XI, a new aminoglycoside 3-N-acetyltransferase from Corynebacterium striatum. Antimicrob. Agents Chemother. 2015, 59, 5647–5653. [Google Scholar] [CrossRef]
- Wright, G.D.; Thompson, P.R. Aminoglycoside phosphotransferases: Proteins, structure, and mechanism. Front. Biosci. 1999, 4, D9–D21. [Google Scholar]
- Zhang, G.; Leclercq, S.O.; Tian, J.; Wang, C.; Yahara, K.; Ai, G.; Liu, S.; Feng, J. A new subclass of intrinsic aminoglycoside nucleotidyltransferases, ANT(3′′)-II, is horizontally transferred among Acinetobacter spp. by homologous recombination. PLoS Genet. 2017, 13, e1006602. [Google Scholar] [CrossRef]
- Kocaman, S.; Selvaraj, B.; Wright, E.; Cuneo, M.; Serpersu, E. Investigation of the molecular mechanisms which result in aminoglycoside nucleotidyltransferase 4′(ANT4) variants with different levels of thermostability. Biophys. J. 2018, 114, 52a. [Google Scholar] [CrossRef]
- Wang, C.-M.; Zhao, F.-L.; Zhang, L.; Chai, X.-Y.; Meng, Q.-G. Synthesis and antibacterial evaluation of a series of 11, 12-cyclic carbonate azithromycin-3-o-descladinosyl-3-o-carbamoyl glycosyl derivatives. Molecules 2017, 22, 2146. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Tomich, A.D.; McElheny, C.L.; Cooper, V.S.; Tait-Kamradt, A.; Wang, M.; Hu, F.; Rice, L.B.; Sluis-Cremer, N.; Doi, Y. High-level fosfomycin resistance in vancomycin-resistant Enterococcus faecium. Emerg. Infect. Dis. 2017, 23, 1902. [Google Scholar] [CrossRef] [PubMed]
- Falagas, M.E.; Athanasaki, F.; Voulgaris, G.L.; Triarides, N.A.; Vardakas, K.Z. Resistance to fosfomycin: Mechanisms, frequency and clinical consequences. Int. J. Antimicrob. Agents 2018. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.S.; Tolmasky, M.E. Aminoglycoside Modifying Enzymes. Drug Resist. Update. 2010, 13, 151–171. [Google Scholar] [CrossRef]
- Jana, S.; Deb, J. Molecular understanding of aminoglycoside action and resistance. Appl. Microbiol. Biotechnol. 2006, 70, 140–150. [Google Scholar] [CrossRef]
- Azucena, E.; Mobashery, S. Aminoglycoside-modifying enzymes: Mechanisms of catalytic processes and inhibition. Drug Resist. Update. 2001, 4, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Shaw, K.; Rather, P.; Hare, R.; Miller, G. Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol. Rev. 1993, 57, 138–163. [Google Scholar]
- Wright, G.D. Aminoglycoside-modifying enzymes. Curr. Opin. Microbiol. 1999, 2, 499–503. [Google Scholar] [CrossRef]
- Petinaki, E.; Guérin-Faublée, V.; Pichereau, V.; Villers, C.; Achard, A.; Malbruny, B.; Leclercq, R. Lincomycin resistance gene lnu(D) in Streptococcus uberis. Antimicrob. Agents Chemother. 2008, 52, 626–630. [Google Scholar] [CrossRef]
- Robicsek, A.; Strahilevitz, J.; Jacoby, G.A.; Macielag, M.; Abbanat, D.; Park, C.H.; Bush, K.; Hooper, D.C. Fluoroquinolone-modifying enzyme: A new adaptation of a common aminoglycoside acetyltransferase. Nat. Med. 2006, 12, 83. [Google Scholar] [CrossRef] [PubMed]
- Speer, B.S.; Salyers, A.A. Characterization of a novel tetracycline resistance that functions only in aerobically grown Escherichia coli. J. Bacteriol. 1988, 170, 1423–1429. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; LaPara, T.M.; Sadowsky, M.J. Transformation of tetracycline by TetX and its subsequent degradation in a heterologous host. FEMS Microbiol. Ecol. 2015, 91. [Google Scholar] [CrossRef] [Green Version]
- Volkers, G.; Palm, G.J.; Weiss, M.S.; Wright, G.D.; Hinrichs, W. Structural basis for a new tetracycline resistance mechanism relying on the TetX monooxygenase. FEBS Lett. 2011, 585, 1061–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naamala, J.; Jaiswal, S.K.; Dakora, F.D. Antibiotics resistance in Rhizobium: Type, process, mechanism and benefit for agriculture. Curr Microbiol 2016, 72, 804–816. [Google Scholar] [CrossRef] [PubMed]
- Ainsworth, G.; Brown, A.M.; Brownlee, G. ‘Aerosporin’, an antibiotic produced by Bacillus aerosporus. Nature 1947, 160, 263. [Google Scholar] [CrossRef]
- Benedict, R.; Langlykke, A. Antibiotic activity of Bacillus polymyxa. J. Bacteriol. 1947, 54, 24. [Google Scholar]
- Wolinsky, E.; Hines, J.D. Neurotoxic and nephrotoxic effects of colistin in patients with renal disease. N. Engl. J. Med. 1962, 266, 759–762. [Google Scholar] [CrossRef]
- Dai, C.; Li, J.; Li, J. New insight in colistin induced neurotoxicity with the mitochondrial dysfunction in mice central nervous tissues. Exp. Toxicol. Pathol. 2013, 65, 941–948. [Google Scholar] [CrossRef]
- Dai, C.S.; Li, J.C.; Tang, S.S.; Li, J.; Xiao, X.L. Colistin-induced nephrotoxicity in mice involves the mitochondrial, death receptor, and endoplasmic reticulum pathways. Antimicrob. Agents Chemother. 2014, 58, 4075–4085. [Google Scholar] [CrossRef]
- Falagas, M.E.; Kasiakou, S.K.; Saravolatz, L.D. Colistin: The revival of polymyxins for the management of multidrug-resistant Gram-negative bacterial infections. Clin. Infect. Dis. 2005, 40, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Nation, R.L.; Turnidge, J.D.; Milne, R.W.; Coulthard, K.; Rayner, C.R.; Paterson, D.L. Colistin: The re-emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet. Infect. Dis. 2006, 6, 589–601. [Google Scholar] [CrossRef]
- Velkov, T.; Dai, C.; Ciccotosto, G.D.; Cappai, R.; Hoyer, D.; Li, J. Polymyxins for Cns infections: Pharmacology and neurotoxicity. Pharmacol. Ther. 2018, 181, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Al-Tawfiq, J.A.; Laxminarayan, R.; Mendelson, M. How should we respond to the emergence of plasmid-mediated colistin resistance in humans and animals? Int. J. Infect. Dis. 2017, 54, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Liu, F.; Lin, I.Y.; Gao, G.F.; Zhu, B. Dissemination of the mcr-1 colistin resistance gene. Lancet. Infect. Dis. 2016, 16, 146–147. [Google Scholar] [CrossRef]
- Yang, Y.Q.; Li, Y.X.; Lei, C.W.; Zhang, A.Y.; Wang, H.N. Novel plasmid-mediated colistin resistance gene mcr-7.1 in Klebsiella pneumoniae. J. Antimicrob. Chemother. 2018, 73, 1791–1795. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Zhou, Y.; Li, J.; Yin, W.; Wang, S.; Zhang, S.; Shen, J.; Shen, Z.; Wang, Y. Emergence of a novel mobile colistin resistance gene, mcr-8, in NDM-producing Klebsiella pneumoniae. Emerg. Microbes Infec. 2018, 7, 122. [Google Scholar] [CrossRef]
- Yin, W.; Li, H.; Shen, Y.; Liu, Z.; Wang, S.; Shen, Z.; Zhang, R.; Walsh, T.R.; Shen, J.; Wang, Y. Novel plasmid-mediated colistin resistance gene mcr-3 in Escherichia coli. mBio 2017, 8, e00543-17. [Google Scholar] [CrossRef]
- Carattoli, A.; Villa, L.; Feudi, C.; Curcio, L.; Orsini, S.; Luppi, A.; Pezzotti, G.; Magistrali, C.F. Novel plasmid-mediated colistin resistance mcr-4 gene in Salmonella and Escherichia coli, Italy 2013, Spain and Belgium, 2015 to 2016. Eurosurveillance 2017, 22. [Google Scholar] [CrossRef]
- Borowiak, M.; Fischer, J.; Hammerl, J.A.; Hendriksen, R.S.; Szabo, I.; Malorny, B. Identification of a novel transposon-associated phosphoethanolamine transferase gene, mcr-5, conferring colistin resistance in d-tartrate fermenting Salmonella enterica subsp. enterica serovar Paratyphi B. J. Antimicrob. Chemother. 2017, 72, 3317–3324. [Google Scholar] [CrossRef]
- Abuoun, M.; Stubberfield, E.J.; Duggett, N.A.; Kirchner, M.; Dormer, L.; Nunezgarcia, J.; Randall, L.P.; Lemma, F.; Crook, D.W.; Teale, C. mcr-1 and mcr-2 variant genes identified in Moraxella species isolated from pigs in Great Britain from 2014 to 2015. J. Antimicrob. Chemother. 2017, 72, 2745–2749. [Google Scholar] [CrossRef] [PubMed]
- Xavier, B.B.; Lammens, C.; Ruhal, R.; Kumar-Singh, S.; Butaye, P.; Goossens, H.; Malhotra-Kumar, S. Identification of a novel plasmid-mediated colistin-resistance gene, mcr-2, in Escherichia coli, Belgium, June 2016. Eurosurveillance 2016, 21, 30280. [Google Scholar] [CrossRef] [PubMed]
- Kaminska, K.H.; Purta, E.; Hansen, L.H.; Bujnicki, J.M.; Vester, B.; Long, K.S. Insights into the structure, function and evolution of the radical-SAM 23S rRNA methyltransferase Cfr that confers antibiotic resistance in bacteria. Nucleic Acids Res. 2009, 38, 1652–1663. [Google Scholar] [CrossRef] [Green Version]
- Arias, C.A.; Vallejo, M.; Reyes, J.; Panesso, D.; Moreno, J.; Castañeda, E.; Villegas, M.V.; Murray, B.E.; Quinn, J.P. Clinical and microbiological aspects of linezolid resistance mediated by the cfr gene encoding a 23S rRNA methyltransferase. J. Clin. Microbiol. 2008, 46, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Long, K.S.; Poehlsgaard, J.; Kehrenberg, C.; Schwarz, S.; Vester, B. The Cfr rRNA methyltransferase confers resistance to phenicols, lincosamides, oxazolidinones, pleuromutilins, and streptogramin A antibiotics. Antimicrob. Agents Chemother. 2006, 50, 2500–2505. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.; Kiratisin, P.; Mendes, R.; Panesso, D.; Singh, K.V.; Arias, C.A. Transferable plasmid-mediated resistance to linezolid due to cfr in a human clinical isolate of Enterococcus faecalis. Antimicrob. Agents Chemother. 2012, 56, 3917–3922. [Google Scholar] [CrossRef]
- Shen, J.; Wang, Y.; Schwarz, S. Presence and dissemination of the multiresistance gene cfr in Gram-positive and Gram-negative bacteria. J. Antimicrob. Chemother. 2013, 68, 1697–1706. [Google Scholar] [CrossRef]
- Locke, J.B.; Finn, J.; Hilgers, M.; Morales, G.; Rahawi, S.; Kedar, G.; Picazo, J.J.; Im, W.; Shaw, K.J.; Stein, J.L. Structure-activity relationships of diverse oxazolidinones for linezolid-resistant Staphylococcus aureus strains possessing the cfr methyltransferase gene or ribosomal mutations. Antimicrob. Agents Chemother. 2010, 54, 5337–5343. [Google Scholar] [CrossRef]
- Yang, Q.; Li, M.; Spiller, O.B.; Andrey, D.O.; Hinchliffe, P.; Li, H.; MacLean, C.; Niumsup, P.; Powell, L.; Pritchard, M. Balancing mcr-1 expression and bacterial survival is a delicate equilibrium between essential cellular defence mechanisms. Nat. Commun. 2017, 8, 2054. [Google Scholar] [CrossRef]
- Howarth, T.T.; Brown, A.G.; King, T.J. Clavulanic acid, a novel β-lactam isolated from Streptomyces clavuligerus; X-ray crystal structure analysis. Chem. Commun. 1976, 266–267. [Google Scholar] [CrossRef]
- Neu, H.C.; Fu, K.P. Clavulanic acid, a novel inhibitor of β-lactamases. Antimicrob. Agents Chemother. 1978, 14, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Vons, C.; Barry, C.; Maitre, S.; Pautrat, K.; Leconte, M.; Costaglioli, B.; Karoui, M.; Alves, A.; Dousset, B.; Valleur, P. Amoxicillin plus clavulanic acid versus appendicectomy for treatment of acute uncomplicated appendicitis: An open-label, non-inferiority, randomised controlled trial. Lancet 2011, 377, 1573–1579. [Google Scholar] [CrossRef]
- Lodise Jr, T.P.; Lomaestro, B.; Drusano, G.L. Piperacillin-tazobactam for Pseudomonas aeruginosa infection: Clinical implications of an extended-infusion dosing strategy. Clin. Infect. Dis. 2007, 44, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.S.; Levy, C.E.; Manrique, A.E.I.; Medeiros, E.A.; Costa, S.F. Severe nosocomial infections with imipenem-resistant Acinetobacter baumannii treated with ampicillin/sulbactam. Int. J. Antimicrob. Agents 2003, 21, 58–62. [Google Scholar] [CrossRef]
- Jacoby, G.A. AmpC β-lactamases. Clin. Microbiol. Rev. 2009, 22, 161–182. [Google Scholar] [CrossRef] [PubMed]
- BA, E.; SG, A. OXA β-Lactamases. Clin. Microbiol. Rev. 2014, 27, 241–263. [Google Scholar]
- Coleman, K. Diazabicyclooctanes (DBOs): A potent new class of non-β-lactam β-lactamase inhibitors. Curr. Opin. Microbiol. 2011, 14, 550–555. [Google Scholar] [CrossRef]
- Ehmann, D.E.; Jahić, H.; Ross, P.L.; Gu, R.-F.; Hu, J.; Kern, G.; Walkup, G.K.; Fisher, S.L. Avibactam is a covalent, reversible, non–β-lactam β-lactamase inhibitor. Proc. Natl. Acad. Sci. USA 2012, 109, 11663–11668. [Google Scholar] [CrossRef]
- Bonnet, R. Growing group of extended-spectrum β-lactamases: The CTX-M enzymes. Antimicrob. Agents Chemother. 2004, 48, 1–14. [Google Scholar] [CrossRef]
- Cantón, R.; Coque, T.M. The CTX-M β-lactamase pandemic. Curr. Opin. Microbiol. 2006, 9, 466–475. [Google Scholar] [CrossRef]
- Cantón, R.; González-Alba, J.M.; Galán, J.C. CTX-M enzymes: Origin and diffusion. Front. Microbiol. 2012, 3, 110. [Google Scholar] [CrossRef] [PubMed]
- Yigit, H.; Queenan, A.M.; Anderson, G.J.; Domenech-Sanchez, A.; Biddle, J.W.; Steward, C.D.; Alberti, S.; Bush, K.; Tenover, F.C. Novel carbapenem-hydrolyzing β-lactamase, KPC-1, from a carbapenem-resistant strain of Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2001, 45, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, P.; Cuzon, G.; Naas, T. The real threat of Klebsiella pneumoniae carbapenemase-producing bacteria. Lancet. Infect. Dis. 2009, 9, 228–236. [Google Scholar] [CrossRef]
- US Food and Drug Administration, FDA Approves New Antibacterial Drug Avycaz. US FDA 2015. Available online: https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugInnovation/ucm474696.htm (accessed on 22 December 2018).
- Lucasti, C.; Vasile, L.; Sandesc, D.; Venskutonis, D.; McLeroth, P.; Lala, M.; Rizk, M.L.; Brown, M.L.; Losada, M.C.; Pedley, A. Phase 2, dose-ranging study of relebactam with imipenem/cilastatin in subjects with complicated intra-abdominal infection. Antimicrob. Agents Chemother. 2016, 60, 6234–6243. [Google Scholar] [CrossRef] [PubMed]
- Lapuebla, A.; Abdallah, M.; Olafisoye, O.; Cortes, C.; Urban, C.; Landman, D.; Quale, J. Activity of imipenem with relebactam against Gram-negative pathogens from New York City. Antimicrob. Agents Chemother. 2015, 59, 5029–5031. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, S.D.; Mangani, S.; Jahić, H.; Benvenuti, M.; Durand-Reville, T.F.; De Luca, F.; Ehmann, D.E.; Rossolini, G.M.; Alm, R.A.; Docquier, J.-D. Molecular basis of selective inhibition and slow reversibility of avibactam against class D carbapenemases: A structure-guided study of OXA-24 and OXA-48. ACS Chem. Biol. 2014, 10, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Durandréville, T.F.; Guler, S.; Comitaprevoir, J.; Chen, B.; Bifulco, N.; Huynh, H.; Lahiri, S.; Shapiro, A.B.; Mcleod, S.M.; Carter, N.M. ETX2514 is a broad-spectrum β-lactamase inhibitor for the treatment of drug-resistant Gram-negative bacteria including Acinetobacter baumannii. Nat. Microbiol. 2017, 2, 17104. [Google Scholar] [CrossRef]
- Castanheira, M.; Huband, M.D.; Mendes, R.E.; Flamm, R.K. Meropenem-vaborbactam tested against contemporary Gram-negative isolates collected worldwide during 2014, including carbapenem-resistant, KPC-producing, multidrug-resistant, and extensively drug-resistant Enterobacteriaceae. Antimicrob. Agents Chemother. 2017, 62, e01904-17. [Google Scholar] [CrossRef]
- Brem, J.; van Berkel, S.S.; Zollman, D.; Lee, S.Y.; Gileadi, O.; McHugh, P.J.; Walsh, T.R.; McDonough, M.A.; Schofield, C.J. Structural basis of metallo-β-lactamase inhibition by captopril stereoisomers. Antimicrob. Agents Chemother. 2016, 60, 142–150. [Google Scholar] [CrossRef]
- García-Sáez, I.; Hopkins, J.; Papamicael, C.; Franceschini, N.; Amicosante, G.; Rossolini, G.M.; Galleni, M.; Frère, J.-M.; Dideberg, O. The 1.5-Å structure of Chryseobacterium meningosepticum zinc β-lactamase in complex with the inhibitor, D-captopril. J. Biol. Chem. 2003, 278, 23868–23873. [Google Scholar] [CrossRef]
- Klingler, F.-M.; Wichelhaus, T.A.; Frank, D.; Cuesta-Bernal, J.; El-Delik, J.; Möller, H.F.; Sjuts, H.; Göttig, S.; Koenigs, A.; Pos, K.M. Approved drugs containing thiols as inhibitors of metallo-β-lactamases: Strategy to combat multidrug-resistant bacteria. J. Med. Chem. 2015, 58, 3626–3630. [Google Scholar] [CrossRef] [PubMed]
- Li, G.-B.; Abboud, M.I.; Brem, J.; Someya, H.; Lohans, C.T.; Yang, S.-Y.; Spencer, J.; Wareham, D.W.; McDonough, M.A.; Schofield, C.J. NMR-filtered virtual screening leads to non-metal chelating metallo-β-lactamase inhibitors. Chem. Sci. 2017, 8, 928–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CM, R.; GD, W. Inhibitors of metallo-β-lactamases. Curr. Opin. Microbiol. 2017, 39, 96. [Google Scholar]
- Bach, E.; Christensen, S.; Dalgaard, L.; Larsen, P.; Olsen, C.; Smedegård-Petersen, V. Structures, properties and relationship to the aspergillomarasmines of toxins produced by Pyrenophora teres. Physiol. Plant Pathol. 1979, 14, 41–46. [Google Scholar] [CrossRef]
- Kalinka, K.; King, A.M.; Capretta, A.; Wright, G.D. Total synthesis and activity of the metallo-β-lactamase inhibitor Aspergillomarasmine A. Angew. Chem. Int. Ed. 2016, 55, 2210–2212. [Google Scholar]
- King, A.M.; Reid-Yu, S.A.; Wang, W.; King, D.T.; De Pascale, G.; Strynadka, N.C.; Walsh, T.R.; Coombes, B.K.; Wright, G.D. Aspergillomarasmine A overcomes metallo-β-lactamase antibiotic resistance. Nature 2014, 510, 503–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigsby, R.E.; Rife, C.L.; Fillgrove, K.L.; Newcomer, M.E.; Armstrong, R.N. Phosphonoformate: A minimal transition state analogue inhibitor of the fosfomycin resistance protein, FosA. Biochemistry 2004, 43, 13666–13673. [Google Scholar] [CrossRef]
- Fourmy, D.; Recht, M.I.; Blanchard, S.C.; Puglisi, J.D. Structure of the A site of Escherichia coli 16S ribosomal RNA complexed with an aminoglycoside antibiotic. Science 1996, 274, 1367–1371. [Google Scholar] [CrossRef]
- Benveniste, R.; Davies, J. Structure-activity relationships among the aminoglycoside antibiotics: Role of hydroxyl and amino groups. Antimicrob. Agents Chemother. 1973, 4, 402–409. [Google Scholar] [CrossRef]
- Hirsch, D.R.; Cox, G.; D’Erasmo, M.P.; Shakya, T.; Meck, C.; Mohd, N.; Wright, G.D.; Murelli, R.P. Inhibition of the ANT(2″)-Ia resistance enzyme and rescue of aminoglycoside antibiotic activity by synthetic α-hydroxytropolones. Bioorg. Med. Chem. Lett. 2014, 24, 4943–4947. [Google Scholar] [CrossRef] [Green Version]
- Welch, K.T.; Virga, K.G.; Whittemore, N.A.; Özen, C.; Wright, E.; Brown, C.L.; Lee, R.E.; Serpersu, E.H. Discovery of non-carbohydrate inhibitors of aminoglycoside-modifying enzymes. Bioorg. Med. Chem. 2005, 13, 6252–6263. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Yan, X.; Shakya, T.; Baettig, O.M.; Ait-Mohand-Brunet, S.; Berghuis, A.M.; Wright, G.D.; Auclair, K. Synthesis and structure-activity relationships of truncated bisubstrate inhibitors of aminoglycoside 6′-N-acetyltransferases. J. Med. Chem. 2006, 49, 5273–5281. [Google Scholar] [CrossRef] [PubMed]
- Daigle, D.M.; Mckay, G.A.; Wright, G.D. Inhibition of aminoglycoside antibiotic resistance enzymes by protein kinase inhibitors. J. Biol. Chem. 1997, 272, 24755. [Google Scholar] [CrossRef] [PubMed]
- Shakya, T.; Stogios, P.J.; Waglechner, N.; Evdokimova, E.; Ejim, L.; Blanchard, J.E.; McArthur, A.G.; Savchenko, A.; Wright, G.D. A small molecule discrimination map of the antibiotic resistance kinome. Chem. Biol. 2011, 18, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Sampei, K.; Nakagawa, M.; Uchiumi, N.; Amanuma, T.; Aiba, S.; Oikawa, M.; Mizuno, K. Damnacanthal, an effective inhibitor of LIM-kinase, inhibits cell migration and invasion. Mol. Biol. Cell 2014, 25, 828–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Jiang, N.; Wu, J.; Dai, W.; Rosenblum, J.S. Polo-like kinases inhibited by wortmannin Labeling site and downstream effects. J. Biol. Chem. 2007, 282, 2505–2511. [Google Scholar] [CrossRef] [PubMed]
- Stogios, P.J.; Spanogiannopoulos, P.; Evdokimova, E.; Egorova, O.; Shakya, T.; Todorovic, N.; Capretta, A.; Wright, G.D.; Savchenko, A. Structure-guided optimization of protein kinase inhibitors reverses aminoglycoside antibiotic resistance. Biochem. J. 2013, 454, 191–200. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug. Discov. 2004, 3, 935. [Google Scholar] [CrossRef]
- Alonso, H.; Bliznyuk, A.A.; Gready, J.E. Combining docking and molecular dynamic simulations in drug design. Med. Res. Rev. 2006, 26, 531–568. [Google Scholar] [CrossRef]
- Moroy, G.; Martiny, V.Y.; Vayer, P.; Villoutreix, B.O.; Miteva, M.A. Toward in silico structure-based ADMET prediction in drug discovery. Drug Discov. Today 2012, 17, 44–55. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177. [Google Scholar] [CrossRef] [PubMed]
- Rohit, T.; Kiran, M.; Ryan, P.; Chenglong, L.; Werner, T. Carborane clusters in computational drug design: A comparative docking evaluation using AutoDock, FlexX, Glide, and Surflex. J. Chem. Inf. Model. 2009, 49, 1581–1589. [Google Scholar]
- Sato, H.; Shewchuk, L.M.; Tang, J. Prediction of multiple binding modes of the CDK2 inhibitors, anilinopyrazoles, using the automated docking programs GOLD, FlexX, and LigandFit: An evaluation of performance. J. Chem. Inf. Model. 2006, 46, 2552. [Google Scholar] [CrossRef] [PubMed]
- Schellhammer, I.; Rarey, M. FlexX-Scan: Fast, structure-based virtual screening. Proteins 2004, 57, 504–517. [Google Scholar] [CrossRef]
- Stahl, M. Modifications of the scoring function in FlexX for virtual screening applications. Perspect. Drug Discov. 2000, 20, 83–98. [Google Scholar] [CrossRef]
- Jain, A.N. Scoring functions for protein-ligand docking. Curr. Protein Pept. Sci. 2006, 7, 407–420. [Google Scholar]
- Jain, A.N. Surflex: Fully automatic flexible molecular docking using a molecular similarity-based search engine. J. Med. Chem. 2003, 46, 499–511. [Google Scholar] [CrossRef]
- Chiem, K.; Jani, S.; Fuentes, B.; Lin, D.L.; Rasche, M.E.; Tolmasky, M.E. Identification of an inhibitor of the aminoglycoside 6′-N-acetyltransferase type Ib [AAC(6′)-Ib] by glide molecular docking. Medchemcommun 2016, 7, 184–189. [Google Scholar] [CrossRef]
- Green, K.D.; Chen, W.; Garneautsodikova, S. Identification and characterization of inhibitors of the aminoglycoside resistance acetyltransferase Eis from Mycobacterium tuberculosis. Chemmedchem 2012, 7, 73–77. [Google Scholar] [CrossRef]
- Suga, T.; Ishii, T.; Iwatsuki, M.; Yamamoto, T.; Nonaka, K.; Masuma, R.; Matsui, H.; Hanaki, H.; Ōmura, S.; Shiomi, K. Aranorosin circumvents arbekacin-resistance in MRSA by inhibiting the bifunctional enzyme AAC (6′)/APH (2 ′′). J. Antibiot. 2012, 65, 527. [Google Scholar] [CrossRef]
- Mccormack, D.; Mcfadden, D. Pterostilbene and cancer: Current review. J. Surg. Res. 2012, 173, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.G.; Alosi, J.A.; Mcdonald, D.E.; Mcfadden, D.W. Pterostilbene inhibits lung cancer through induction of apoptosis. J. Surg. Res. 2010, 161, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Remsberg, C.M.; Yáñez, J.A.; Ohgami, Y.; Vega-Villa, K.R.; Rimando, A.M.; Davies, N.M. Pharmacometrics of pterostilbene: Preclinical pharmacokinetics and metabolism, anticancer, antiinflammatory, antioxidant and analgesic activity. Phytother. Res. 2008, 22, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Rimando, A.M.; Cuendet, M.; Desmarchelier, C.; Mehta, R.G.; Pezzuto, J.M.; Duke, S.O. Cancer chemopreventive and antioxidant activities of pterostilbene, a naturally occurring analogue of resveratrol. J. Agric. Food. Chem. 2002, 50, 3453–3457. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liu, S.; Wang, T.; Li, H.; Tang, S.; Wang, J.; Wang, Y.; Deng, X. Pterostilbene, a potential MCR-1 inhibitor that enhances the efficacy of polymyxin B. Antimicrob. Agents Chemother. 2018, 62, e02146-17. [Google Scholar] [CrossRef]
- Good, L.; Awasthi, S.K.; Dryselius, R.; Larsson, O.; Nielsen, P.E. Bactericidal antisense effects of peptide-PNA conjugates. Nat. Biotechnol. 2001, 19, 360–364. [Google Scholar] [CrossRef]
- Hatamoto, M.; Ohashi, A.; Imachi, H. Peptide nucleic acids (PNAs) antisense effect to bacterial growth and their application potentiality in biotechnology. Appl. Microbiol. Biotechnol. 2010, 86, 397–402. [Google Scholar] [CrossRef]
- Górska, A.; Markowska-Zagrajek, A.; Równicki, M.; Trylska, J. Scanning of 16S ribosomal RNA for peptide nucleic acid targets. J. Phys. Chem. B 2016, 120, 8369–8378. [Google Scholar] [CrossRef]
- Good, L.; Sandberg, R.; Larsson, O.; Nielsen, P.E.; Wahlestedt, C. Antisense PNA effects in Escherichia coli are limited by the outer-membrane LPS layer. Microbiology 2000, 146, 2665–2670. [Google Scholar] [CrossRef] [PubMed]
- Gryko, D.; Giedyk, M.; Jackowska, A.; Równicki, M.; Kolanowska, M.; Trylska, J. Vitamin B12 transports modified RNA into E. coli and S. typhimurium cells. Chem. Commun. 2018. [Google Scholar] [CrossRef]
- Forier, K.; Raemdonck, K.; De Smedt, S.C.; Demeester, J.; Coenye, T.; Braeckmans, K. Lipid and polymer nanoparticles for drug delivery to bacterial biofilms. J. Control. Release 2014, 190, 607–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abushahba, M.F.; Mohammad, H.; Thangamani, S.; Hussein, A.A.; Seleem, M.N. Impact of different cell penetrating peptides on the efficacy of antisense therapeutics for targeting intracellular pathogens. Sci. Rep. 2016, 6, 20832. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.Y.; Mao, X.G.; Li, Z.; Chen, Z.; Zhou, Y.; Hou, Z.; Li, M.K.; Meng, J.R.; Luo, X.X. A potent and selective antimicrobial poly(amidoamine) dendrimer conjugate with LED209 targeting QseC receptor to inhibit the virulence genes of Gram negative bacteria. Nanomed. Nanotechnol. 2015, 11, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Huschka, R.; Barhoumi, A.; Liu, Q.; Roth, J.A.; Ji, L.; Halas, N.J. Gene silencing by gold nanoshell-mediated delivery and laser-triggered release of antisense oligonucleotide and siRNA. ACS Nano 2012, 6, 7681–7691. [Google Scholar] [CrossRef] [PubMed]
- Daly, S.M.; Sturge, C.R.; Felder-Scott, C.F.; Geller, B.L.; Greenberg, D.E. MCR-1 inhibition with peptide-conjugated Phosphorodiamidate morpholino oligomers restores sensitivity to polymyxin in Escherichia coli. mBio 2017, 8, e01315-17. [Google Scholar] [CrossRef]
- Thomason, M.K.; Storz, G. Bacterial antisense RNAs: How many are there, and what are they doing? Annu. Rev. Genet. 2010, 44, 167–188. [Google Scholar] [CrossRef]
- Ryszard, K.; Krainer, A.R.; Sidney, A. RNA therapeutics: Beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug. Discov. 2012, 11, 125–140. [Google Scholar]
- Bennett, C.F.; Swayze, E.E. RNA targeting therapeutics: Molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 259–293. [Google Scholar] [CrossRef]
- Geller, B.L.; Deere, J.D.; Stein, D.A.; Kroeker, A.D.; Moulton, H.M.; Iversen, P.L. Inhibition of gene expression in Escherichia coli by antisense phosphorodiamidate morpholino oligomers. Antimicrob. Agents Chemother. 2003, 47, 3233–3239. [Google Scholar] [CrossRef] [PubMed]
- Mellbye, B.L.; Puckett SETilley, L.D. Variations in amino acid composition of antisense peptide-phosphorodiamidate morpholino oligomer affect potency against Escherichia coli in vitro and in vivo. Antimicrob. Agents Chemother. 2009, 53, 525–530. [Google Scholar] [CrossRef]
- Mellbye, B.L.; Weller, D.D.; Hassinger, J.N.; Reeves, M.D.; Lovejoy, C.E.; Iversen, P.L.; Geller, B.L. Cationic phosphorodiamidate morpholino oligomers efficiently prevent growth of Escherichia coli in vitro and in vivo. J. Antimicrob. Chemother. 2010, 65, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.J.; Sturge, C.R.; Moustafa, D.A.; Daly, S.M.; Marshallbatty, K.R.; Felder, C.F.; Zamora, D.; Yabegill, M.; Labandeirarey, M.; Bailey, S.M. Inhibition of Pseudomonas aeruginosa by peptide-conjugated phosphorodiamidate morpholino oligomers. Antimicrob. Agents Chemother. 2017, 61, e01938-16. [Google Scholar] [CrossRef]
- Sully, E.K.; Geller, B.L.; Li, L.; Moody, C.M.; Bailey, S.M.; Moore, A.L.; Wong, M.; Nordmann, P.; Daly, S.M.; Sturge, C.R. Peptide-conjugated phosphorodiamidate morpholino oligomer (PPMO) restores carbapenem susceptibility to NDM-1-positive pathogens in vitro and in vivo. J Antimicrob Chemother 2017, 72, 782. [Google Scholar] [PubMed]
- Ji, Y.; Marra, A.; Rosenberg, M.; Woodnutt, G. Regulated antisense RNA eliminates alpha-toxin virulence in Staphylococcus aureus infection. J. Bacteriol. 1999, 181, 6585–6590. [Google Scholar] [PubMed]
- Chi, N.T.; Giangrossi, M.; Prosseda, G.; Brandi, A.; Martino, M.L.D.; Colonna, B.; Falconi, M. A multifactor regulatory circuit involving H-NS, VirF and an antisense RNA modulates transcription of the virulence gene icsA of Shigella flexneri. Nucleic Acids Res. 2011, 39, 8122–8134. [Google Scholar]
- Eun-Jin, L.; Groisman, E.A. An antisense RNA that governs the expression kinetics of a multifunctional virulence gene. Mol. Microbiol. 2010, 76, 1020–1033. [Google Scholar] [Green Version]
- Mara, G.; Gianni, P.; Nhan, T.C.; Anna, B.; Bianca, C.; Maurizio, F. A novel antisense RNA regulates at transcriptional level the virulence gene icsA of Shigella flexneri. Nucleic Acids Res. 2010, 38, 3362–3375. [Google Scholar] [Green Version]
- Panthee, S.; Paudel, A.; Hamamoto, H.; Sekimizu, K. Advantages of the silkworm as an animal model for developing novel antimicrobial agents. Front. Microbiol. 2017, 8, 373. [Google Scholar] [CrossRef]
Figure 1.
Scheme of antibiotic resistance mechanisms mediated by bacterial resistance enzymes. (A) Resistance enzymes hydrolyze the antibiotics and confer resistance. (B) Resistance enzymes modify the structure of antibiotics or antibiotic targets, preventing the antibiotics from binding to their targets and conferring resistance.
Figure 1.
Scheme of antibiotic resistance mechanisms mediated by bacterial resistance enzymes. (A) Resistance enzymes hydrolyze the antibiotics and confer resistance. (B) Resistance enzymes modify the structure of antibiotics or antibiotic targets, preventing the antibiotics from binding to their targets and conferring resistance.

Figure 2.
β-Lactams are hydrolized by (i) Ser-β-lactamase and (ii) metallo-β-lactamase [21]. The hydrolysis site on the antibiotic is marked by red arrows.
Figure 2.
β-Lactams are hydrolized by (i) Ser-β-lactamase and (ii) metallo-β-lactamase [21]. The hydrolysis site on the antibiotic is marked by red arrows.

Figure 3.
Ring-opening reactions catalyzed by (A) macrolide esterases [73] and (B) fosfomycin-resistance enzymes FosX/A/B [77]. The hydrolysis sites on the antibiotics are marked by red arrows.

Figure 4.
Modification of kanamycin B by aminoglycoside nucleotidyltransferases (ANTs), phosphotransferases (APHs), and acetyltransferases (AACs) [87,88,89,90,91].

Figure 5.
Inactivation of lincomycin/clindamycin and chloramphenicol by nucleotidyltransferases or acetyltransferases. LinA/A’/B: Lincomycin nucleotidyltransferases A/A’/B.
Figure 5.
Inactivation of lincomycin/clindamycin and chloramphenicol by nucleotidyltransferases or acetyltransferases. LinA/A’/B: Lincomycin nucleotidyltransferases A/A’/B.

Figure 6.
Enzyme-catalyzed inactivation of macrolides by phosphotransferases and glycosyltransferases.
Figure 6.
Enzyme-catalyzed inactivation of macrolides by phosphotransferases and glycosyltransferases.


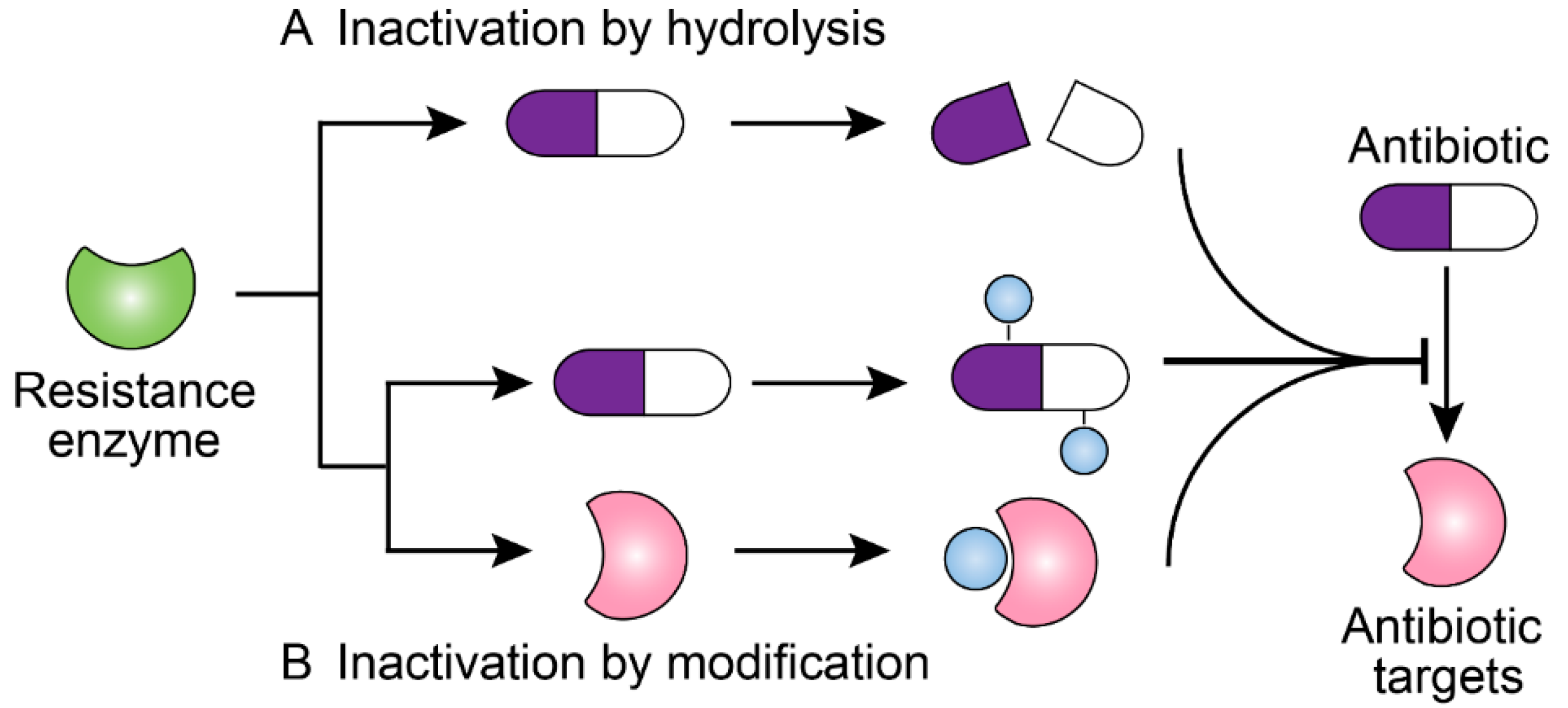{kind=link}
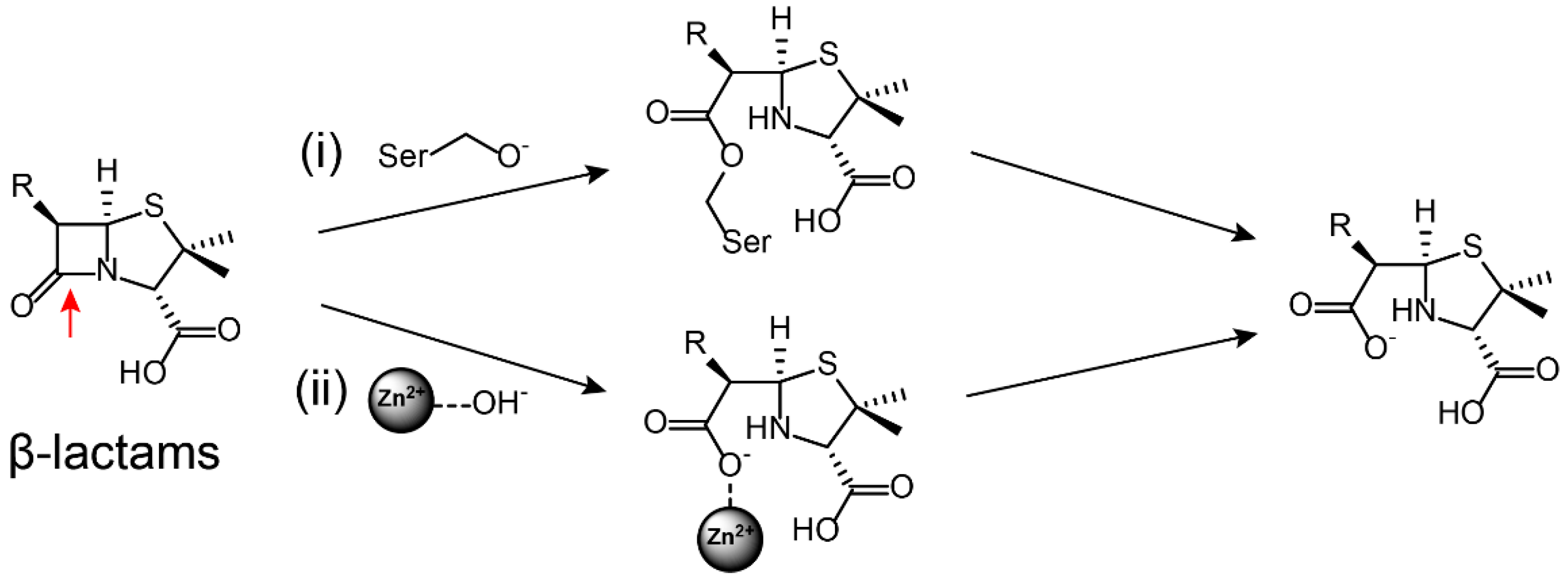{kind=link}
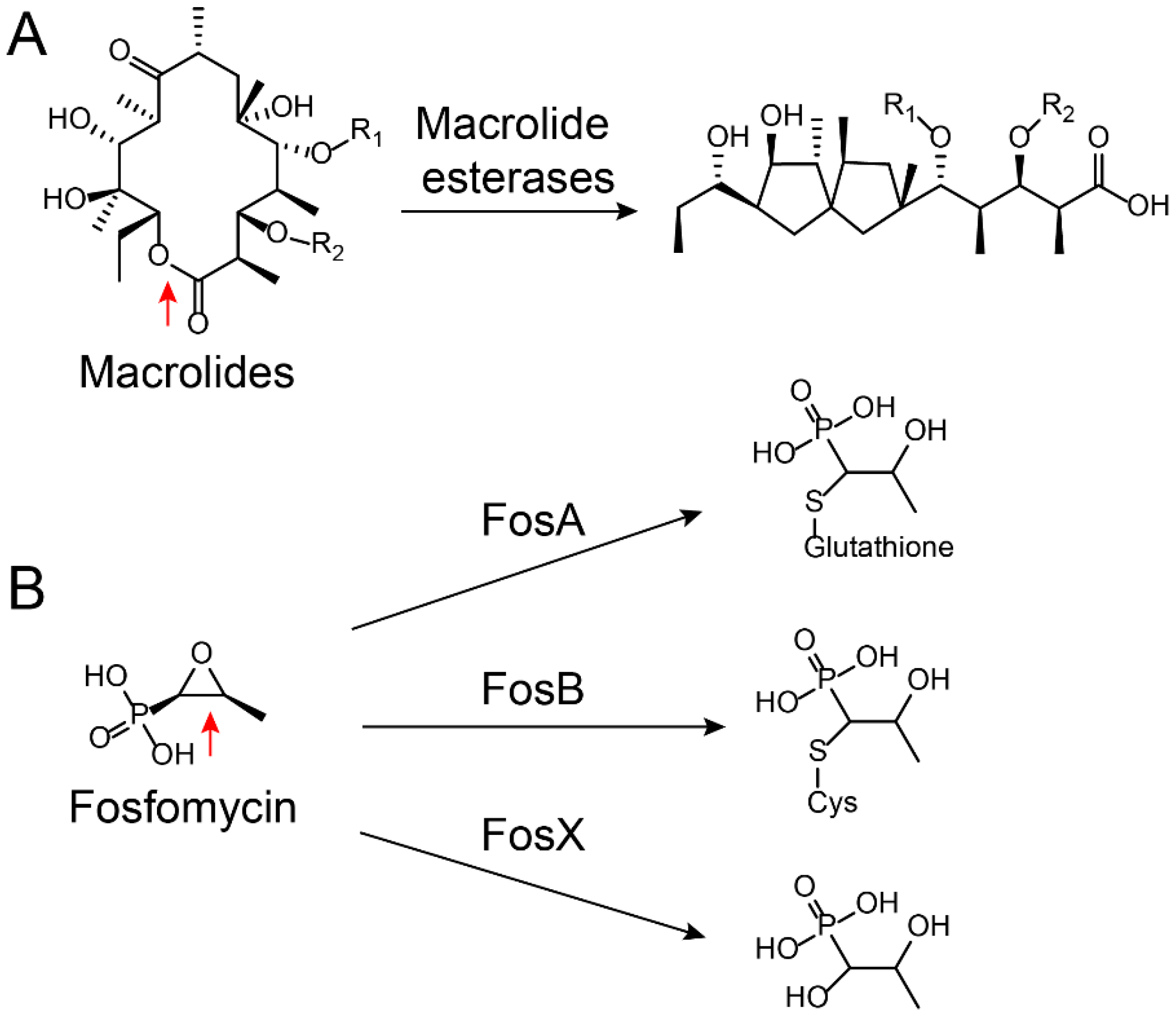{kind=link}
{kind=link}
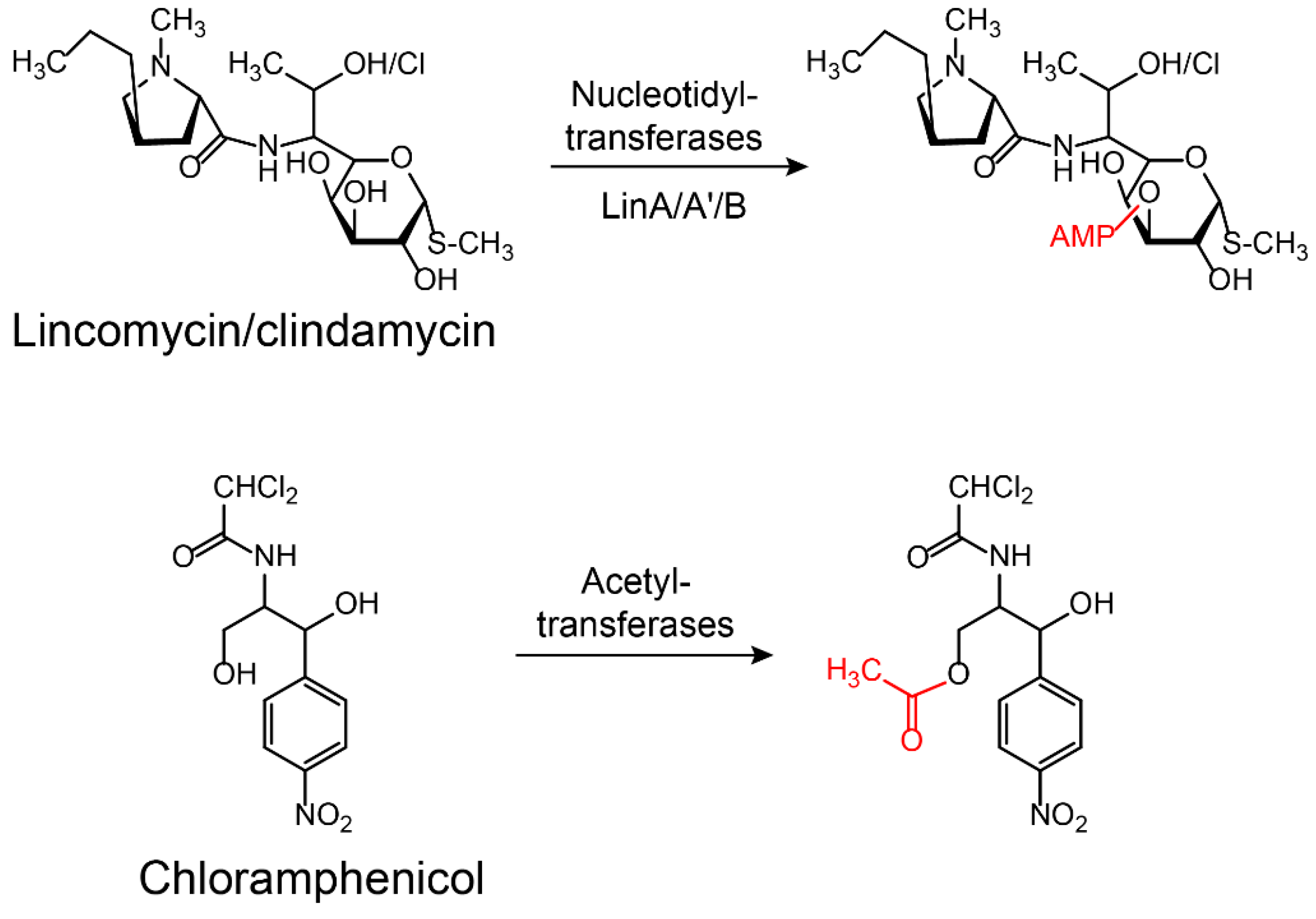{kind=link}
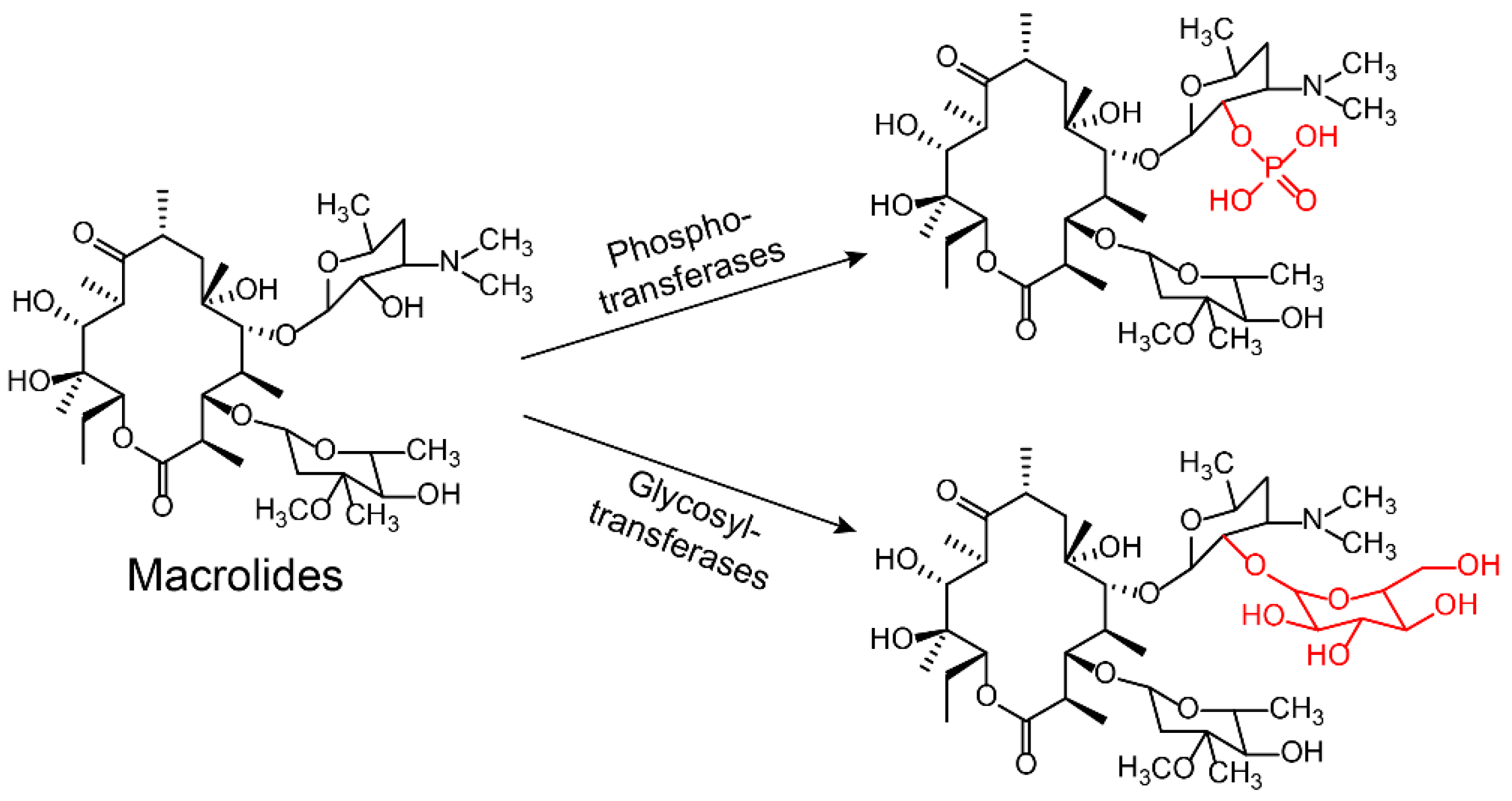{kind=link}
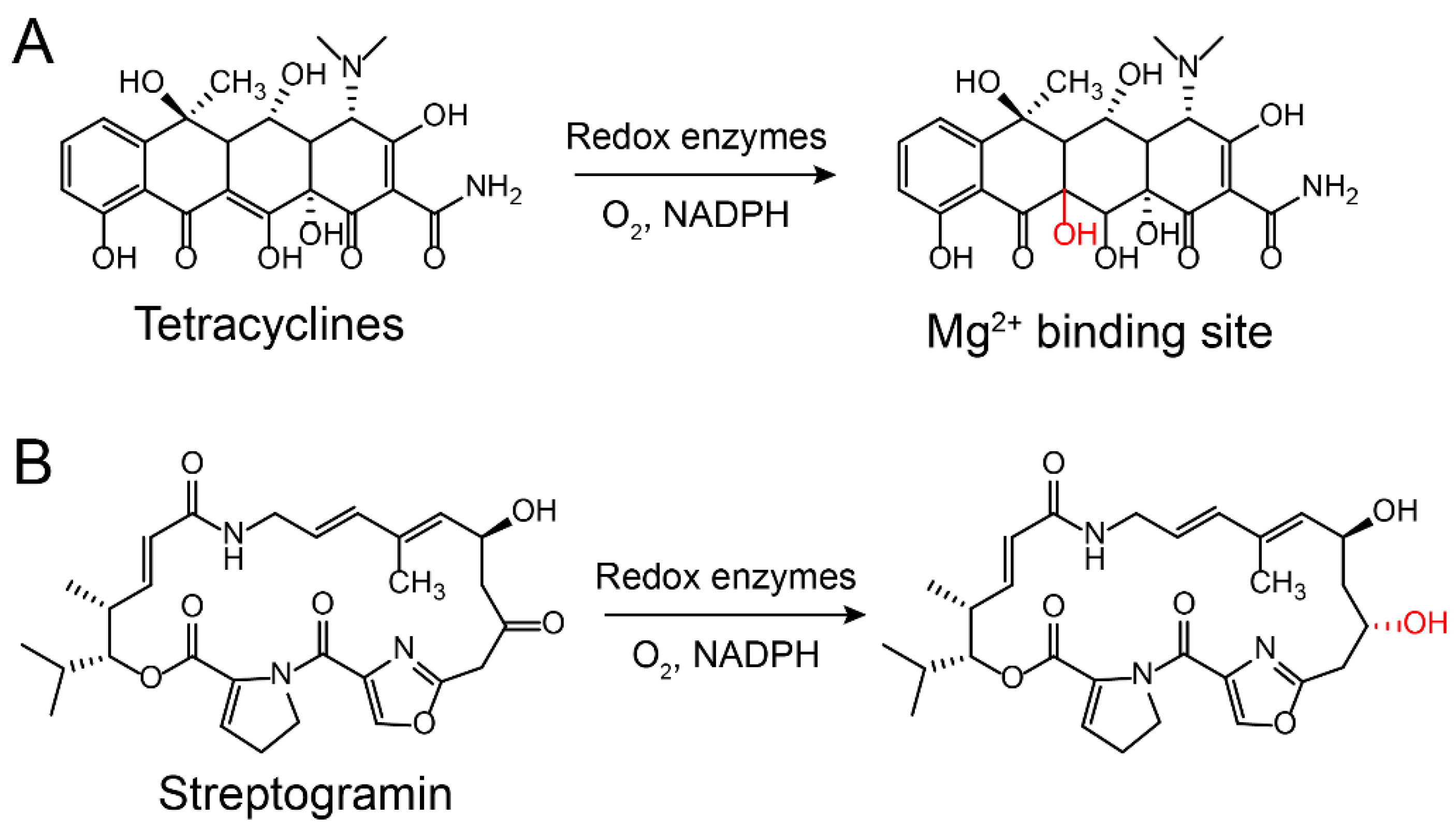{kind=link}
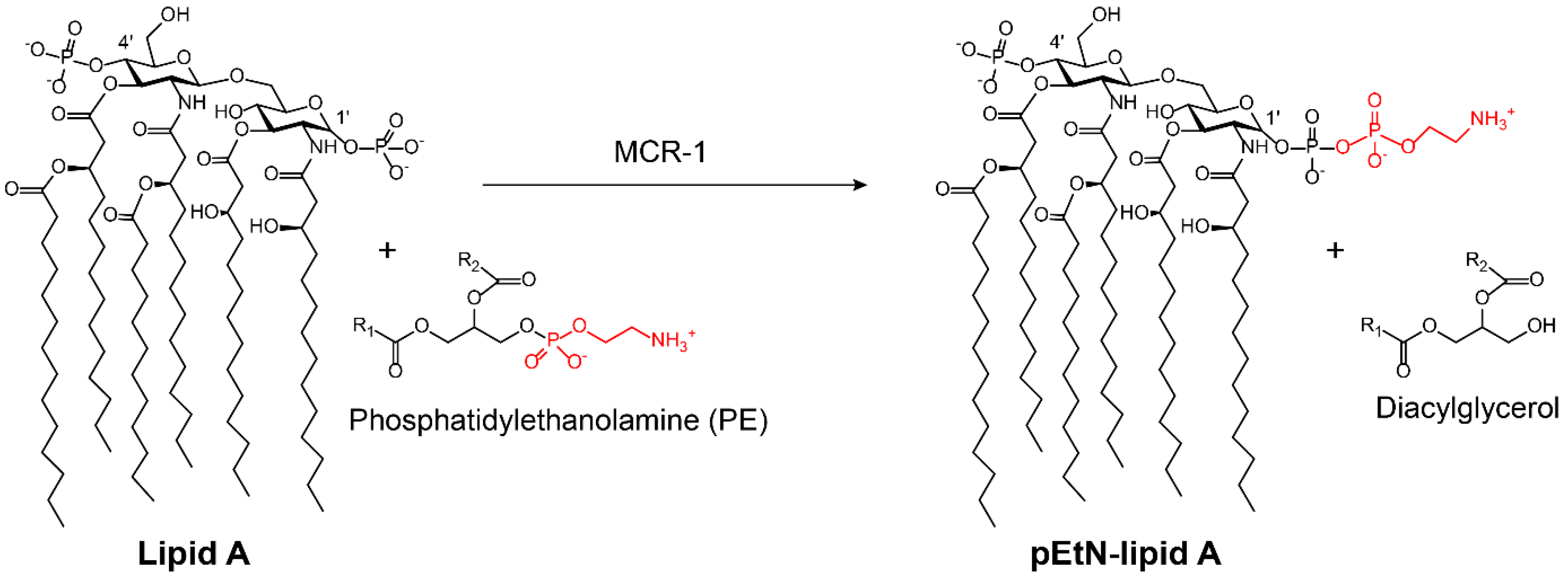{kind=link}
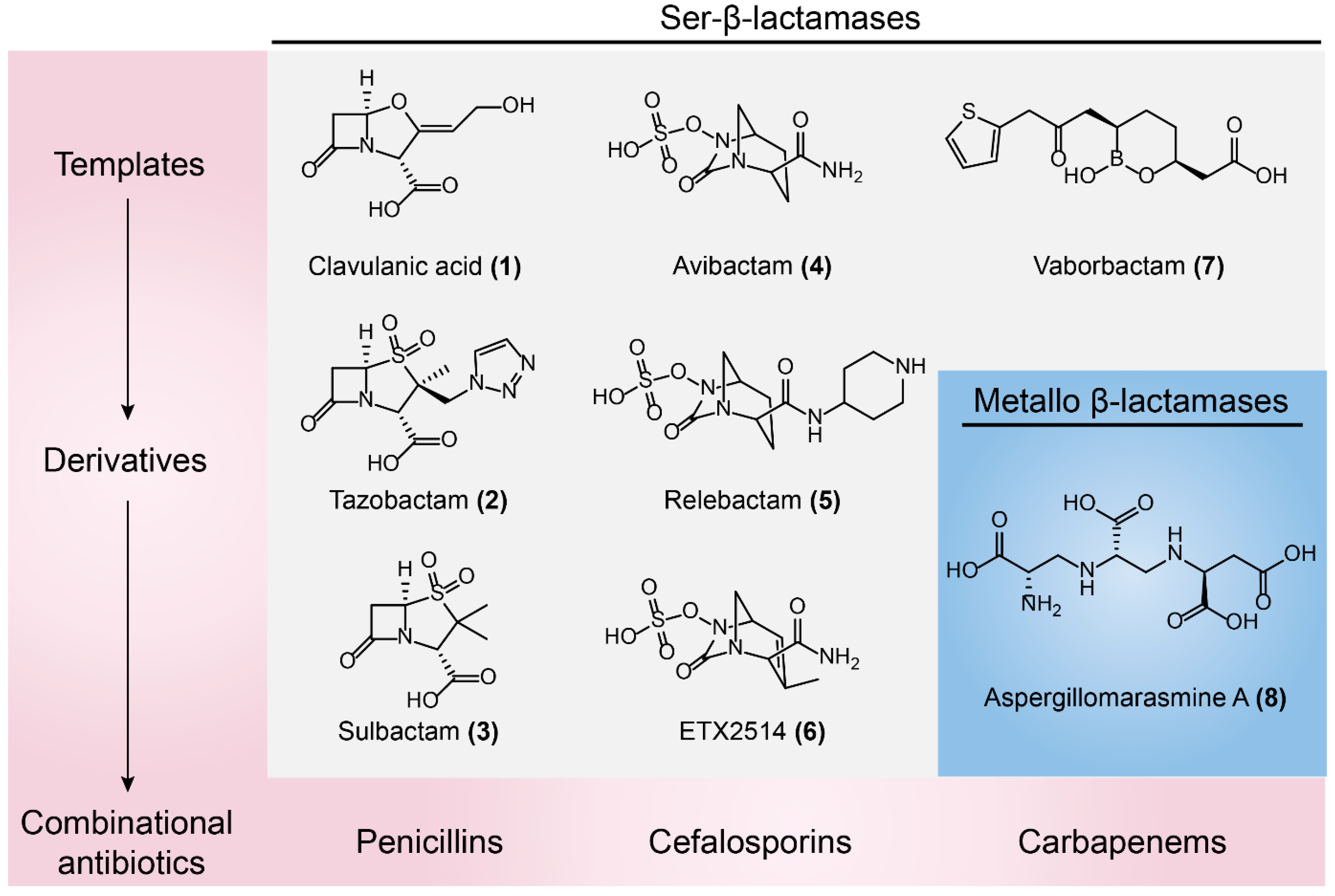{kind=link}
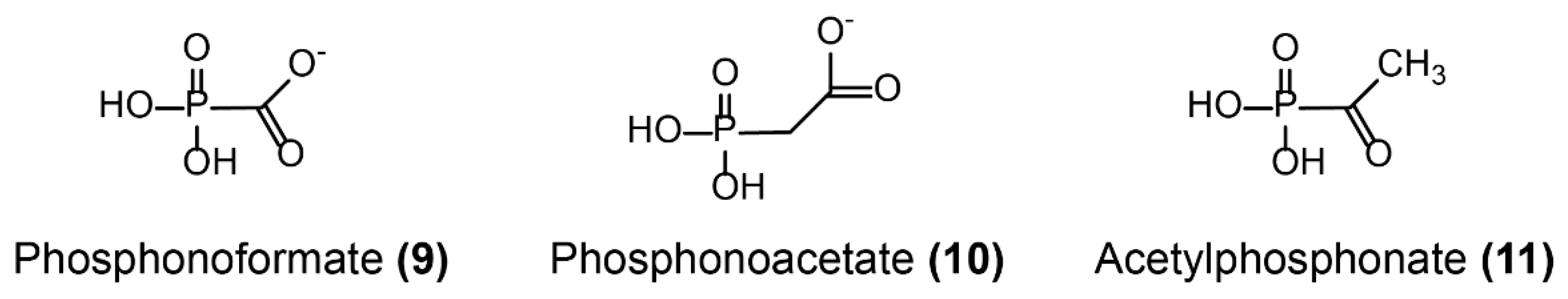{kind=link}
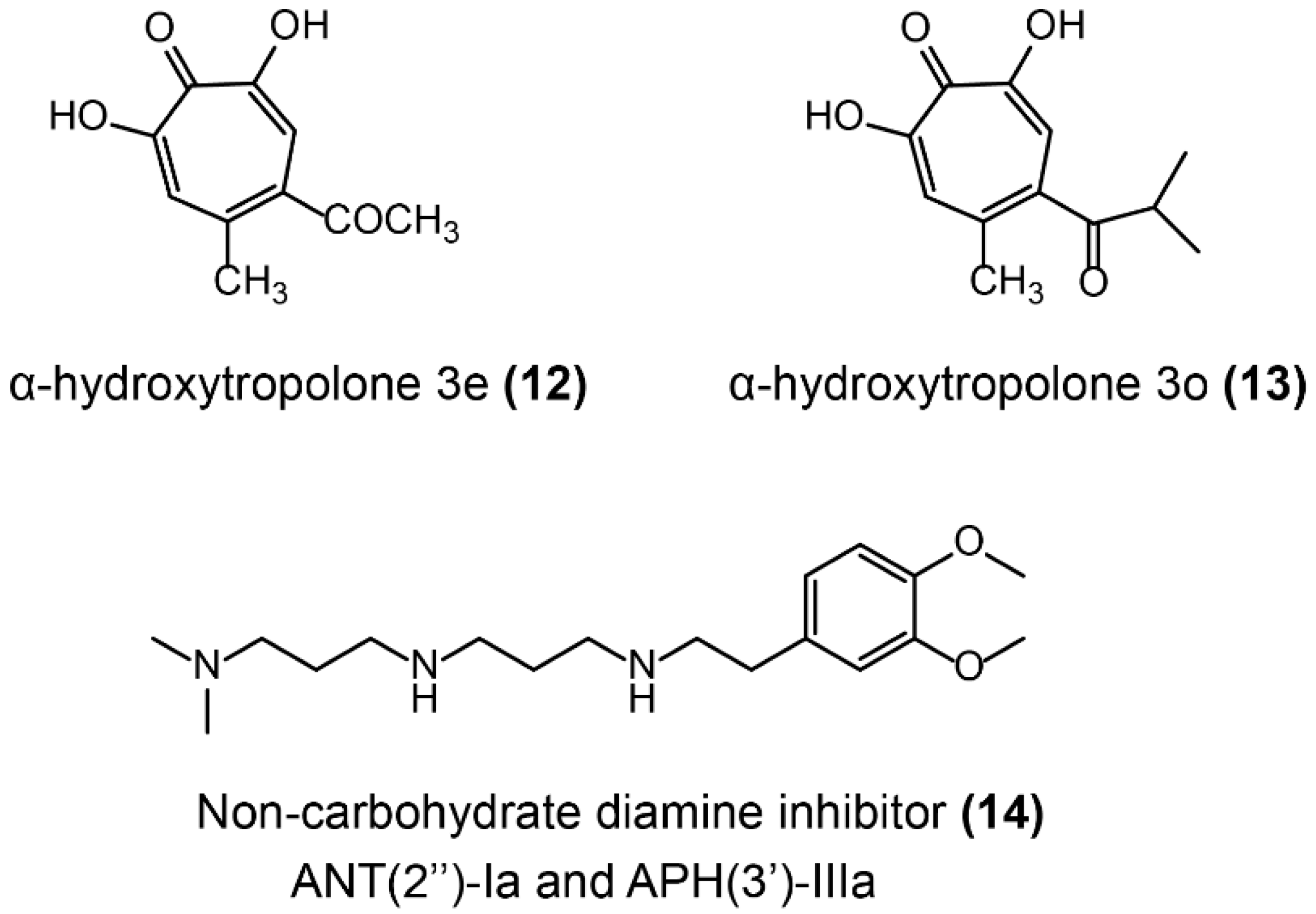{kind=link}
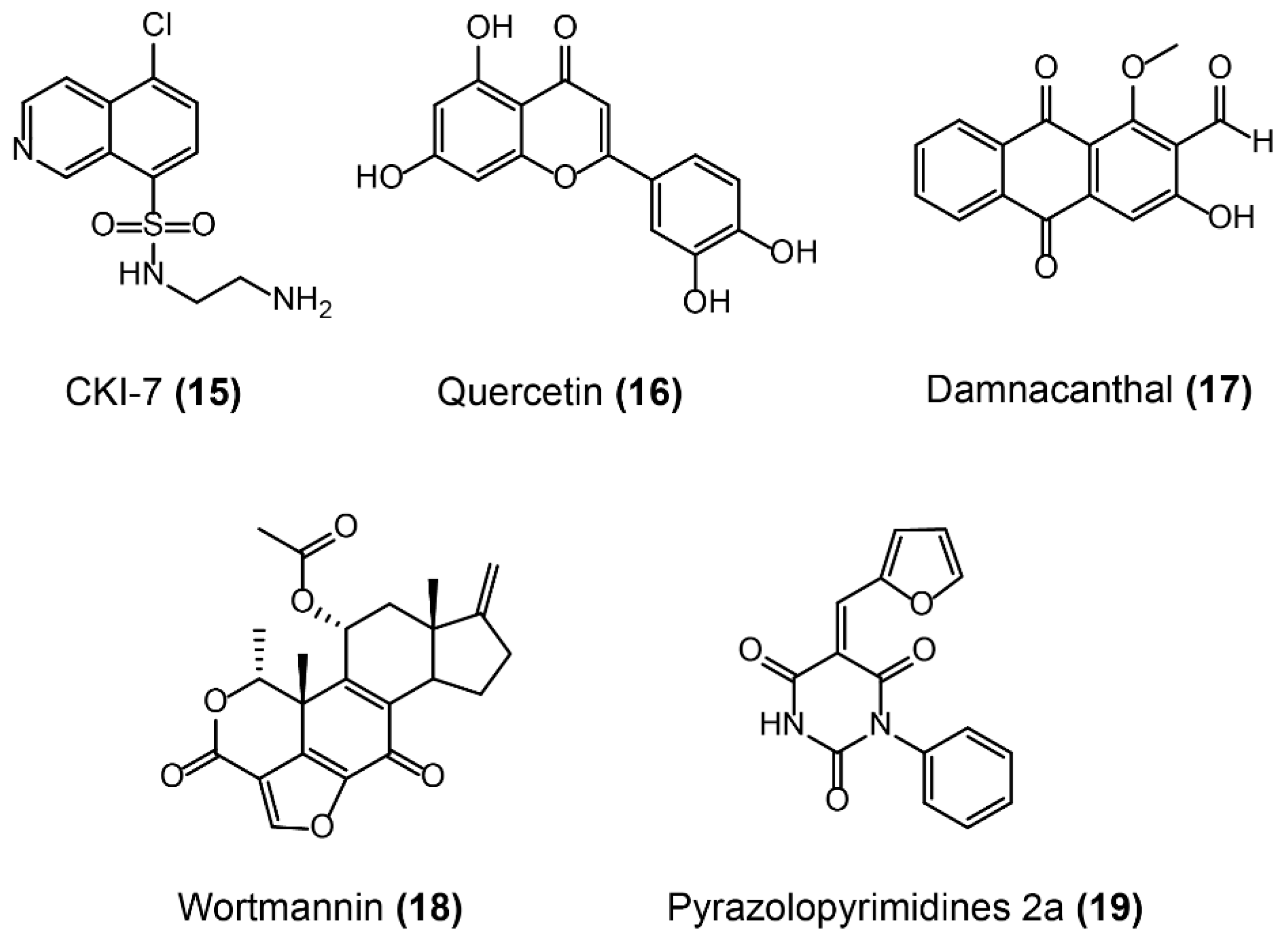{kind=link}
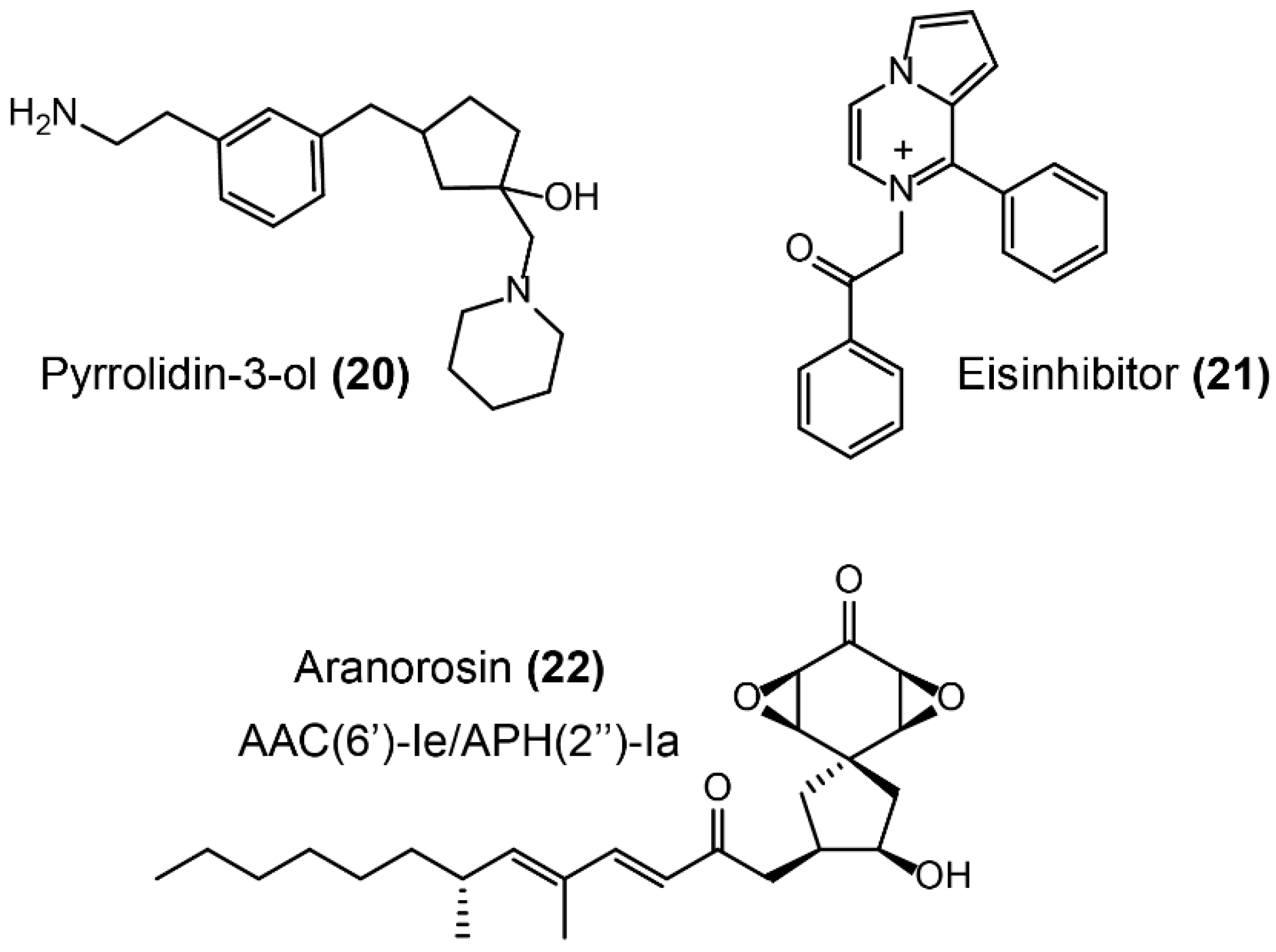{kind=link}
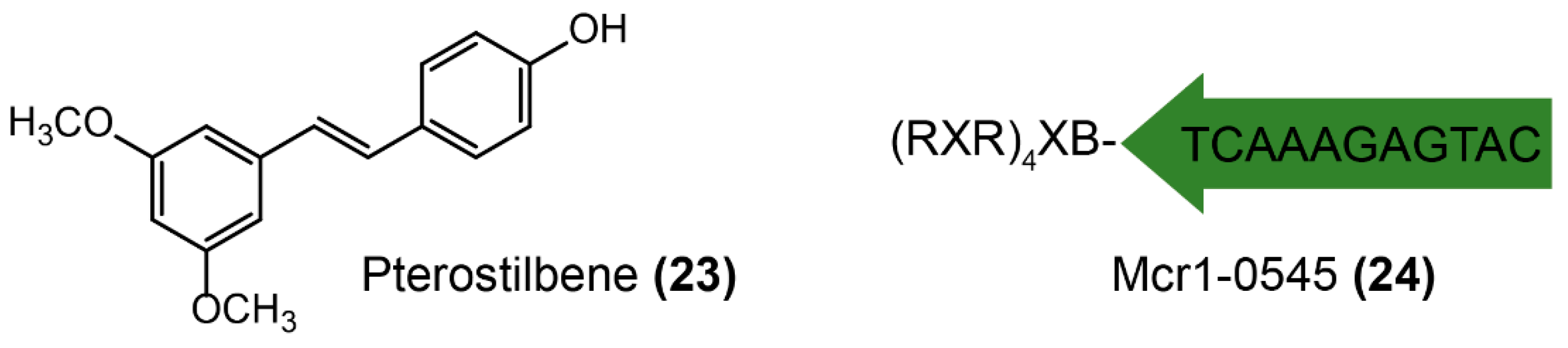{kind=link}
Figure 8.
Scheme of bacterial lipid A modified by phosphatidylethanolamine (PE) through MCR-1 [121]. The modified groups on the antibiotic are in red; pEtN: phosphoethanolamine.
Figure 8.
Scheme of bacterial lipid A modified by phosphatidylethanolamine (PE) through MCR-1 [121]. The modified groups on the antibiotic are in red; pEtN: phosphoethanolamine.

Figure 9.
Inhibitors of Ser-β-lactamases and metallo-β-lactamases. The chemical structures, templates, derivatives of β-lactamases inhibitors, and commonly paired antibiotics are shown.
Figure 9.
Inhibitors of Ser-β-lactamases and metallo-β-lactamases. The chemical structures, templates, derivatives of β-lactamases inhibitors, and commonly paired antibiotics are shown.

Figure 10.
Inhibitors of the fosfomycin-resistance enzyme FosA.

Figure 11.
Inhibitors of aminoglycoside ANTs.

Figure 12.
Inhibitors of APHs.

Figure 13.
Inhibitors of AACs.

Figure 14.
Inhibition of MCR-1 by molecules directly interacting with resistance enzymes or by targeting its mRNA.
Figure 14.
Inhibition of MCR-1 by molecules directly interacting with resistance enzymes or by targeting its mRNA.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Liu, Y.; Li, R.; Xiao, X.; Wang, Z. Molecules that Inhibit Bacterial Resistance Enzymes. Molecules 2019, 24, 43. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24010043
AMA Style
Liu Y, Li R, Xiao X, Wang Z. Molecules that Inhibit Bacterial Resistance Enzymes. Molecules. 2019; 24(1):43. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24010043
Chicago/Turabian StyleLiu, Yuan, Ruichao Li, Xia Xiao, and Zhiqiang Wang. 2019. "Molecules that Inhibit Bacterial Resistance Enzymes" Molecules 24, no. 1: 43. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24010043