An Eco-Friendly Technique: Solvent-Free Microwave Synthesis and Docking Studies of Some New Pyridine Nucleosides and Their Pharmacological Significance


Abstract
:1. Introduction

2. Results and Discussion
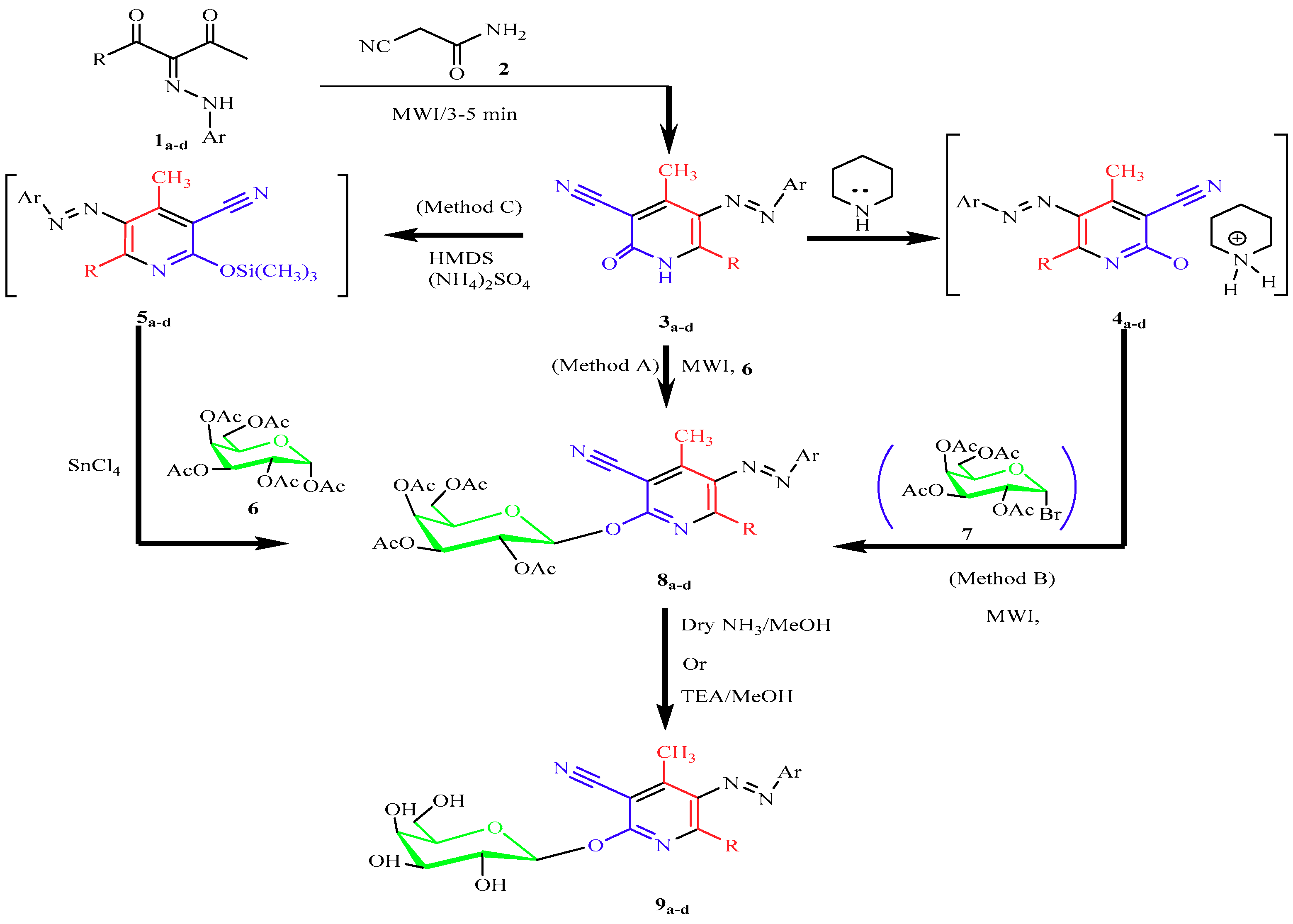
2.1. Chemistry
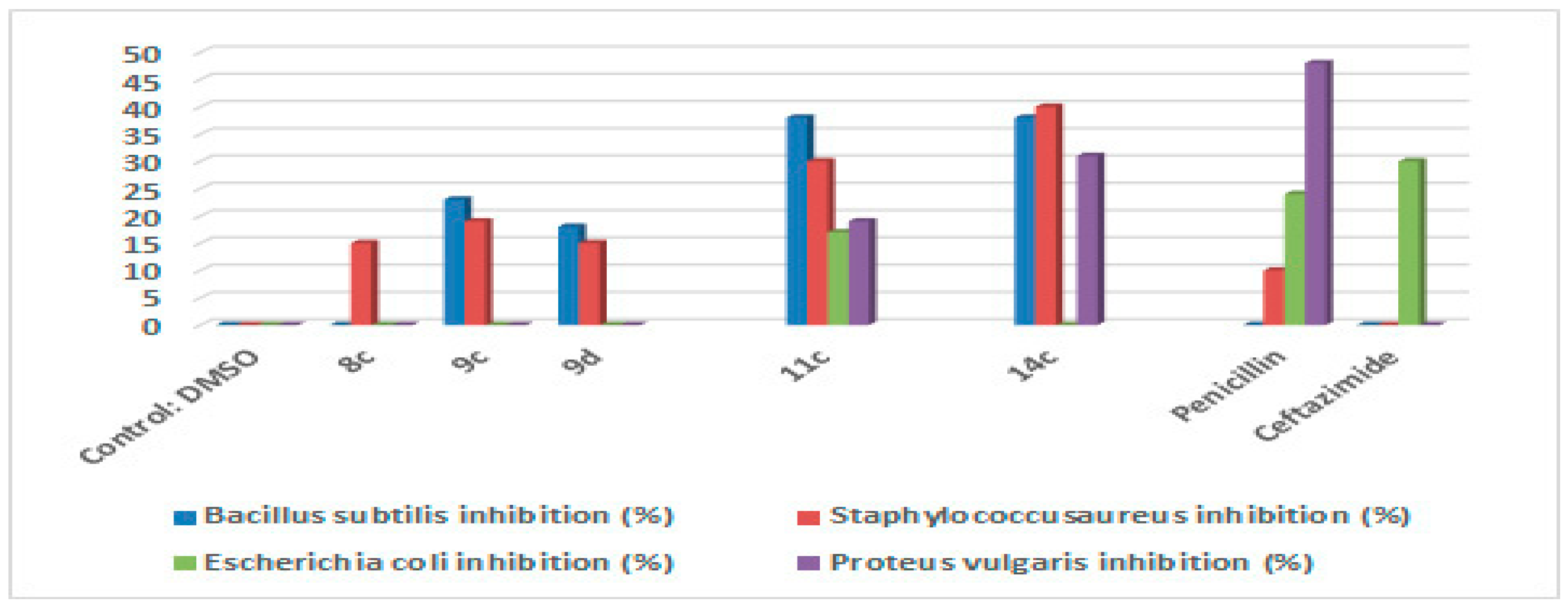
2.2. Antimicrobial Activity
2.3. Anticancer Activity
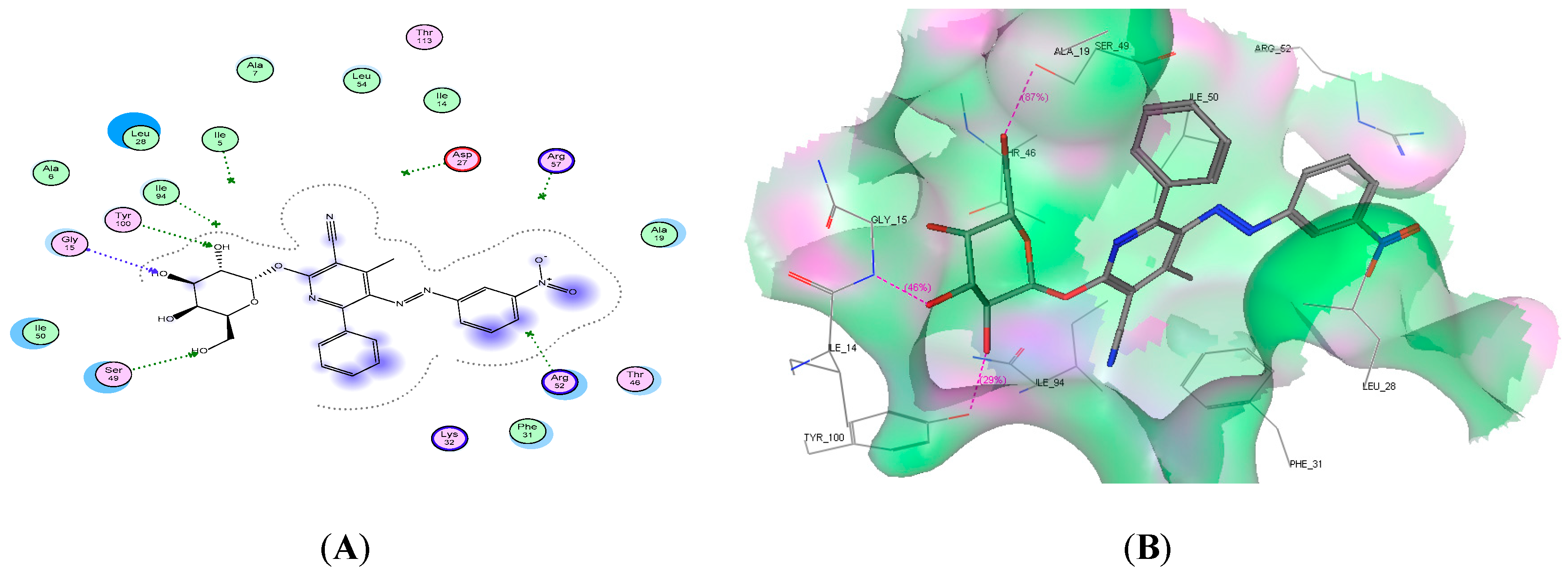
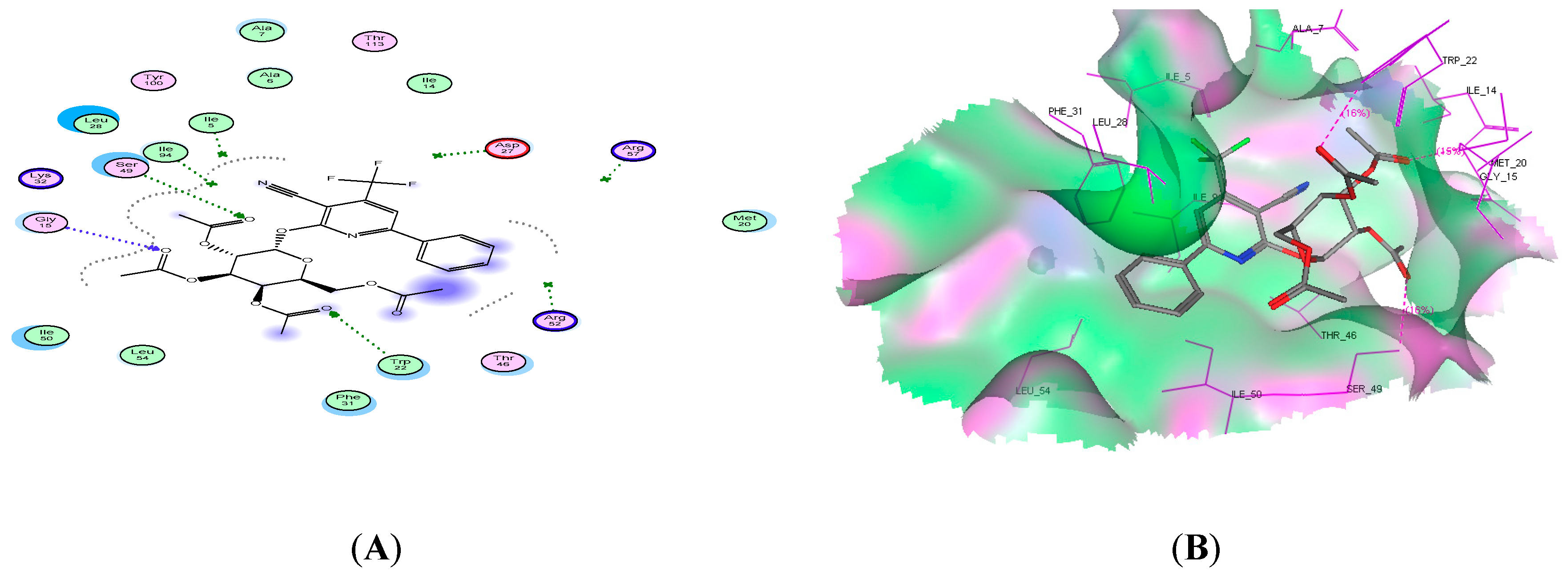
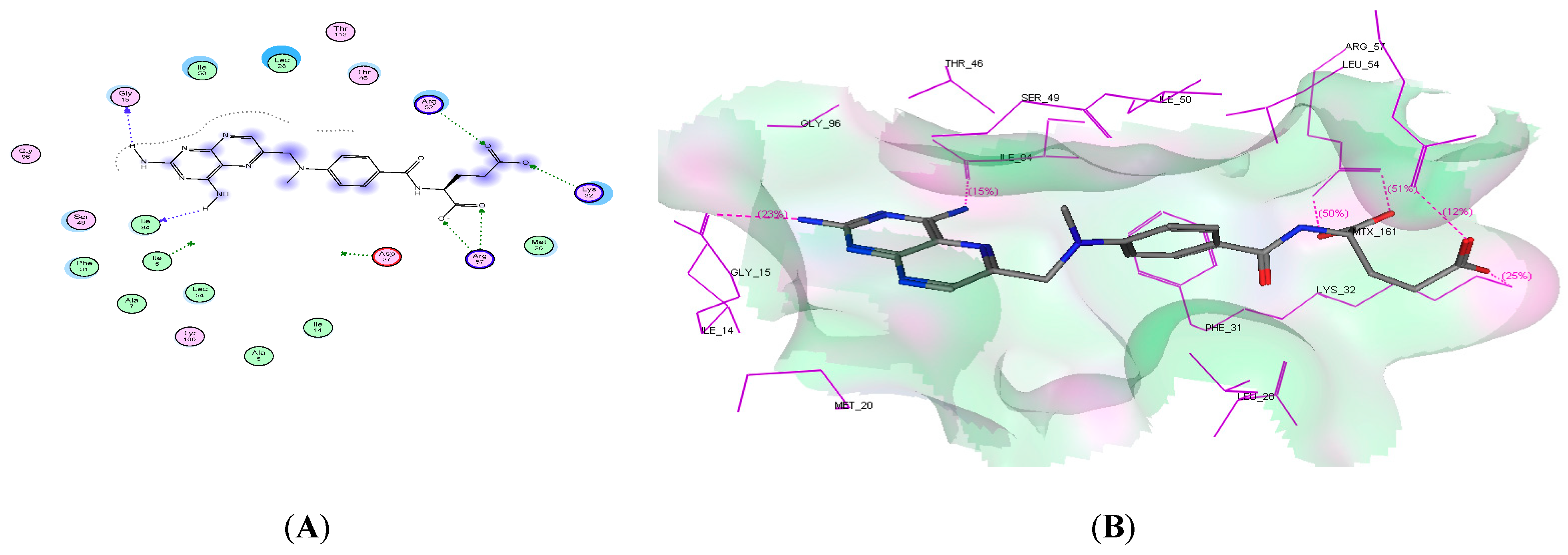
2.4. Molecular Docking Studies
3. Conclusion
4. Materials and Methods
4.1. Chemistry
4.1.1. General Procedure for the Synthesis of 3-Cyano-2-(2″,3″,4″,6″-tetra-O-acetyl-β-d-galactopyranosyloxo)-pyridines (8a–d, 11a–c)
Microwave Methods A (Solvent-Free Method)
Microwave Methods B (Catalyst-Free Method)
Conventional Synthesis Method C (Silyl Method)
4.1.2. General Procedure for the Synthesis of 3-cyano-2-(β-d-galactopyranosyloxy)-pyridines (9a–d, 14a–c).
General Procedure for Nucleoside Deactylation Method A
General Procedure for Nucleoside Deactylation Method B
4.2. Biological Activity Evaluations
4.2.1. Antimicrobial Activity
Materials and Methods
Agar Disc-Diffusion Method
4.2.2. Anticancer Activity
Materials and Methods: Cell Culture and Viability Assay
4.2.3. Molecular Docking
Author Contributions
Funding
Conflicts of Interest
References
- Li, Q.; Mitscher, L.; Shen, L. The 2-pyridone antibacterial agents: Bacterial topoisomerase inhibitors. Med. Res. Rev. 2000, 20, 231–293. [Google Scholar] [CrossRef]
- Fujita, Y.; Oguri, H.; Oikawa, H. Biosynthetic studies on the antibiotics PF1140: A novel pathway for a 2-pyridone framework. Tetrahedron Lett. 2005, 46, 5885–5888. [Google Scholar] [CrossRef]
- Fassihi, A.; Abedi, D.; Saghaie, L.; Sabet, R.; Fazeli, H.; Bostaki, G.; Deilami, O.; Sadinpou, H. Synthesis, antimicrobial evaluation and QSAR study of some 3-hydroxypyridine-4-one and 3-hydroxypyran-4-one derivatives. Eur. J. Med. Chem. 2009, 44, 2145–2157. [Google Scholar] [CrossRef]
- Semple, G.; Andersson, B.; Chhajlani, V.; Georgsson, J.; Johansson, M.; Rosenquist, Å. Swanson, Synthesis and Biological activity of kappa opioid receptor agonists. Part 2: Preparation of 3-aryl-2-pyridone analogues generated by solution- and solid-phase parallel synthesis methods. Bioorg. Med. Chem. Lett. 2003, 13, 1141–1145. [Google Scholar] [CrossRef]
- Parreira, R.; Abrahão, O.; Galembeck, E. Conformational preferences of non-nucleoside HIV-1 reverse transcriptase inhibitors. Tetrahedron 2001, 57, 243–3253. [Google Scholar] [CrossRef]
- Dragovich, P.; Prins, T.; Zhou, R.; Brown, E.; Maldonado, F.; Fuhrman, S.; Zalman, S.; Tuntland, T.; Lee, C.; Patick, A.; et al. Structure-Based Design, Synthesis, and Biological Evaluation of Irreversible Human Rhinovirus 3C Protease Inhibitors. 6. Structure−Activity Studies of Orally Bioavailable, 2-Pyridone-Containing Peptidomimetics. J. Med. Chem. 2002, 45, 1607–1623. [Google Scholar] [CrossRef]
- Hasvold, L.; Wang, W.; Gwaltney, S.; Rockway, T.; Nelson, L.; Mantei, R.; Fakhoury, S.; Sullivan, G.; Li, Q.; Lin, N.; et al. Pyridonecontaining farnesyltransferase inhibitors: Synthesis and biological evaluation. Bioorg. Med. Chem. Lett. 2003, 13, 4001–4005. [Google Scholar] [CrossRef]
- Parlow, J.; Kurumbail, R.; Stegeman, R.; Stevens, A.; Stallings, W.; South, M. Design, Synthesis, and Crystal Structure of Selective 2-Pyridone Tissue Factor VIIa Inhibitors. J. Med. Chem. 2003, 46, 4696–4701. [Google Scholar] [CrossRef]
- Parlow, J.; South, M. Synthesis of 2-pyridones as tissue factor VIIa inhibitors. Tetrahedron 2003, 59, 7695–7701. [Google Scholar] [CrossRef]
- Mijin, D.; Ušćumlić, G.; Valentić, N.; Marinković, A. Sinteza arilazo piridonskih boja. Hem. Ind. 2011, 65, 517–532. [Google Scholar] [CrossRef]
- Litvinov, V.; Krivokolysko, S.; Dyachenko, V. Synthesis and properties of 3-cyanopyridine-2(1H)-chalcogenones. Review. Chem. Heterocyclic Comp. 1999, 35, 509–540. [Google Scholar] [CrossRef]
- Nesnow, S.; Miyazaki, T.; Kawaja, T.; Meyer, B. Pyridine nucleosides related to 5-fluorouracil and thymine. J. Med. Chem. 1973, 16, 524–528. [Google Scholar] [CrossRef]
- Imai, K.I.; Nohara, A.; Honjo, M. Reaction of Steroidal Diosphenols with Manganese Dioxide. Chem. Pharm. Bull. 1966, 14, 1377–1381. [Google Scholar] [CrossRef]
- Onodera, K.; Hirano, S.F. Syntheses of O-Glycosides, Glycosylamines and Purine Nucleosides by Fusion Reaction in the Presence of Polyphosphoric Acid or Ethyl Polyphosphate as New catalyst. Agr. Biol. Chem. 1964, 28, 173–178. [Google Scholar]
- Abdou, I.; Rateb, N.; Eldeab, H. Fast and efficient microwave synthetic methods for some new 2(1H)-pyridone arabinosides. Heterocycl. Commun. 2012, 18, 135–141. [Google Scholar] [CrossRef]
- Rateb, N.; Eldeab, H.; Abdou, I. Antimicrobial Evaluation of New Synthesized Pyridine Nucleosides under Solvent-Free Conditions. Nucleosides, Nucleotides and Nucleic Acids 2013, 32, 493–509. [Google Scholar] [CrossRef]
- Abdellattif, M.; Maghrabi, I.; Areef, M.; ElDeab, H.; Mouneirand, S.; Belal, A. Efficient Microwave-Assisted Solvent-Free Synthesis and Molecular Docking Studies of 2-pyridone derivatives as Anticancer Agents and Evaluation of Cytotoxic Effects. J. Adv. Chem. 2016, 12, 4351–4364. [Google Scholar] [CrossRef]
- Maghrabi, I.; Alghamdi, S.; Alrobaian, M.; Eldeab, H. Green Technique-Solvent Free Microwave Synthesis and Antimicrobial Evaluation of New Thiopyridine Arabinosides. Molecules 2016, 21, 477. [Google Scholar] [CrossRef]
- Abdou, I.; Salem, A.; Adem, A.; Zohdy, H.; Eldeab, H. Substituted Pyridine Derivatives Useful in the Treatment of Cancer. U.S. Patent 9,051,271B2, 9 June 2015. [Google Scholar]
- Eldeab, H.; Greish, Y.; Thomas, S.; Karam, S. Pyridine Compound, Making, and Use Thereof. WO2017115321A1, 6 July 2017. [Google Scholar]
- Eldeab, H. Ecofriendly microwave assisted synthesis of some new pyridine glycosides. Nucleosides Nucleotides Nucleic Acids 2019, 38, 509–520. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef]
- Sharma, M.; Chauhan, M. Dihydrofolate reductase as a therapeutic target for infectious diseases: Opportunities and challenges. Future Med. Chem. 2012, 4, 1335–1365. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment. Available online: https://www.chemcomp.com/MOE-Molecular_Operating_Environment.htm (accessed on 20 December 2018).
- Protein Data Bank. Available online: http://www.rcsb.org/pdb/explore.do?structureId=4dfr (accessed on 18 December 2018).
- Atlas, R.M. Handbook of Microbiological Media; CRC Press: London, UK, 2004; p. 1226. [Google Scholar]
- Stoddart, M. Mammalian Cell Viability, (Methods in Molecular Biology Series); Humana Press: New York, NY, USA, 2011. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound No. | R | Ar | Microwave Synthesis | Conventional Synthesis | |
|---|---|---|---|---|---|
| Reaction Time/min Yield (%) | Reaction Time/h Yield (%) | ||||
| Method A | Method B | Method C | |||
| 8a | CH3 | 4-ClC6H4 | 2 (92) | 5 (86) | 53 (50) |
| 8b | CH3 | 3-NO2C6H4 | 3 (90) | 5 (81) | 53 (49) |
| 8c | C6H5 | 4-ClC6H4 | 2 (95) | 4 (85) | 51 (44) |
| 8d | C6H5 | 3-NO2C6H4 | 2 (93) | 4 (83) | 51 (51) |
| Compound No. | R1 | R2 | Microwave Synthesis | Conventional Synthesis | |
|---|---|---|---|---|---|
| Reaction Time/min Yield (%) | Reaction Time/h Yield (%) | ||||
| Method A | Method B | Method C | |||
| 11a | CH3 | CH3 | 3 (87) | 7 (80) | 62 (61) |
| 11b | C6H5 | CH3 | 2 (91) | 7 (83) | 67 (70) |
| 11c | C6H5 | CF3 | 2 (94) | 6 (85) | 65 (61) |
| Comound No. | R | Ar | Method A Yield % | Method B Yield % |
|---|---|---|---|---|
| 9a | CH3 | 4-ClC6H4 | 90 | 83 |
| 9b | CH3 | 3-NO2C6H4 | 88 | 87 |
| 9c | C6H5 | 4-ClC6H4 | 87 | 83 |
| 9d | C6H5 | 3-NO2C6H4 | 89 | 80 |
| Comound No. | R1 | R2 | Method A Yield % | Method B Yield % |
|---|---|---|---|---|
| 14a | CH3 | CH3 | 85 | 81 |
| 14b | C6H5 | CH3 | 87 | 81 |
| 14c | C6H5 | CF3 | 90 | 83 |
| Compound No. | Docking Score (kcal/mol) | No. of H-Bonds | Amino Acid Residue | Interacting Group |
|---|---|---|---|---|
| 8d | −21.86 | 2 | Met20 and Trp22 | 2 C=O |
| 9d | −19.91 | 3 | Gly15, Ser49, and Tyr100 | 3 OH |
| 11c | −19.09 | 3 | Ser49, Tre22, and Gly15 | 3 C=O |
| 14c | −19.59 | 3 | Trp22, Asp27, and lle94 | CN, 2OH |
| Ligand | −19.54 | 6 | Gly15, lle94, Arg57, Lys32, and Arg52 | 2NH2 and 2 COO |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alrobaian, M.; Azwari, S.A.; Belal, A.; Eldeab, H.A. An Eco-Friendly Technique: Solvent-Free Microwave Synthesis and Docking Studies of Some New Pyridine Nucleosides and Their Pharmacological Significance. Molecules 2019, 24, 1969. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24101969
Alrobaian M, Azwari SA, Belal A, Eldeab HA. An Eco-Friendly Technique: Solvent-Free Microwave Synthesis and Docking Studies of Some New Pyridine Nucleosides and Their Pharmacological Significance. Molecules. 2019; 24(10):1969. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24101969
Chicago/Turabian StyleAlrobaian, Majed, Sana Al Azwari, Amany Belal, and Hany A. Eldeab. 2019. "An Eco-Friendly Technique: Solvent-Free Microwave Synthesis and Docking Studies of Some New Pyridine Nucleosides and Their Pharmacological Significance" Molecules 24, no. 10: 1969. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24101969