Machine Learning Models Combined with Virtual Screening and Molecular Docking to Predict Human Topoisomerase I Inhibitors

Abstract
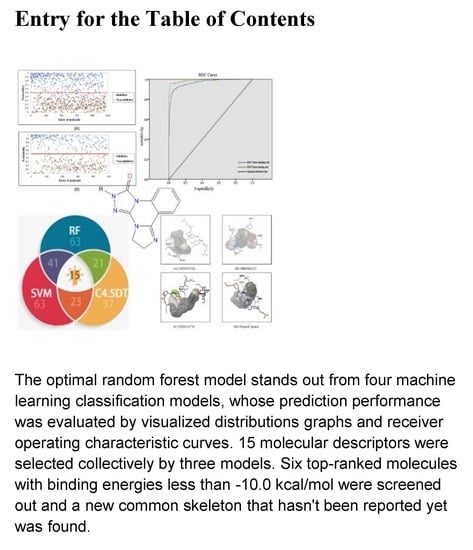
:
1. Introduction
2. Results and Discussion
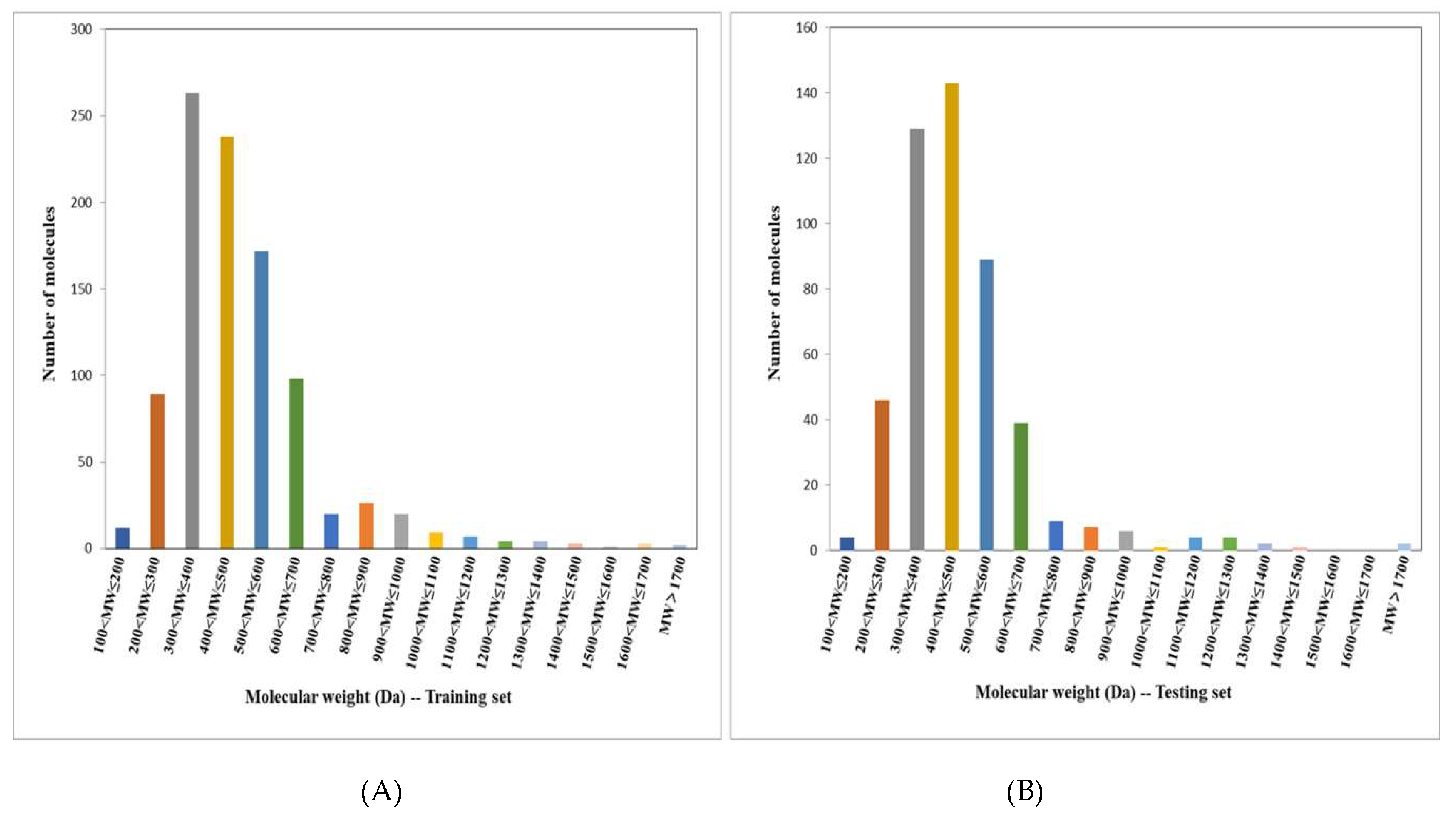
2.1. Structural Diversity Analysis
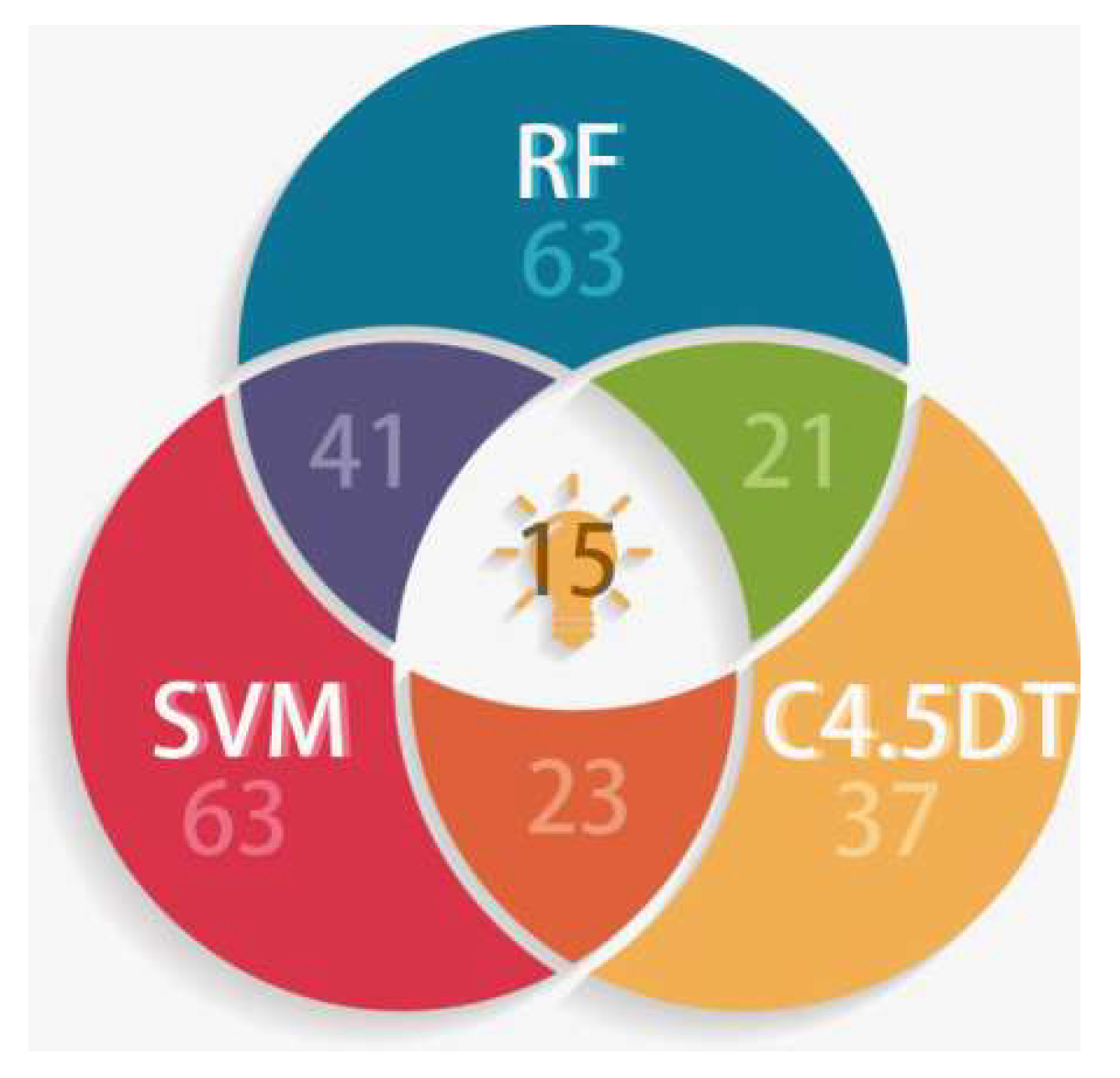
2.2. Comparison on Prediction Accuracies of Four Machine Learning Methods
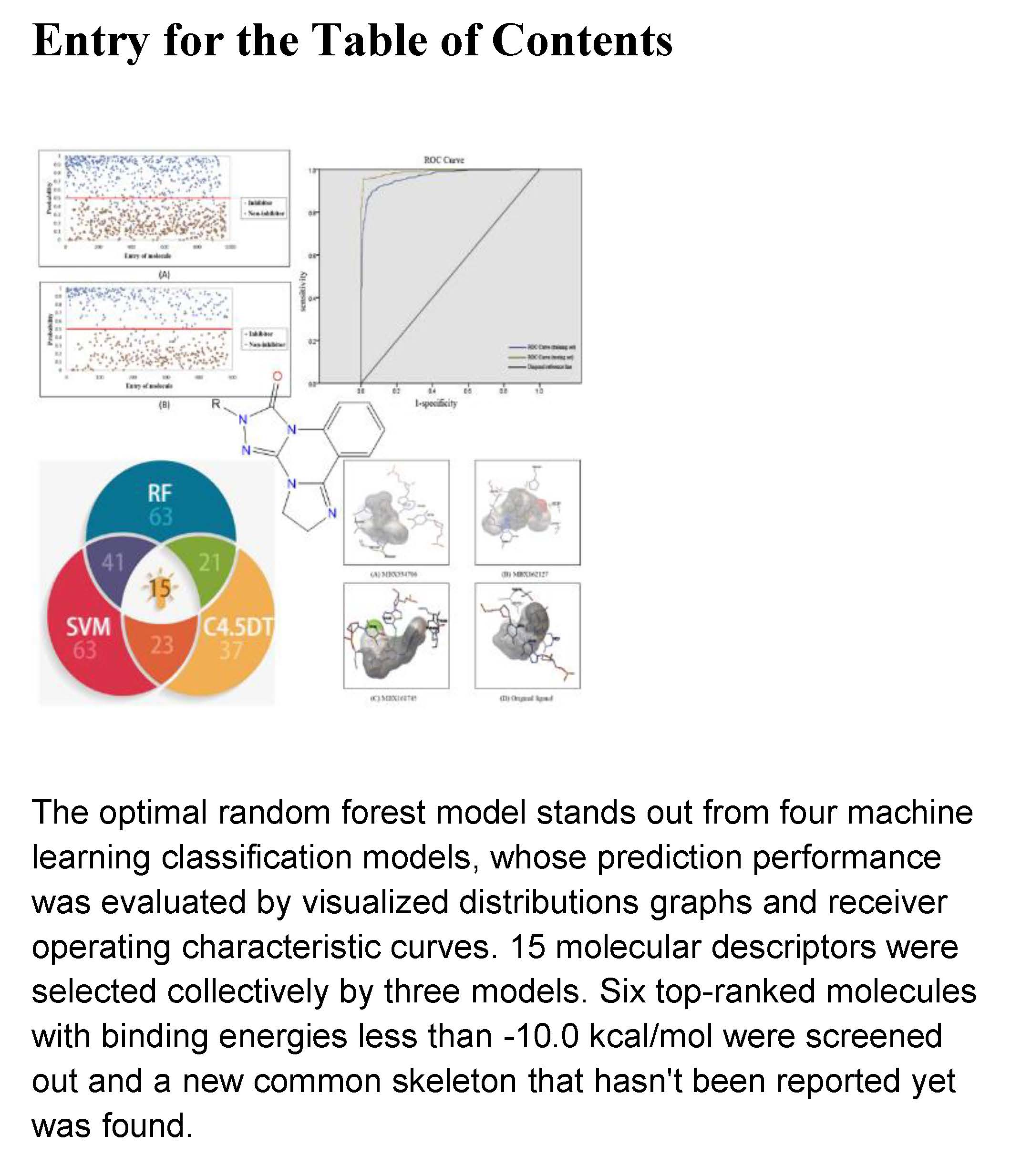
2.3. Optimization of RF Model Parameters
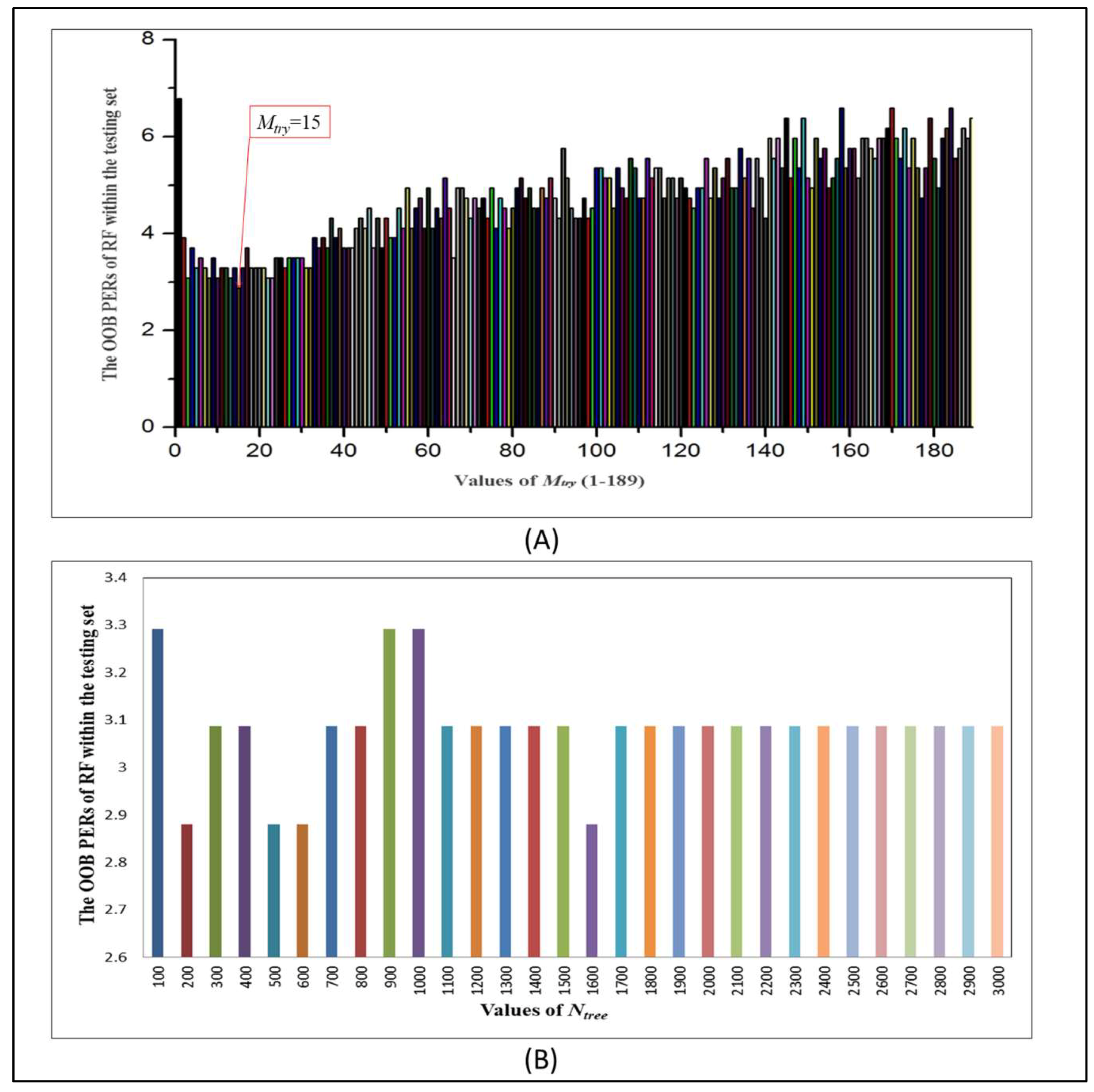
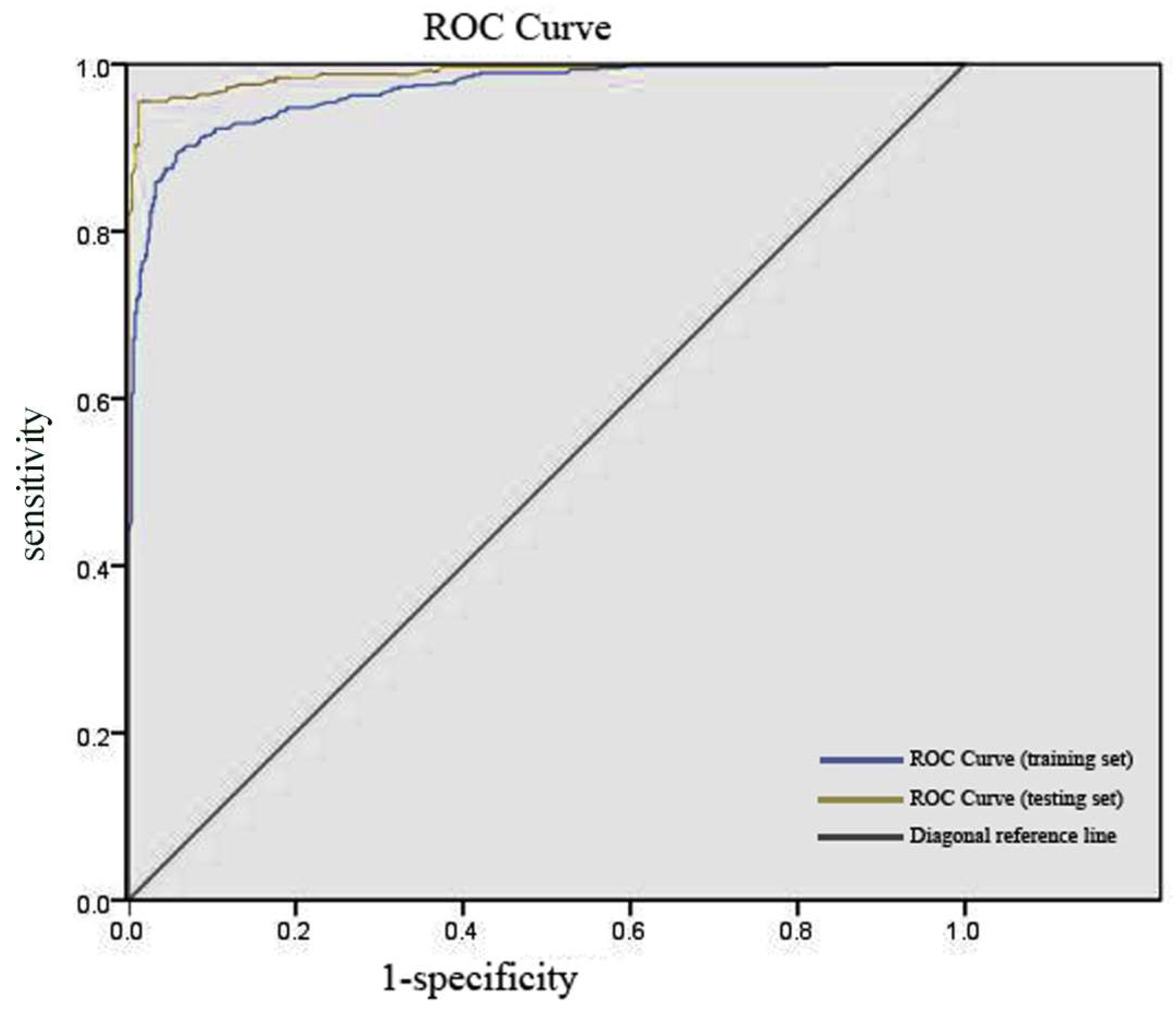
2.4. Evaluation of RF Optimal Model
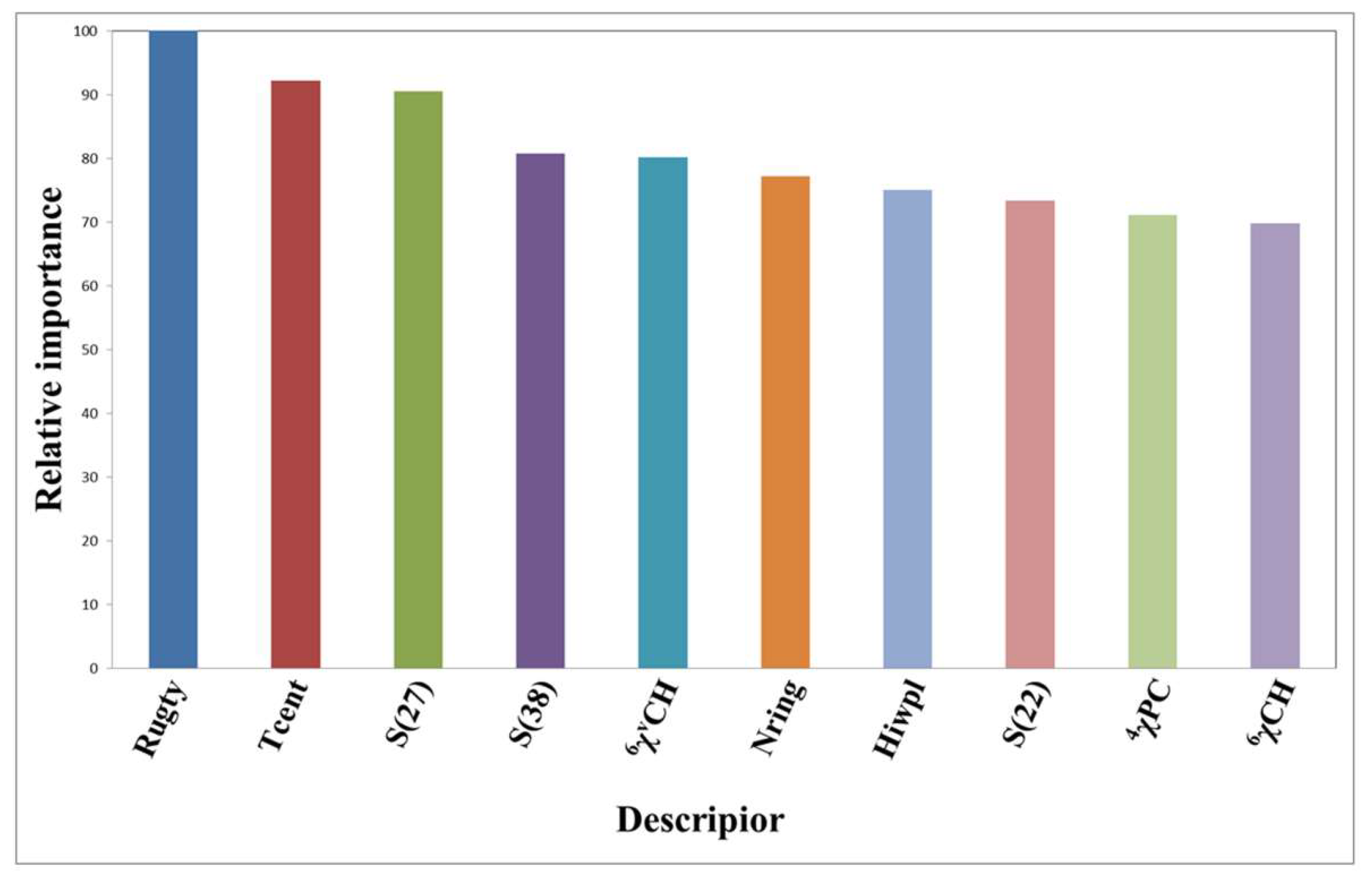
2.5. Features Selection
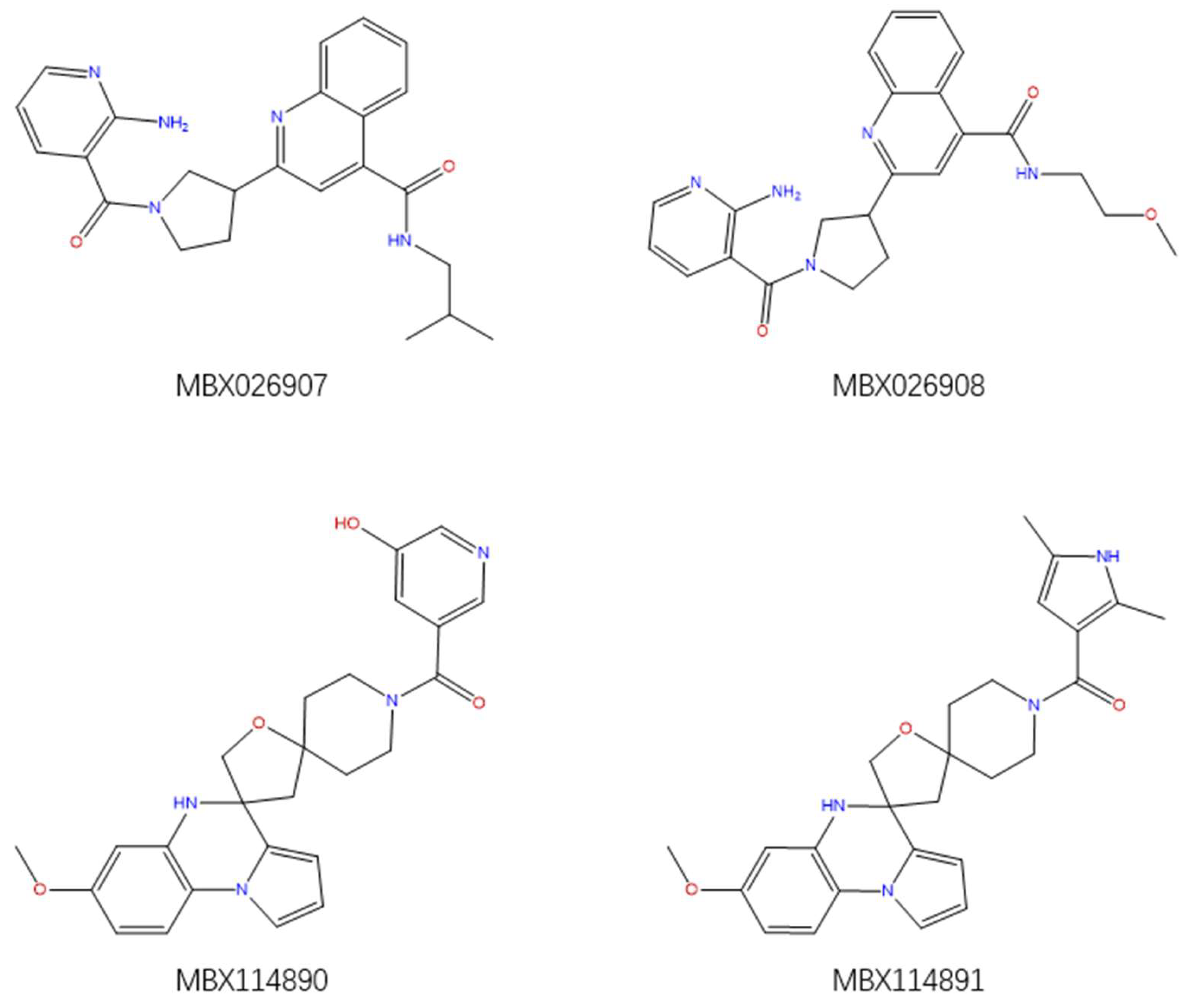
2.6. Virtual Screening
2.7. Docking Calculation
3. Materials and Methods
3.1. Data Sources
3.2. Molecular Descriptors
3.3. Machine Learning Methods
3.4. Virtual Screening
3.5. Molecular Docking
3.6. Targets Selection and Preparation
3.7. Ligands Preparation
4. Conclusion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wtorek, K.; Długosz, A.; Janecka, A. Drug resistance in topoisomerase-targeting therapy. Postep. Hig. Med. Dosw. 2018, 72, 1073–1083. [Google Scholar] [CrossRef]
- Corless, S.; Gilbert, N. Investigating DNA supercoiling in eukaryotic genomes. Brief. Funct. Genomics 2017, 16, 379–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, N.; Jinks-Robertson, S. The Top1 paradox: Friend and foe of the eukaryotic genome. DNA Repair 2017, 56, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Sun, Y.; Huang, S.-y.N.; Nitiss, J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C. Cellular roles of DNA topoisomerases: A molecular perspective. Nat. Rev. Mol. Cell Biol. 2002, 3, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C. DNA topoisomerases. Annu. Rev. Biochem. 1996, 65, 635–692. [Google Scholar] [CrossRef] [PubMed]
- Champoux, J.J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef]
- Stewart, L.; Redinbo, M.R.; Qiu, X.; Hol, W.G.J.; Champoux, J.J. A model for the mechanism of human topoisomerase I. Science 1998, 279, 1534–1541. [Google Scholar] [CrossRef]
- Champoux, J.J. Domains of human topoisomerase I and associated functions. In Progress in Nucleic Acid Research and Molecular Biology; Moldave, K., Ed.; Academic Press: Pittsburgh, PA, USA, 1998; Volume 60, pp. 111–132. [Google Scholar]
- Hevener, K.; Verstak, T.A.; Lutat, K.E.; Riggsbee, D.L.; Mooney, J.W. Recent developments in topoisomerase-targeted cancer chemotherapy. Acta Pharm. Sin. B 2018, 8, 844–861. [Google Scholar] [CrossRef]
- Capranico, G.; Marinello, J.; Chillemi, G. Type I DNA topoisomerases. J. Med. Chem. 2017, 60, 2169–2192. [Google Scholar] [CrossRef]
- Pommier, Y. Drugging topoisomerases: Lessons and challenges. ACS Chem. Biol. 2013, 8, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Reinhold, W.C.; Mergny, J.-L.; Liu, H.; Ryan, M.; Pfister, T.D.; Kinders, R.; Parchment, R.; Doroshow, J.; Weinstein, J.N.; Pommier, Y. Exon array analyses across the NCI-60 reveal potential regulation of TOP1 by transcription pausing at guanosine quartets in the first intron. Cancer Research 2010, 70, 2191–2203. [Google Scholar] [CrossRef] [PubMed]
- Pfister, T.D.; Reinhold, W.C.; Agama, K.; Gupta, S.; Khin, S.A.; Kinders, R.J.; Parchment, R.E.; Tomaszewski, J.E.; Doroshow, J.H.; Pommier, Y. Topoisomerase I levels in the NCI-60 cancer cell line panel determined by validated ELISA and microarray analysis and correlation with indenoisoquinoline sensitivity. Mol. Cancer Ther. 2009, 8, 1878–1884. [Google Scholar] [CrossRef] [PubMed]
- Khadka, D.B.; Park, S.; Jin, Y.; Han, J.; Kwon, Y.; Cho, W.-J. Design, synthesis, and biological evaluation of 1,3-diarylisoquinolines as novel topoisomerase I catalytic inhibitors. Eur. J. Med. Chem. 2018, 143, 200–215. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Ghosh, A.; Paul Chowdhuri, S.; Halder, N.; Rehman, I.; Sengupta, S.; Sahoo, K.C.; Rath, H.; Das, B.B. Neutral porphyrin derivative exerts anticancer activity by targeting cellular topoisomerase I (Top1) and promotes apoptotic cell death without stabilizing Top1-DNA cleavage complexes. J. Med. Chem. 2018, 61, 804–817. [Google Scholar] [CrossRef] [PubMed]
- Ryan, A.J.; Squires, S.; Strutt, H.L.; Johnson, R.T. Camptothecin cytotoxicity in mammalian cells is associated with the induction of persistent double strand breaks in replicating DNA. Nucleic Acids Res. 1991, 19, 3295–3300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pommier, Y. DNA topoisomerase I inhibitors: Chemistry, biology, and interfacial inhibition. Chem. Rev. 2009, 109, 2894–2902. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y. Topoisomerase I inhibitors: Camptothecins and beyond. Nat. Rev. Cancer 2006, 6, 789–802. [Google Scholar] [CrossRef]
- Pan, P.; Li, Y.; Yu, H.; Sun, H.; Hou, T. Molecular principle of topotecan resistance by topoisomerase I mutations through molecular modeling approaches. J. Chem. Inf. Model. 2013, 53, 997–1006. [Google Scholar] [CrossRef]
- Cinelli, M.A. Topoisomerase 1B poisons: Over a half-century of drug leads, clinical candidates, and serendipitous discoveries. Med. Res. Rev. 2018. [Google Scholar] [CrossRef]
- Staker, B.L.; Hjerrild, K.; Feese, M.D.; Behnke, C.A.; Burgin, A.B.; Stewart, L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc. Natl. Acad. Sci. 2002, 99, 15387–15392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, K.; Ren, Y.; Li, R. Combined pharmacophore-guided 3D-QSAR, molecular docking and molecular dynamics studies for evodiamine analogs as DNA topoisomerase I inhibitors. J. Taiwan Inst. Chem. Eng. 2017, 78, 81–95. [Google Scholar] [CrossRef]
- Thai, K.-M.; Nguyen, T.-Q.; Ngo, T.-D.; Tran, T.-D.; Huynh, T.-N.-P. A support vector machine classification model for benzo[c]phenathridine analogues with topoisomerase-I inhibitory activity. Molecules 2012, 17, 5690–5712. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Vapnik, V.N. The vicinal risk minimization principle and the SVMs. In The Nature of Statistical Learning Theory; Vapnik, V.N., Ed.; Springer: New York, NY, USA, 2000; pp. 267–290. [Google Scholar]
- Dhanabal, S.; Chandramathi, S. A review of various k-nearest neighbor query processing techniques. Int. J. Comput. Appl. 2011, 31, 14–22. [Google Scholar]
- Rokach, L.; Maimon, O. Top-down induction of decision trees classifiers - a survey. IEEE Trans. Syst. Man Cybern. Part C Appl. Rev. 2005, 35, 476–487. [Google Scholar] [CrossRef]
- Dong, G.; Fang, Y.; Liu, Y.; Liu, N.; Wu, S.; Zhang, W.; Sheng, C. Design, synthesis and evaluation of 4-substituted anthra [2,1-c][1,2,5]thiadiazole-6,11-dione derivatives as novel non-camptothecin topoisomerase I inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 1929–1933. [Google Scholar] [CrossRef]
- Li, B.; Hu, L.; Xue, Y.; Yang, M.; Huang, L.; Zhang, Z.; Liu, J.; Deng, G. Prediction of matrix metal proteinases-12 inhibitors by machine learning approaches. J. Biomol. Struct. Dyn. 2019, 37, 2627–2640. [Google Scholar] [CrossRef]
- Li, B.-K.; Cong, Y.; Yang, X.-G.; Xue, Y.; Chen, Y.-Z. In silico prediction of spleen tyrosine kinase inhibitors using machine learning approaches and an optimized molecular descriptor subset generated by recursive feature elimination method. Comput. Biol. Med. 2013, 43, 395–404. [Google Scholar] [CrossRef]
- Yang, X.-G.; Lv, W.; Chen, Y.-Z.; Xue, Y. In silico prediction and screening of γ-secretase inhibitors by molecular descriptors and machine learning methods. J. Comput. Chem. 2010, 31, 1249–1258. [Google Scholar]
- Trott, O.; Olson, A.J. Autodock vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K. Data clustering: 50 years beyond K-means. Pattern Recognit. Lett. 2010, 31, 651–666. [Google Scholar] [CrossRef]
- Xue, Y.; Yap, C.W.; Sun, L.Z.; Cao, Z.W.; Wang, J.F.; Chen, Y.Z. Prediction of P-glycoprotein substrates by a support vector machine approach. J. Chem. Inf. Comp. Sci. 2004, 44, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of Topoisomerase I Inhibitors are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Sets | Number of Compounds | D(A) Values |
|---|---|---|
| The whole data set in this work | 1457 | 0.4716 |
| The training set in this work | 971 | 0.4824 |
| The testing set in this work | 486 | 0.4494 |
| The external validation set in recent literature [29] | 55 | 0.0936 |
| Molecules picked out by virtual screening with RF scores greater than 0.7 in this work | 67 | 0.1281 |
| The HDAC_Library in the Maybridge database | 4107 | 0.1550 |
| The whole data set in our previous work [30] | 283 | 0.3537 |
| Methods | Parameters | Top1is | non-Top1is | Q (%) | MCC | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FN | SE (%) | TN | FP | SP (%) | ||||
| RF | Mtry = 15 | 237 | 11 | 95.56 | 235 | 3 | 98.73 | 97.12 | 0.9429 |
| SVM | σ = 0.2 | 228 | 20 | 91.94 | 226 | 12 | 94.96 | 93.41 | 0.8688 |
| k-NN | k = 6pr | 223 | 25 | 89.92 | 221 | 17 | 92.86 | 91.36 | 0.8277 |
| C4.5 DT | / | 223 | 26 | 89.56 | 212 | 25 | 89.84 | 89.70 | 0.7939 |
| Name | Binding Energy kcal/mol | Structural Formula |
|---|---|---|
| MBX534706 | −11.4 |  |
| MBX162127 | −10.5 |  |
| MBX209152 | −10.3 |  |
| MBX161748 | −10.2 |  |
| MBX161745 | −10.1 |  |
| MBX190732 | −10.1 |  |
| The original ligand | −9.0 |  |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, B.; Kang, X.; Zhao, D.; Zou, Y.; Huang, X.; Wang, J.; Zhang, C. Machine Learning Models Combined with Virtual Screening and Molecular Docking to Predict Human Topoisomerase I Inhibitors. Molecules 2019, 24, 2107. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24112107
Li B, Kang X, Zhao D, Zou Y, Huang X, Wang J, Zhang C. Machine Learning Models Combined with Virtual Screening and Molecular Docking to Predict Human Topoisomerase I Inhibitors. Molecules. 2019; 24(11):2107. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24112107
Chicago/Turabian StyleLi, Bingke, Xiaokang Kang, Dan Zhao, Yurong Zou, Xudong Huang, Jiexue Wang, and Chenghua Zhang. 2019. "Machine Learning Models Combined with Virtual Screening and Molecular Docking to Predict Human Topoisomerase I Inhibitors" Molecules 24, no. 11: 2107. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24112107