
Computational Investigation of Bisphosphate Inhibitors of 3-Deoxy-d-manno-octulosonate 8-phosphate Synthase


Abstract
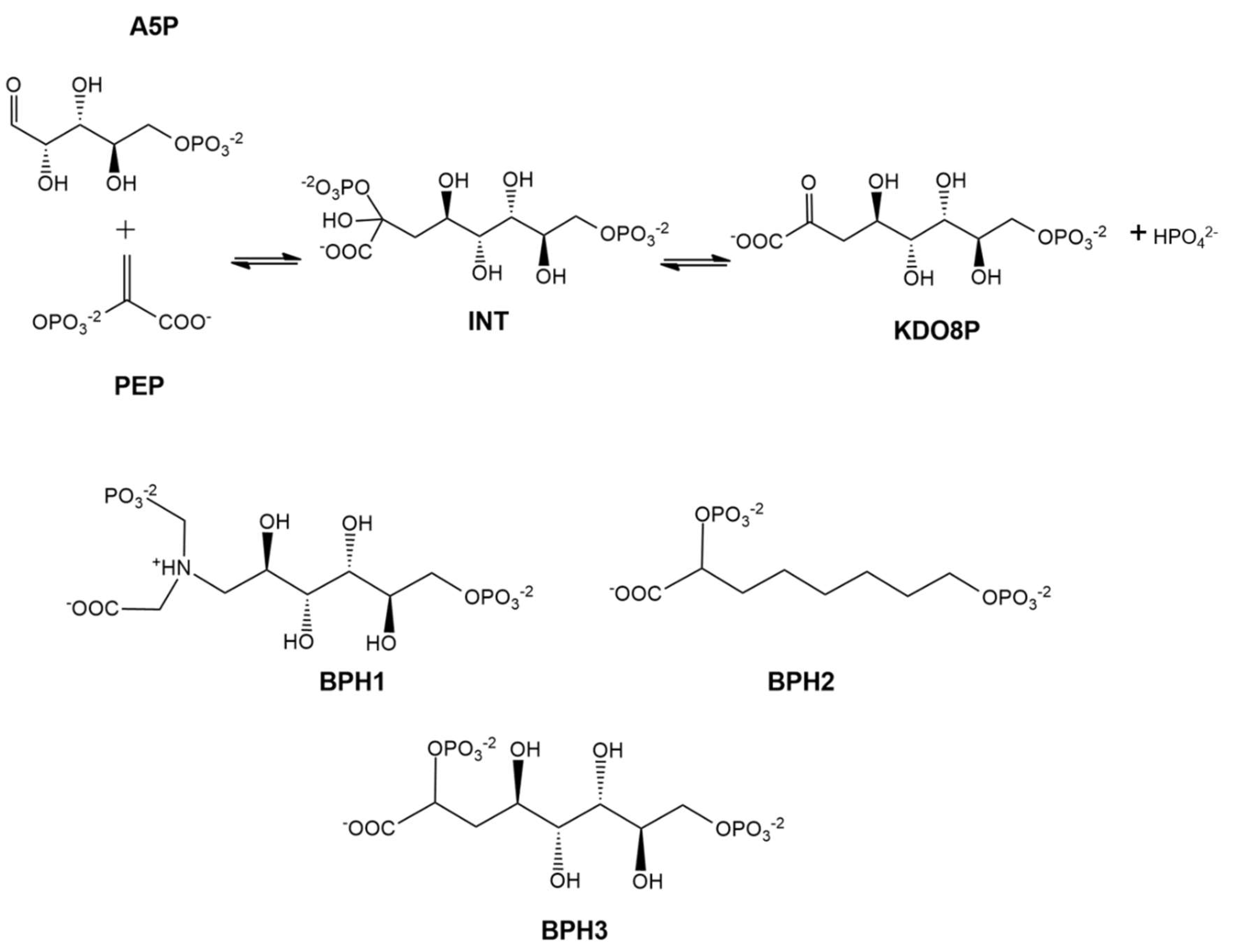
:1. Introduction
2. Results and Discussion
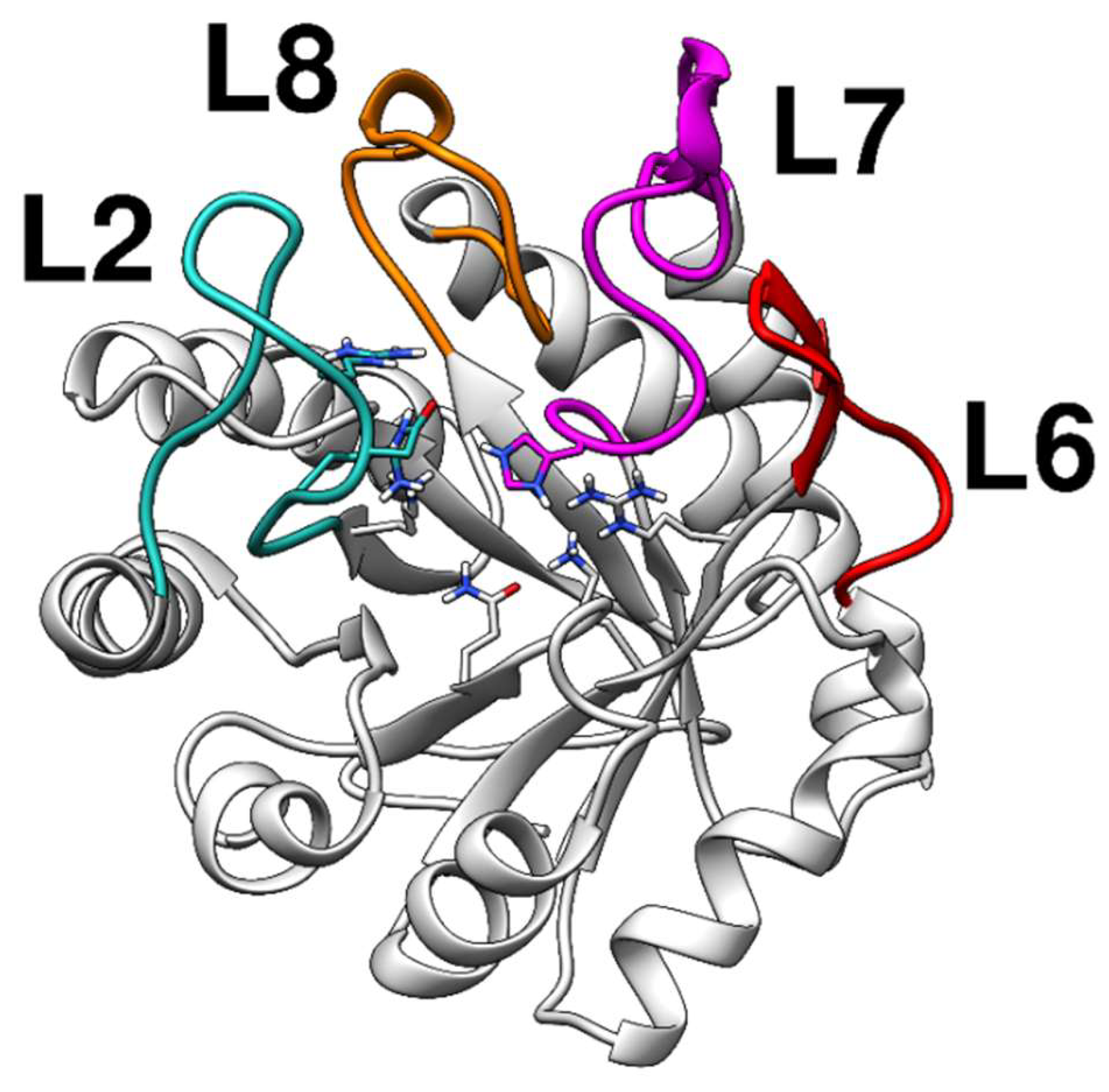
2.1. Homology Modeling
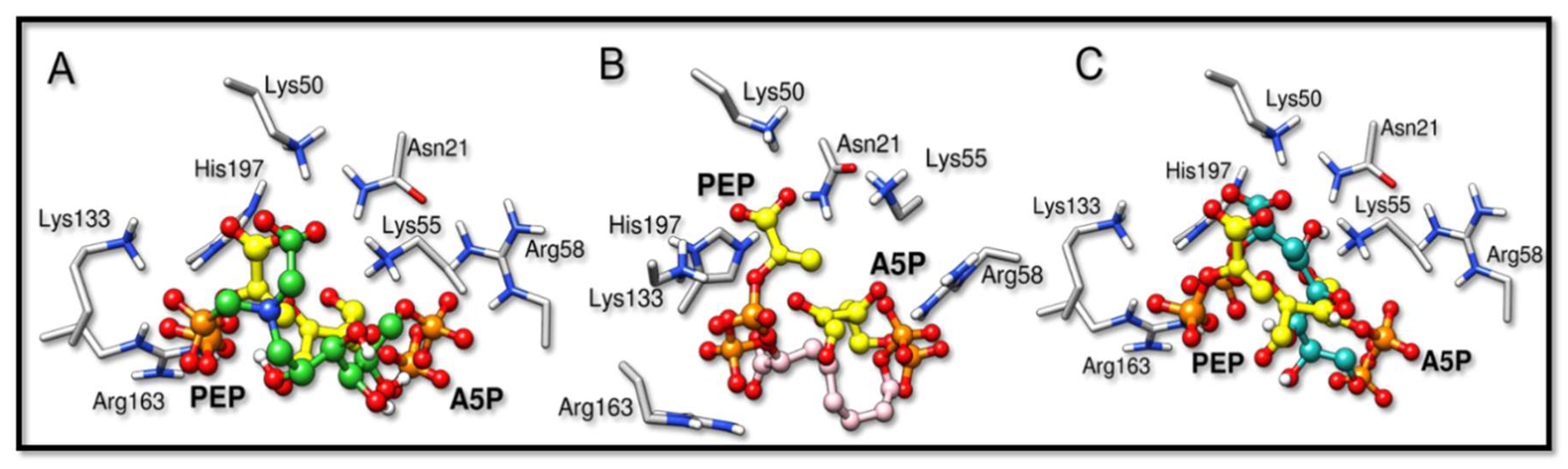
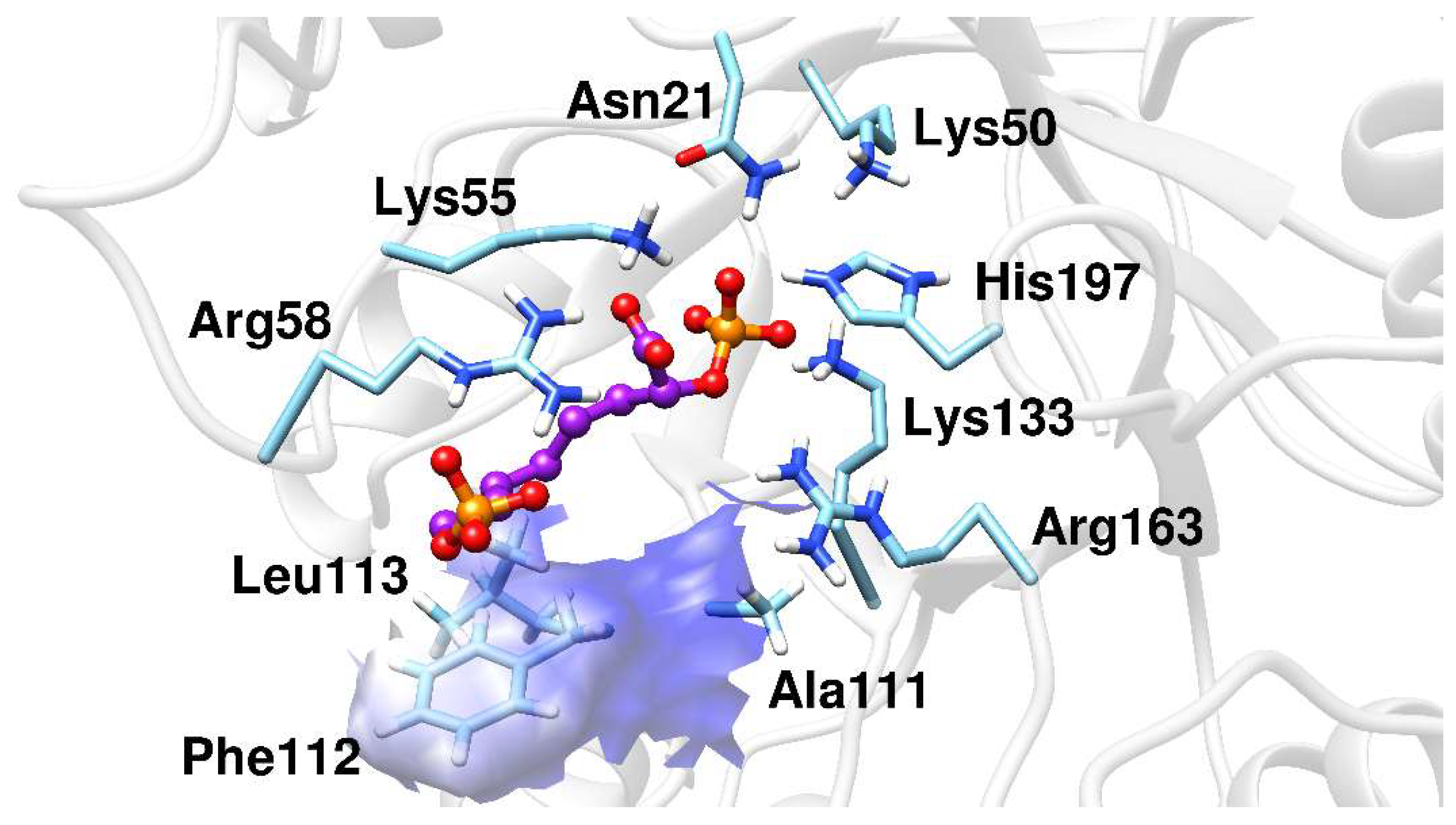
2.2. Docking Analysis
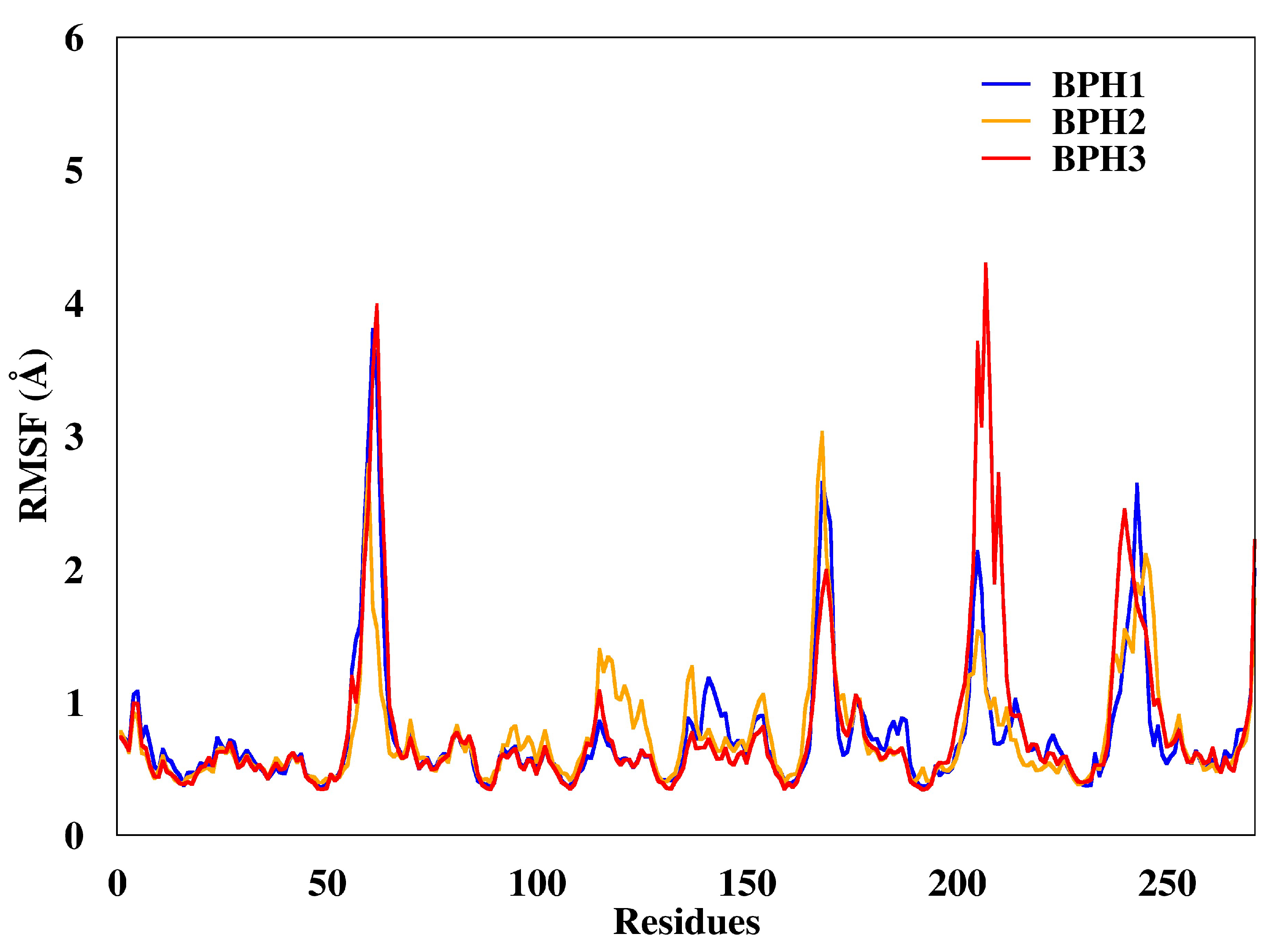
2.3. Molecular Dynamics Simulations
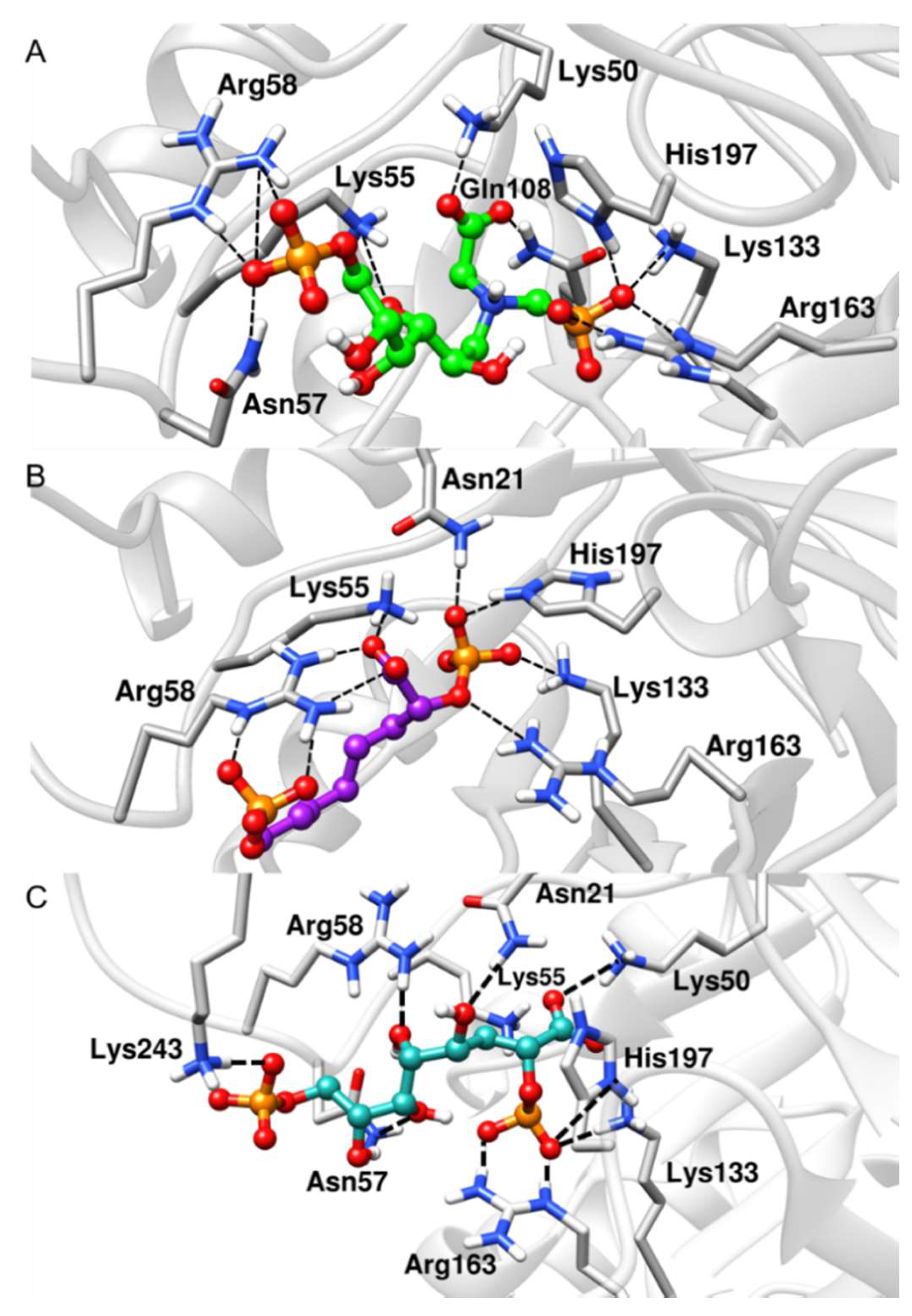
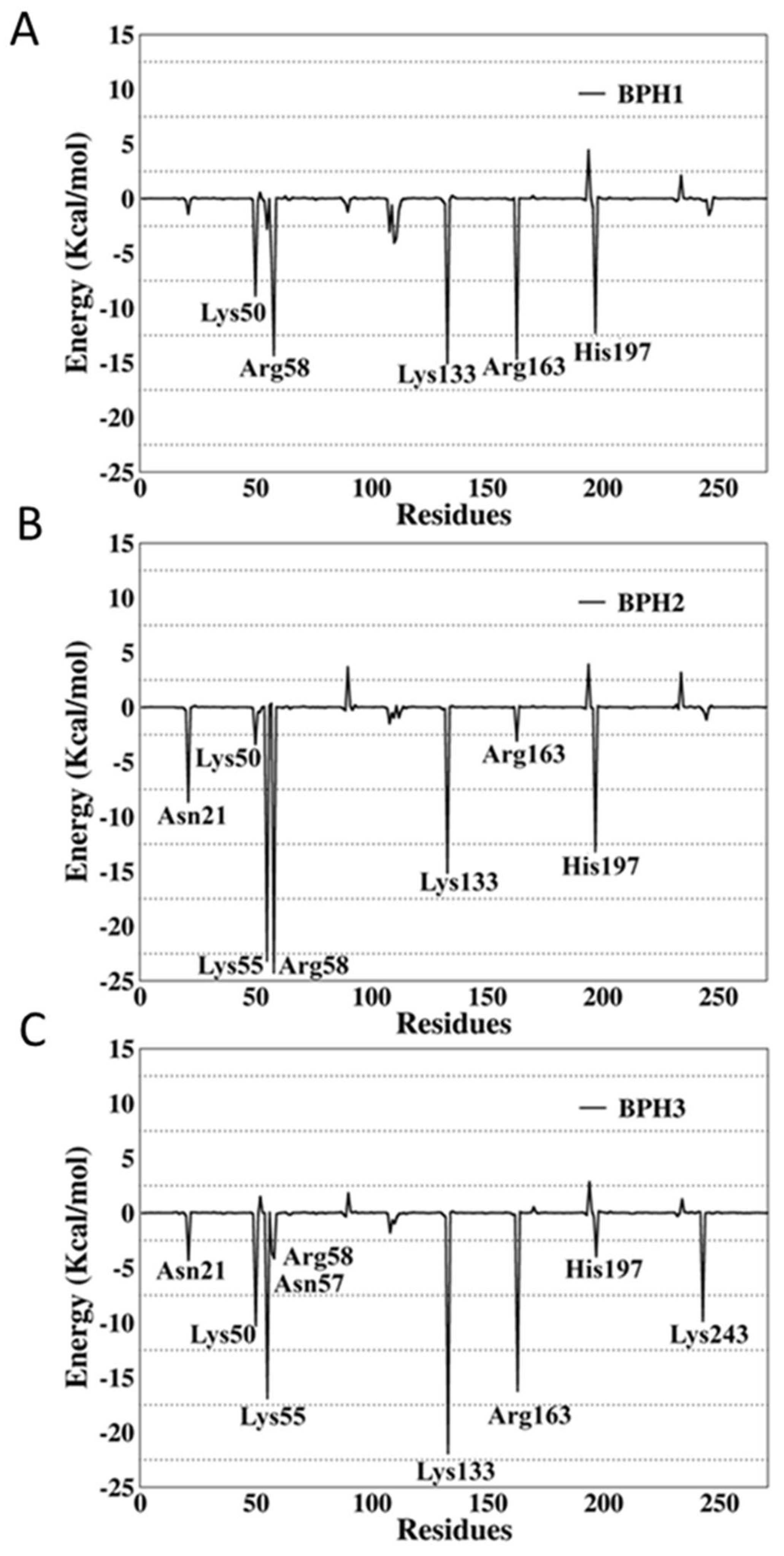
2.4. Binding Free-Energy Analysis
3. Materials and Methods
3.1. Homology Modeling
3.2. Molecular Docking Study
3.3. MD Simulations
3.4. Free Energy Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Raetz, C.R.H.; Whitfield, C. Lipopolysaccharide Endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, P.H. Purification and characterization of 3-deoxy-d-manno-octulosonate 8-phosphate synthetase from Escherichia coli. J. Bacteriol. 1980, 141, 635–644. [Google Scholar] [PubMed]
- Brennan, P.J.; Nikaido, H. The Envelope of Mycobacteria. Annu. Rev. Biochem. 1995, 64, 29–63. [Google Scholar] [CrossRef] [PubMed]
- Raetz, C. Biochemistry of Endotoxins. Annu. Rev. Biochem. 1990, 59, 129–170. [Google Scholar] [CrossRef] [PubMed]
- Rick, P.D.; Young, D.A. Isolation and characterization of a temperature-sensitive lethal mutant of Salmonella typhimurium that is conditionally defective in 3-deoxy-d-manno-octulosonate-8-phosphate synthesis. J. Bacteriol. 1982, 150, 447–455. [Google Scholar] [PubMed]
- Maldonado, R.F.; Sá-Correia, I.; Valvano, M.A. Lipopolysaccharide modification in Gram-negative bacteria during chronic infection. FEMS Microbiol. Rev. 2016, 40, 480–493. [Google Scholar] [CrossRef] [PubMed]
- Raetz, C.R.H.; Guan, Z.; Ingram, B.O.; Six, D.A.; Song, F.; Wang, X.; Zhao, J. Discovery of new biosynthetic pathways: The lipid A story. J. Lipid Res. 2009, 50, S103–S108. [Google Scholar] [CrossRef] [PubMed]
- Cipolla, L.; Polissi, A.; Airoldi, C.; Galliani, P.; Sperandeo, P.; Nicotra, F. The Kdo Biosynthetic Pathway Toward OM Biogenesis as Target in Antibacterial Drug Design and Development. Curr. Drug Discov. Technol. 2009, 6, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Duewel, H.S.; Radaev, S.; Wang, J.; Woodard, R.W.; Gatti, D.L. Substrate and metal complexes of 3-deoxy-d-manno-octulosonate-8-phosphate synthase from Aquifex aeolicus at 1.9-Å Resolution: Implications for the condensation mechanism. J. Biol. Chem. 2001, 276, 8393–8402. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, F.C.; Cookson, T.V.M.; Jameson, G.B.; Parker, E.J. Reversing Evolution: Re-establishing Obligate Metal Ion Dependence in a Metal-independent KDO8P Synthase. J. Mol. Biol. 2009, 390, 646–661. [Google Scholar] [CrossRef]
- Wagner, T.; Kretsinger, R.H.; Bauerle, R.; Tolbert, W.D. 3-deoxy-d-manno-octulosonate-8-phosphate synthase from Escherichia coli. Model of binding of phosphoenolpyruvate and D-arabinose-5-phosphate. J. Mol. Biol. 2000, 301, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Asojo, O.; Friedman, J.; Adir, N.; Belakhov, V.; Shoham, Y.; Baasov, T. Crystal structures of KDOP synthase in its binary complexes with the substrate phosphoenolpyruvate and with a mechanism-based inhibitor. Biochemistry 2001, 40, 6326–6334. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.; Cochrane, F.C.; Patchett, M.L.; Parker, E.J. Arabinose 5-phosphate analogues as mechanistic probes for Neisseria meningitidis 3-deoxy-d-manno-octulosonate 8-phosphate synthase. Bioorg. Med. Chem. 2008, 16, 9830–9836. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Duewel, H.S.; Woodard, R.W.; Gatti, D.L. Structures of Aquifex aeolicus KDO8P synthase in complex with R5P and PEP, and with a bisubstrate inhibitor: Role of active site water in catalysis. Biochemistry 2001, 40, 15676–15683. [Google Scholar] [CrossRef] [PubMed]
- Birck, M.R.; Woodard, R.W. Aquifex aeolicus 3-deoxy-d-manno-2-octulosonic acid 8-phosphate synthase: A new class of KDO 8-P synthase? J. Mol. Evol. 2001, 52, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Duewel, H.S.; Woodard, R.W. A metal bridge between two enzyme families: 3-deoxy-d-manno-octulosonate-8-phosphate synthase from Aquifex aeolicus requires a divalent metal for activity. J. Biol. Chem. 2000, 275, 22824–22831. [Google Scholar] [CrossRef] [PubMed]
- Shulami, S.; Yaniv, O.; Rabkin, E.; Shoham, Y.; Baasov, T. Cloning, expression, and biochemical characterization of 3-deoxy-d-manno-2-octulosonate-8-phosphate (KDO8P) synthase from the hyperthermophilic bacterium Aquifex pyrophilus. Extremophiles 2003, 7, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Dotson, G.D.; Dua, R.K.; Clemens, J.C.; Wooten, E.W.; Woodard, R.W. Overproduction and one-step purification of Escherichia coli 3-Deoxy-d-manno-octulosonic acid 8-phosphate synthase and oxygen transfer studies during catalysis using isotopic-shifted heteronuclear NMR. J. Biol. Chem. 1995, 270, 13698–13705. [Google Scholar] [CrossRef] [PubMed]
- Dotson, G.D.; Nanjappan, P.; Reily, M.D.; Woodard, R.W. Stereochemistry of 3-Deoxyoctulosonate 8-Phosphate Synthase. Biochemistry 1993, 32, 12392–12397. [Google Scholar] [CrossRef] [PubMed]
- Kohen, A.; Berkovich, R.; Belakhov, V.; Baasov, T. Stereochemistry of the KDO8P synthase. An efficient synthesis of the 3-fluoro analogues of KDO8P. Bioorganic Med. Chem. Lett. 1993, 3, 1577–1582. [Google Scholar] [CrossRef]
- Kona, F.; Xu, X.; Martin, P.; Kuzmic, P.; Gatti, D.L. Structural and mechanistic changes along an engineered path from metallo to nonmetallo 3-deoxy-d-manno-octulosonate 8-phosphate synthases. Biochemistry 2007, 46, 4532–4544. [Google Scholar] [CrossRef] [PubMed]
- Harrison, A.N.; Reichau, S.; Parker, E.J. Synthesis and evaluation of tetrahedral intermediate mimic inhibitors of 3-deoxy-d-manno-octulosonate 8-phosphate synthase. Bioorganic Med. Chem. Lett. 2012, 22, 907–911. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allison, T.M.; Hutton, R.D.; Jiao, W.; Gloyne, B.J.; Nimmo, E.B.; Jameson, G.B.; Parker, E.J. An Extended β7α7 Substrate-Binding Loop Is Essential for Efficient Catalysis by 3-Deoxy-d-manno-Octulosonate 8-Phosphate Synthase. Biochemistry 2011, 50, 9318–9327. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, R.; Christensen, M.H. MolDock: A New Technique for High-Accuracy Molecular Docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef] [PubMed]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Tsipori, H.; Baasov, T. Synthesis and evaluation of putative oxocarbenium intermediate mimic in the KDO8P synthase-catalyzed reaction as a tool for the design of potent inhibitors for lipopolysaccharide biosynthesis. Bioorg. Med. Chem. Lett. 1997, 7, 2469–2472. [Google Scholar] [CrossRef]
- Kona, F.; Tao, P.; Martin, P.; Xu, X.; Gatti, D.L. Electronic Structure of the Metal Center in the Cd2+, Zn2+, and Cu2+ Substituted Forms of KDO8P Synthase: Implications for Catalysis. Biochemistry 2009, 48, 3610–3630. [Google Scholar] [CrossRef]
- Grison, C.; Petek, S.; Finance, C.; Coutrot, P. Synthesis and antibacterial activity of mechanism-based inhibitors of KDO8P synthase and DAH7P synthase. Carbohydr. Res. 2005, 340, 529–537. [Google Scholar] [CrossRef]
- Xu, X.; Wang, J.; Grison, C.; Petek, S.; Coutrot, P.; Birck, M.; Woodard, R.; Gatti, D. Structure-Based Design of Novel Inhibitors of 3-Deoxy-d-manno-octulosonate 8-Phosphate Synthase. Drug Des. Discov. 2003, 18, 91–99. [Google Scholar] [CrossRef]
- Du, S.; Faiger, H.; Belakhov, V.; Baasov, T. Towards the development of novel antibiotics: Synthesis and evaluation of a mechanism-based inhibitor of Kdo8P synthase. Bioorganic Med. Chem. 1999, 7, 2671–2682. [Google Scholar] [CrossRef]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinforma 2006, 22, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Vainer, R.; Belakhov, V.; Rabkin, E.; Baasov, T.; Adir, N. Crystal Structures of Escherichia coli KDO8P Synthase Complexes Reveal the Source of Catalytic Irreversibility. J. Mol. Biol. 2005, 351, 641–652. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Morris, A.L.; MacArthur, M.W.; Hutchinson, E.G.; Thornton, J.M. Stereochemical quality of protein structure coordinates. Proteins Struct. Funct. Bioinform. 1992, 12, 345–364. [Google Scholar] [CrossRef]
- Melo, F.; Devos, D.; Depiereux, E.; Feytmans, E. ANOLEA: A www Server to Assess Protein Structures. In Proceedings of the 5th International Conference on Intelligent Systems for Molecular Biology; AAAI Press: Halkidiki, Greece, 1997; pp. 187–190. [Google Scholar]
- Benkert, P.; Schwede, T.; Tosatto, S.C.E. QMEANclust: Estimation of protein model quality by combining a composite scoring function with structural density information. BMC Struct. Biol. 2009, 9, 35. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- De Azevedo, W.F. MolDock applied to structure-based virtual screening. Curr. Drug Targets 2010, 11, 327–334. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A. V H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, V.B.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian09; Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Naïm, M.; Bhat, S.; Rankin, K.N.; Dennis, S.; Chowdhury, S.F.; Siddiqi, I.; Drabik, P.; Sulea, T.; Bayly, C.I.; Jakalian, A.; et al. Solvated Interaction Energy (SIE) for Scoring Protein−Ligand Binding Affinities. 1. Exploring the Parameter Space. J. Chem. Inf. Model. 2007, 47, 122–133. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KDO8P Synthase (N. Meningitidis) | |||||||
|---|---|---|---|---|---|---|---|
| BPH1 (P1) | BPH2 (P1) | BPH3 (P1) | BPH1 (P2) | BPH2 (P2) | BPH3 (P2) | ||
| Arg58 (NH2) | 2.8 (± 0.1) | 2.8 (± 0.1) | 5.8 (± 0.1) | Lys133 (NZ) | 2.8 (± 0.1) | 2.8 (± 0.1) | 2.7 (± 0.1) |
| Asn57 (ND2) | 2.9 (± 0.1) | 7.6 (± 0.1) | 6.7 (± 0.1) | Arg163 (NE) | 2.7 (± 0.1) | 3.2 (± 0.2) | 2.8 (± 0.1) |
| His197 (NE2) | 2.7 (± 0.1) | 2.7 (± 0.1) | 2.7 (± 0.1) | ||||
| BPH1 | BPH2 | BPH3 | |
|---|---|---|---|
| Experimental | −8.82 a,b | −7.0 a,c | (n/d) |
| MM/GBSA | −96.07 | −71.72 | −82.96 |
| MM/PBSA | −107.09 | −82.64 | −110.73 |
| SIE | −13.53 | −13.49 | −14.37 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Araújo, J.d.O.; dos Santos, A.M.; Lameira, J.; Alves, C.N.; Lima, A.H. Computational Investigation of Bisphosphate Inhibitors of 3-Deoxy-d-manno-octulosonate 8-phosphate Synthase. Molecules 2019, 24, 2370. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24132370
Araújo JdO, dos Santos AM, Lameira J, Alves CN, Lima AH. Computational Investigation of Bisphosphate Inhibitors of 3-Deoxy-d-manno-octulosonate 8-phosphate Synthase. Molecules. 2019; 24(13):2370. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24132370
Chicago/Turabian StyleAraújo, Jéssica de Oliveira, Alberto Monteiro dos Santos, Jerônimo Lameira, Cláudio Nahum Alves, and Anderson Henrique Lima. 2019. "Computational Investigation of Bisphosphate Inhibitors of 3-Deoxy-d-manno-octulosonate 8-phosphate Synthase" Molecules 24, no. 13: 2370. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24132370